Chitosan-GPTMS-Silica Hybrid Mesoporous Aerogels for Bone Tissue Engineering

 ,
,  , , , , ,
, , , , ,
Abstract
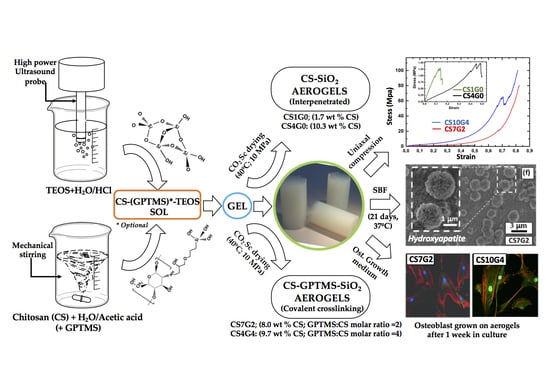
1. Introduction
2. Materials and Methods
2.1. Materials
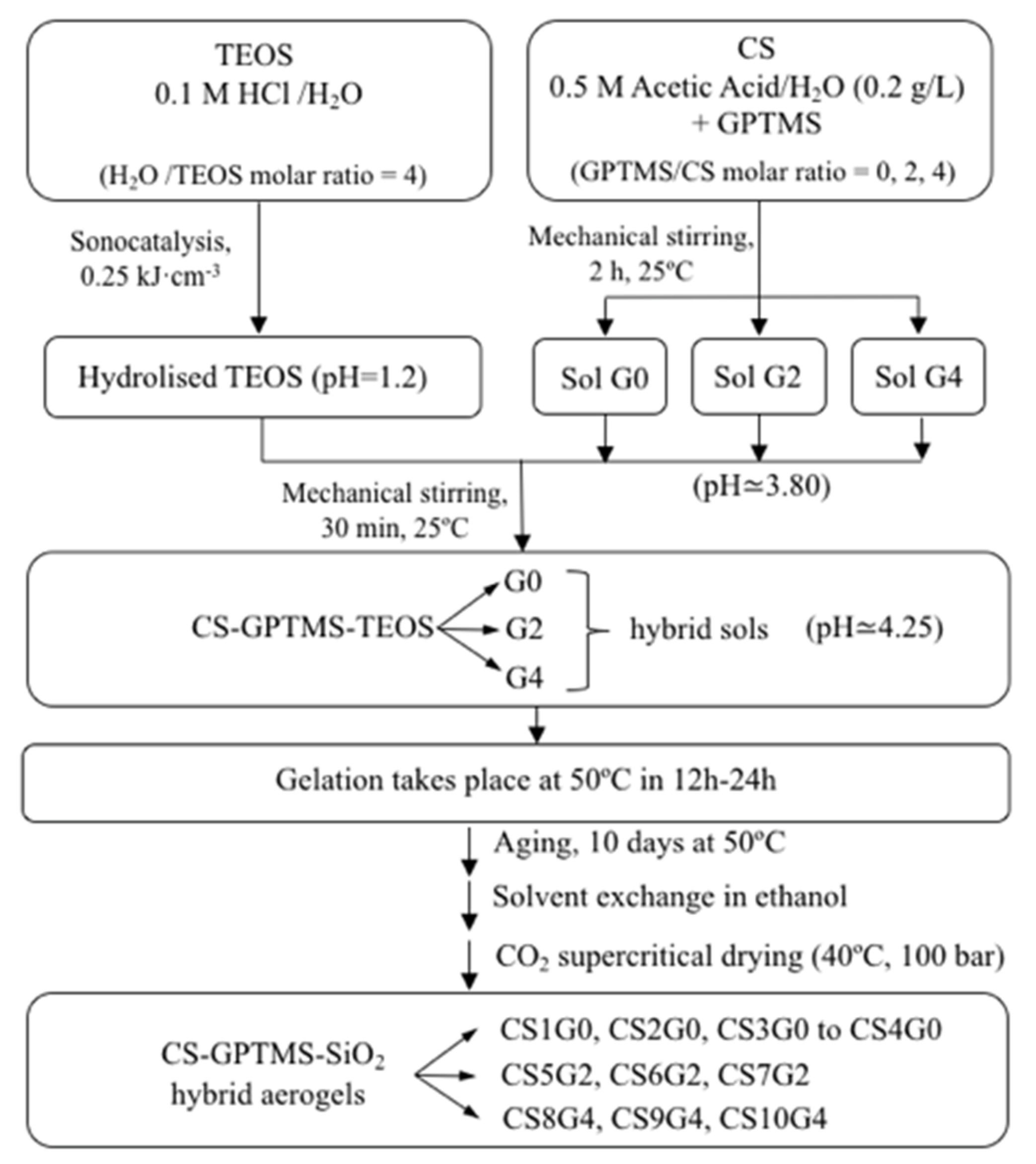
2.2. Synthesis of TEOS and CS-GPTMS Precursor Sols
2.3. Synthesis of CS-GPTMS-Silica Hybrid Aerogels Monolith
2.4. Physical and Textural Characterisation
2.5. Thermal Characterization
2.6. FT-IR Spectroscopy
2.7. Swelling Behavior-PBS Swelling Capacity
2.8. Swelling Kinetics
2.9. Mechanical Properties
2.10. In Vitro Bioactivity
2.11. In Vitro Biocompatibility
2.11.1. Cell Morphology and Spreading
2.11.2. Actin Cytoskeletal Organization and Vinculin Expression
2.11.3. Confocal Examination
2.11.4. Image Analysis
3. Results and Discussion
3.1. Synthesis of CS-GPTMS-Silica Hybrid Aerogels
3.2. Bulk Density and Textural Properties
3.3. Fourier Transformed Infra-Red (FTIR) Spectral Analysis
3.4. Thermogravimetric Analysis (TGA)
3.5. Swelling Behaviour-PBS Absorption
3.6. Absorption Kinetics
3.7. Mechanical Properties-Uniaxial Compression
3.8. In Vitro Bioactivity Experiments
3.9. Osteoblast Response In Vitro
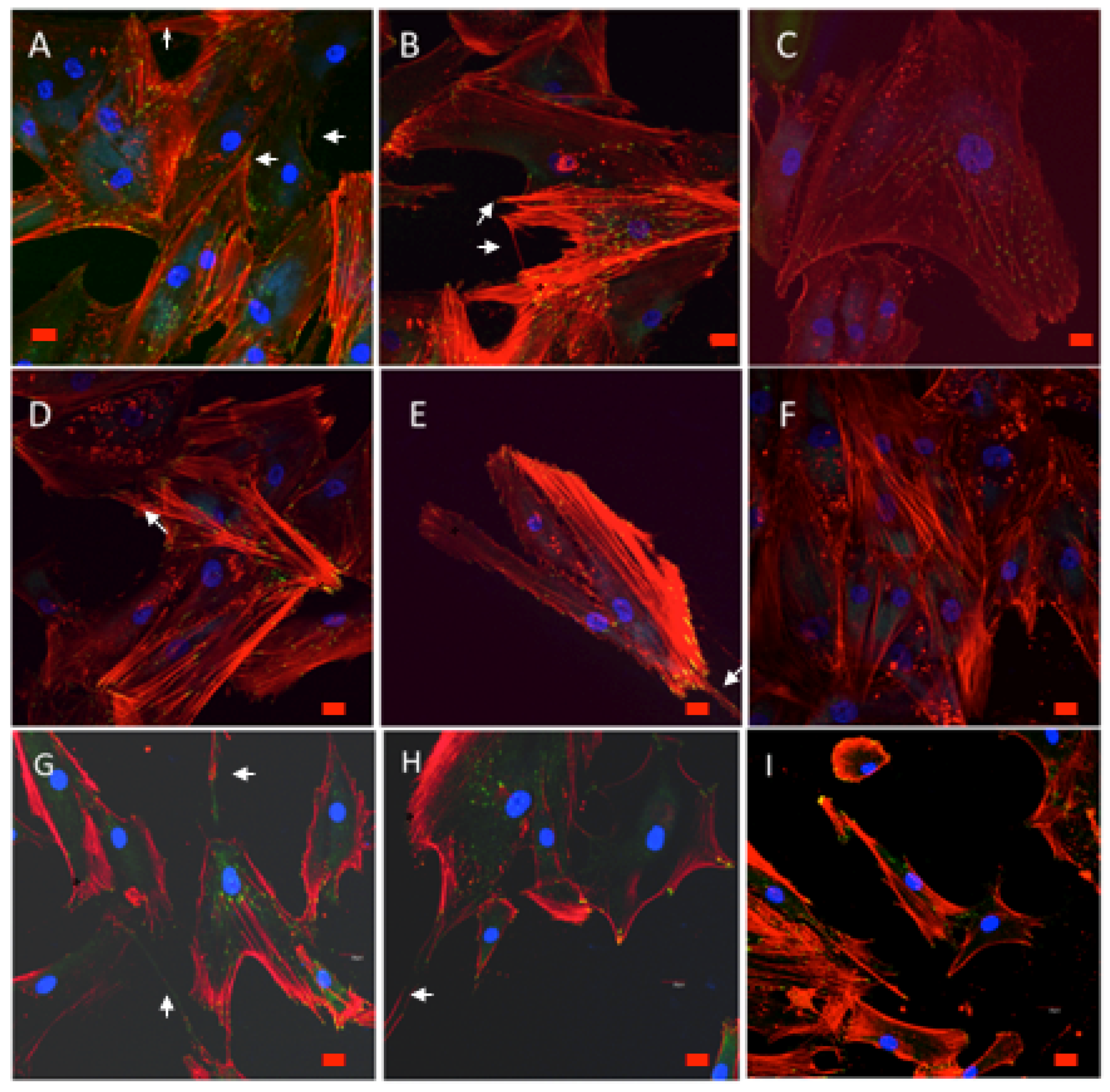
3.9.1. Cell Morphology and Spreading
3.9.2. Cytoskeletal Organization and Focal Adhesions
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- García-González, C.A.; Budtova, T.; Durães, L.; Erkey, C.; Del Gaudio, P.; Gurikov, P.; Koebel, M.; Liebner, F.; Neagu, M.; Smirnova, I. An opinion paper on aerogels for biomedical and environmental applications. Molecules 2019, 24, 1815. [Google Scholar] [CrossRef]
- Maleki, H.; Durães, L.; García-González, C.A.; del Gaudio, P.; Portugal, A.; Mahmoudi, M. Synthesis and biomedical applications of aerogels: Possibilities and challenges. Adv. Colloid Interface Sci. 2016, 236, 1–27. [Google Scholar] [CrossRef]
- Zhao, S.; Malfait, W.J.; Guerrero-Alburquerque, N.; Koebel, M.M.; Nyström, G. Biopolymer Aerogels and Foams: Chemistry, Properties, and Applications. Angew. Chem. Int. Ed. 2018, 57, 7580–7608. [Google Scholar] [CrossRef]
- Soorbaghi, F.P.; Isanejad, M.; Salatin, S.; Ghorbani, M.; Jafari, S.; Derakhshankhah, H. Bioaerogels: Synthesis approaches, cellular uptake, and the biomedical applications. Biomed. Pharmacother. 2019, 111, 964–975. [Google Scholar] [CrossRef]
- Woh, K.K.; Yussof, R. Hybrid Green Aerogels: Processing and Morphology. In Biobased Aerogels: Polysaccharide and Protein-Based Materials; Thomas, S., Pothan, L.A., Mavelil-Sam, R., Eds.; Royal Society of Chemistry: London, UK, 2018; pp. 103–128. ISBN 978-1-78262-765-4. [Google Scholar]
- Nita, L.E.; Ghilan, A.; Rusu, A.G.; Neamtu, I.; Chiriac, A.P. New Trends in Bio-Based Aerogels. Pharmaceutics 2020, 12, 449. [Google Scholar] [CrossRef]
- Alhwaige, A.A.; Agag, T.; Ishida, H.; Qutubuddin, S. Biobased chitosan hybrid aerogels with superior adsorption: Role of graphene oxide in CO2 capture. RSC Adv. 2013, 3, 16011–16020. [Google Scholar] [CrossRef]
- Hasan, M.; Gopakumar, D.A.; Arumughan, V.; Pottathara, Y.B.; Seantier, B.; Nzihou, A.; Thomas, S.; Rizal, S.; Hps, A.K. Robust Superhydrophobic Cellulose Nanofiber Aerogel for Multifunctional. Polymers 2019, 11, 495. [Google Scholar] [CrossRef]
- Ulker, Z.; Erkey, C. An emerging platform for drug delivery: Aerogel based systems. J. Control. Release 2014, 177, 51–63. [Google Scholar] [CrossRef]
- Govindarajan, D.; Duraipandy, N.; Srivatsan, K.V.; Lakra, R.; Korapatti, P.S.; Jayavel, R.; Kiran, M.S. Fabrication of Hybrid Collagen Aerogels Reinforced with Wheat Grass Bioactives as Instructive Scaffolds for Collagen Turnover and Angiogenesis for Wound Healing Applications. ACS Appl. Mater. Interfaces 2017, 9, 16939–16950. [Google Scholar] [CrossRef]
- Mahony, O.; Tsigkou, O.; Ionescu, C.; Minelli, C.; Ling, L.; Hanly, R.; Smith, M.E.; Stevens, M.M.; Jones, J.R. Silica-gelatin hybrids with tailorable degradation and mechanical properties for tissue regeneration. Adv. Funct. Mater. 2010, 20, 3835–3845. [Google Scholar] [CrossRef]
- Betz, M.; García-González, C.A.; Subrahmanyam, R.P.; Smirnova, I.; Kulozik, U. Preparation of novel whey protein-based aerogels as drug carriers for life science applications. J. Supercrit. Fluids 2012, 72, 111–119. [Google Scholar] [CrossRef]
- Ganesan, K.; Budtova, T.; Ratke, L.; Gurikov, P.; Baudron, V.; Preibisch, I.; Niemeyer, P.; Smirnova, I.; Milow, B. Review on the production of polysaccharide aerogel particles. Materials (Basel) 2018, 11, 2144. [Google Scholar] [CrossRef]
- Demilecamps, A.; Beauger, C.; Hildenbrand, C.; Rigacci, A.; Budtova, T. Cellulose-silica aerogels. Carbohydr. Polym. 2015, 122, 293–300. [Google Scholar] [CrossRef]
- Ayers, M.R.; Hunt, A.J. Synthesis and properties of chitosan-silica hybrid aerogels. J. Non. Cryst. Solids 2001, 285, 123–127. [Google Scholar] [CrossRef]
- Rudaz, C.; Courson, R.; Bonnet, L.; Calas-Etienne, S.; Sallee, H.; Budtova, T. Aeropectin: Fully Biomass-Based Mechanically Strong and Thermal Superinsulating Aerogel. Biomacromolecules 2014, 15, 2188–2195. [Google Scholar] [CrossRef]
- Ge, J.; Li, M.; Zhang, Q.; Yang, C.Z.; Wooley, P.H.; Chen, X.; Yang, S.Y. Silica aerogel improves the biocompatibility in a poly-ε-caprolactone composite used as a tissue engineering scaffold. Int. J. Polym. Sci. 2013, 2013. [Google Scholar] [CrossRef]
- Zargar, V.; Asghari, M.; Dashti, A. A Review on Chitin and Chitosan Polymers: Structure, Chemistry, Solubility, Derivatives, and Applications. ChemBioEng Rev. 2015, 2, 204–226. [Google Scholar] [CrossRef]
- Rinaudo, M. Chitin and chitosan: Properties and applications. Prog. Polym. Sci. 2006, 31, 603–632. [Google Scholar] [CrossRef]
- Rinki, K.; Dutta, P.K.; Hunt, A.J.; MacQuarrie, D.J.; Clark, J.H. Chitosan aerogels exhibiting high surface area for biomedical application: Preparation, characterization, and antibacterial study. Int. J. Polym. Mater. Polym. Biomater. 2011, 60, 988–999. [Google Scholar] [CrossRef]
- Ji, J.; Wang, L.; Yu, H.; Chen, Y.; Zhao, Y.; Zhang, H.; Amer, W.A.; Sun, Y.; Huang, L.; Saleem, M. Chemical Modifications of Chitosan and Its Applications. Polym. Plast. Technol. Eng. 2014, 53, 1494–1505. [Google Scholar] [CrossRef]
- Alves, N.M.; Mano, J.F. Chitosan derivatives obtained by chemical modifications for biomedical and environmental applications. Int. J. Biol. Macromol. 2008, 43, 401–414. [Google Scholar] [CrossRef] [PubMed]
- Madera-Santana, T.J.; Herrera-Méndez, C.H.; Rodríguez-Núñez, J.R. An overview of the chemical modifications of chitosan and their advantages. Green Mater. 2018, 6, 131–142. [Google Scholar] [CrossRef]
- Sashiwa, H.; Aiba, S.I. Chemically modified chitin and chitosan as biomaterials. Prog. Polym. Sci. 2004, 29, 887–908. [Google Scholar] [CrossRef]
- Trujillo, S.; Pérez-Román, E.; Kyritsis, A.; Gõmez Ribelles, J.L.; Pandis, C. Organic-inorganic bonding in chitosan-silica hybrid networks: Physical properties. J. Polym. Sci. Part B Polym. Phys. 2015, 53, 1391–1400. [Google Scholar] [CrossRef]
- Götz, W.; Tobiasch, E.; Witzleben, S.; Schulze, M. Effects of silicon compounds on biomineralization, osteogenesis, and hard tissue formation. Pharmaceutics 2019, 11, 117. [Google Scholar] [CrossRef]
- Takadama, H.; Kim, H.M.; Miyaji, F.; Kokubo, T.; Nakamura, T. Mechanism of apatite formation induced by silanol groups—TEM observation. J. Ceram. Soc. Jpn. 2000, 108, 118–121. [Google Scholar] [CrossRef]
- Pipattanawarothai, A.; Suksai, C.; Srisook, K.; Trakulsujaritchok, T. Non-cytotoxic hybrid bioscaffolds of chitosan-silica: Sol-gel synthesis, characterization and proposed application. Carbohydr. Polym. 2017, 178, 190–199. [Google Scholar] [CrossRef]
- Horvat, G.; Pantić, M.; Knez, Ž.; Novak, Z. Preparation and characterization of polysaccharide-silica hybrid aerogels. Sci. Rep. 2019, 9, 3–12. [Google Scholar] [CrossRef]
- Shirosaki, Y.; Tsuru, K.; Moribayashi, H.; Hayakawa, S.; Nakamura, Y.; Gibson, I.R.; Osaka, A. Preparation of osteocompatible Si(IV)-enriched chitosan-silicate hybrids. J. Ceram. Soc. Jpn. 2010, 118, 989–992. [Google Scholar] [CrossRef]
- Wang, J.; Zhou, Q.; Song, D.; Qi, B.; Zhang, Y.; Shao, Y.; Shao, Z. Chitosan–silica composite aerogels: Preparation, characterization and Congo red adsorption. J. Sol-Gel Sci. Technol. 2015, 76, 501–509. [Google Scholar] [CrossRef]
- Wang, D.; Liu, W.; Feng, Q.; Dong, C.; Liu, Q.; Duan, L.; Huang, J.; Zhu, W.; Li, Z.; Xiong, J.; et al. Effect of inorganic/organic ratio and chemical coupling on the performance of porous silica/chitosan hybrid scaffolds. Mater. Sci. Eng. C 2017, 70, 969–975. [Google Scholar] [CrossRef] [PubMed]
- Błaszczyński, T.; Ślosarczyk, A.; Morawski, M. Synthesis of silica aerogel by supercritical drying method. Procedia Eng. 2013, 57, 200–206. [Google Scholar] [CrossRef]
- Chang, X.; Chen, D.; Jiap, X. Chitosan-based aerogels with high adsorption performance. J. Phys. Chem. B 2008, 112, 7721–7725. [Google Scholar] [CrossRef]
- Zhao, S.; Malfait, W.J.; Jeong, E.; Fischer, B.; Zhang, Y.; Xu, H.; Angelica, E.; Risen, W.M.; Suggs, J.W.; Koebel, M.M. Facile One-Pot Synthesis of Mechanically Robust Biopolymer-Silica Nanocomposite Aerogel by Cogelation of Silicic Acid with Chitosan in Aqueous Media. ACS Sustain. Chem. Eng. 2016, 4, 5674–5683. [Google Scholar] [CrossRef]
- Stergar, J. Review of aerogel-based materials in biomedical applications. J. Sol-Gel Sci. Technol. 2016, 77, 738–752. [Google Scholar] [CrossRef]
- Pandis, C.; Madeira, S.; Matos, J.; Kyritsis, A.; Mano, J.F.; Ribelles, J.L.G. Chitosan-silica hybrid porous membranes. Mater. Sci. Eng. C 2014, 42, 553–561. [Google Scholar] [CrossRef]
- Guillory, X.; Tessier, A.; Gratien, G.O.; Weiss, P.; Colliec-Jouault, S.; Dubreuil, D.; Lebreton, J.; Le Bideau, J. Glycidyl alkoxysilane reactivities towards simple nucleophiles in organic media for improved molecular structure definition in hybrid materials. RSC Adv. 2016, 6, 74087–74099. [Google Scholar] [CrossRef]
- Gómez-Romero, P.; Sanchez, C. Hybrid Materials, Functional Applications. An Introduction. In Functional Hybrid Materials; Gómez-Romero, P., Sanchez, C., Eds.; Wiley-VCH: Hoboken, NJ, USA, 2005; pp. 1–14. ISBN 3527304843. [Google Scholar]
- Connell, L.S.; Romer, F.; Suárez, M.; Valliant, E.M.; Zhang, Z.; Lee, P.D.; Smith, M.E.; Hanna, J.V.; Jones, J.R. Chemical characterisation and fabrication of chitosan-silica hybrid scaffolds with 3-glycidoxypropyl trimethoxysilane. J. Mater. Chem. B 2014, 2, 668–680. [Google Scholar] [CrossRef]
- Wang, D.; Romer, F.; Connell, L.; Walter, C.; Saiz, E.; Yue, S.; Lee, P.D.; McPhail, D.S.; Hanna, J.V.; Jones, J.R. Highly flexible silica/chitosan hybrid scaffolds with oriented pores for tissue regeneration. J. Mater. Chem. B 2015, 3, 7560–7576. [Google Scholar] [CrossRef]
- García-González, C.A.; Alnaief, M.; Smirnova, I. Polysaccharide-based aerogels—Promising biodegradable carriers for drug delivery systems. Carbohydr. Polym. 2011, 86, 1425–1438. [Google Scholar] [CrossRef]
- Van Vlierberghen, S.; Graulus, G.-J.; Samal, S.K.; Nieuwenhove, I.; Dubruel, P. Porous hydrogel biomedical foam scaffolds for tissue repair. In Biomedical Foams for Tissue Engineering Applications; Netti, P.A., Ed.; Woodhead Publishing: London, UK, 2014; pp. 335–390. ISBN 9780857098047. [Google Scholar]
- Follmann, H.D.M.; Oliveira, O.N.; Lazarin-Bidóia, D.; Nakamura, C.V.; Huang, X.; Asefa, T.; Silva, R. Multifunctional hybrid aerogels: Hyperbranched polymer-trapped mesoporous silica nanoparticles for sustained and prolonged drug release. Nanoscale 2018, 10, 1704–1715. [Google Scholar] [CrossRef]
- Liu, Y.L.; Su, Y.H.; Lai, J.Y. In situ crosslinking of chitosan and formation of chitosan-silica hybrid membranes with using γ-glycidoxypropyltrimethoxysilane as a crosslinking agent. Polymer (Guildf) 2004, 45, 6831–6837. [Google Scholar] [CrossRef]
- Park, S.B.; You, J.O.; Park, H.Y.; Haam, S.J.; Kim, W.S. A novel pH-sensitive membrane from chitosan—TEOS IPN; preparation and its drug permeation characteristics. Biomaterials 2001, 22, 323–330. [Google Scholar] [CrossRef]
- Saguy, I.S.; Marabi, A.; Wallach, R. Liquid imbibition during rehydration of dry porous foods. Innov. Food Sci. Emerg. Technol. 2005, 6, 37–43. [Google Scholar] [CrossRef]
- Kokubo, T.; Takadama, H. How useful is SBF in predicting in vivo bone bioactivity? Biomaterials 2006, 27, 2907–2915. [Google Scholar] [CrossRef] [PubMed]
- Esquivias, L.; Pinero, M.; Morales-Florez, V.; Rosa-Fox, N. Aerogels Synthesis by Sonocatalysis: Sonogels. In Aerogels Handbook; Aegerter, M.A., Leventis, N., Koebel, M.M., Eds.; Springer Link: New York, NY, USA, 2011; pp. 419–445. ISBN 9781441975898. [Google Scholar]
- Thommes, M.; Kaneko, K.; Neimark, A.V.; Olivier, J.P.; Rodriguez-Reinoso, F.; Rouquerol, J.; Sing, K.S.W. Physisorption of gases, with special reference to the evaluation of surface area and pore size distribution. Pure Appl. Chem. 2015, 87, 1052. [Google Scholar] [CrossRef]
- Morales-Florez, V.; Piñero, M.; Braza, V.; del Mar Mesa, M.; Esquivias, L.; de la Rosa-Fox, N. Absorption capacity, kinetics and mechanical behaviour in dry and wet states of hydrophobic DEDMS/TEOS-based silica aerogels. J. Sol-Gel Sci. Technol. 2017, 81. [Google Scholar] [CrossRef]
- Azhary, S.Y.; Purnama, D.; Florena, F.F.; Vanitha, M.; Panatarani, C.; Joni, I.M. Synthesis and characterization of chitosan: SiO2 nanocomposite by ultrasonic spray drying Synthesis and characterization of chitosan: SiO2 nanocomposite by ultrasonic spray drying. Mater. Sci. Eng. 2019, 550, 6. [Google Scholar] [CrossRef]
- Al-Oweini, R.; El-Rassy, H. Synthesis and characterization by FTIR spectroscopy of silica aerogels prepared using several Si(OR)4and R′′Si(OR′)3precursors. J. Mol. Struct. 2009, 919, 140–145. [Google Scholar] [CrossRef]
- Budnyak, T.M.; Pylypchuk, I.V.; Tertykh, V.A.; Yanovska, E.S.; Kolodynska, D. Synthesis and adsorption properties of chitosan-silica nanocomposite prepared by sol-gel method. Nanoscale Res. Lett. 2015, 10, 87–96. [Google Scholar] [CrossRef]
- Lim, S.; Hudson, S.M. Synthesis and antimicrobial activity of a water-soluble chitosan derivative with a fiber-reactive group. Carbohydr. Res. 2004, 339, 313–319. [Google Scholar] [CrossRef] [PubMed]
- Kumirska, J.; Kaczy, Z.; Bychowska, A. Application of Spectroscopic Methods for Structural Analysis of Chitin and Chitosan. Mar. Drugs 2010, 8, 1567–1636. [Google Scholar] [CrossRef] [PubMed]
- Li, C.P.; Weng, M.C.; Huang, S.L. Preparation and Characterization of pH Sensitive Chitosan/3—Glycidyloxypropyl Trimethoxysilane (GPTMS) Hydrogels by Sol—Gel Method. Polymers 2020, 12, 1326. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y. Engineering of Aerogel-Based Biomaterials for Biomedical Applications. Int. J. Nanomed. 2020, 15, 2363–2378. [Google Scholar]
- Taylor, P.; Lai, S.; Yang, A.J.; Chen, W.; Hsiao, J.; Lai, S. Polymer-Plastics Technology and Engineering the Properties and Preparation of Chitosan/Silica Hybrids Using Sol-Gel Process the Properties and Preparation of Chitosan/Silica Hybrids Using Sol-Gel Process. Polym. Plast. Technol. Eng. 2006, 45, 37–41. [Google Scholar] [CrossRef]
- Coyer, S.R.; Singh, A.; Dumbauld, D.W.; Calderwood, D.A.; Craig, S.W.; Delamarche, E.; García, A.J. Nanopatterning reveals an ECM area threshold for focal adhesion assembly and force transmission that is regulated by integrin activation and cytoskeleton tension. J. Cell Sci. 2012, 125, 5110–5123. [Google Scholar] [CrossRef]
- Ciobanasu, C.; Faivre, B.; Le Clainche, C. Integrating actin dynamics, mechanotransduction and integrin activation: The multiple functions of actin binding proteins in focal adhesions. Eur. J. Cell Biol. 2013, 92, 339–348. [Google Scholar] [CrossRef]
- McNamara, L.E.; Sjöström, T.; Burgess, K.E.V.; Kim, J.J.W.; Liu, E.; Gordonov, S.; Moghe, P.V.; Meek, R.M.D.; Oreffo, R.O.C.; Su, B.; et al. Skeletal stem cell physiology on functionally distinct titania nanotopographies. Biomaterials 2011, 32, 7403–7410. [Google Scholar] [CrossRef]
- Mullen, C.A.; Vaughan, T.J.; Voisin, M.C.; Brennan, M.A.; Layrolle, P.; McNamara, L.M. Cell morphology and focal adhesion location alters internal cell stress. J. R. Soc. Interface 2014, 11. [Google Scholar] [CrossRef]
- Terriza, A.; Díaz-Cuenca, A.; Yubero, F.; Barranco, A.; González-Elipe, A.R.; Caballero, J.L.G.; Vilches, J.; Salido, M. Light induced hydrophilicity and osteoblast adhesion promotion on amorphous TiO2. J. Biomed. Mater. Res. Part A 2013, 101A, 1026–1035. [Google Scholar] [CrossRef]
- Lamers, E.; van Horssen, R.; te Riet, J.; van Delft, F.C.M.J.M.; Luttge, R.; Walboomers, X.F.; Jansen, J.A. The influence of nanoscale topographical cues on initial osteoblast morphology and migration. Eur. Cells Mater. 2010, 20, 329–343. [Google Scholar] [CrossRef] [PubMed]
- Matschegewski, C.; Birkholz, H.; Staehlke, S.; Loeffler, R.; Kern, D.P.; Engel, K.; Nebe, J.B. Quantitative analysis of the cellular actin cytoskeleton on geometrically designed surface topography. Mater. Sci. Forum 2012, 706–709, 543–548. [Google Scholar] [CrossRef]
- Sanz-Herrera, J.A.; García-Aznar, J.M.; Doblaré, M. On scaffold designing for bone regeneration: A computational multiscale approach. Acta Biomater. 2009, 5, 219–229. [Google Scholar] [CrossRef] [PubMed]
- Zinger, O.; Anselme, K.; Denzer, A.; Habersetzer, P.; Wieland, M.; Jeanfils, J.; Hardouin, P.; Landolt, D. Time-dependent morphology and adhesion of osteoblastic cells on titanium model surfaces featuring scale-resolved topography. Biomaterials 2004, 25, 2695–2711. [Google Scholar] [CrossRef] [PubMed]
- Maggi, A.; Li, H.; Greer, J.R. Three-dimensional nano-architected scaffolds with tunable stiffness for efficient bone tissue growth. Acta Biomater. 2017, 63, 294–305. [Google Scholar] [CrossRef]
- Geiger, B.; Spatz, J.P.; Bershadsky, A.D. Environmental sensing through focal adhesions. Nat. Rev. Mol. Cell Biol. 2009, 10, 21–33. [Google Scholar] [CrossRef]
- Parsons, J.T.; Horwitz, A.R.; Schwartz, M.A. Cell adhesion: Integrating cytoskeletal dynamics and cellular tension. Nat. Publ. Gr. 2010, 11, 633–643. [Google Scholar] [CrossRef]
- Gardel, M.L.; Schneider, I.C.; Aratyn-schaus, Y.; Waterman, C.M. Mechanical Integration of Actin and Adhesion Dynamics in Cell Migration. Annu. Rev. Cell Dev. Biol. 2010, 26, 315–335. [Google Scholar] [CrossRef]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample Identification | Nominal CS Content | GPTMS/CS | TEOS/CS | Final CS Content from EA 1 |
|---|---|---|---|---|
| (wt%) | (Molar Ratio) | (Molar Ratio) | (wt%) | |
| CS1G0 | 3.8 | 0 | 150 | 1.7 |
| CS2G0 | 7.3 | 75 | 3.3 | |
| CS3G0 | 12.3 | 45 | 7.6 | |
| CS4G0 | 16.7 | 30 | 10.3 | |
| CS5G2 | 3.7 | 2 | 150 | 3.1 |
| CS6G2 | 6.8 | 75 | 5.9 | |
| CS7G2 | 14.2 | 30 | 8.0 | |
| CS8G4 | 6.3 | 4 | 75 | 3.1 |
| CS9G4 | 9.8 | 45 | 5.3 | |
| CS10G4 | 12.3 | 30 | 9.7 |
| Sample | Density (±0.01 g cm−3) | Physisorption | |||
|---|---|---|---|---|---|
| SBET 1 (m2 g−1) | Pore Volume (cm3 g−1) | Pore Size (nm) | Porosity (%) | ||
| CS1G0 | 0.38 | 1184 | 2.6 | 8.1 | 81.9 |
| CS2G0 | 0.26 | 1230 | 3.4 | 10.2 | 87.6 |
| CS3G0 | 0.17 | 811 | 2.3 | 10.2 | 82.4 |
| CS4G0 | 0.17 | 755 | 1.9 | 11.1 | 91.6 |
| CS5G2 | 0.29 | 1042 | 3.1 | 11.2 | 86.1 |
| CS6G2 | 0.22 | 903 | 3.2 | 14 | 89.3 |
| CS7G2 | 0.23 | 771 | 2.2 | 11.6 | 89.1 |
| CS8G4 | 0.31 | 955 | 2.1 | 8.7 | 85.4 |
| CS9G4 | 0.32 | 697 | 1.6 | 8.9 | 82.6 |
| CS10G4 | 0.37 | 695 | 1.7 | 9.8 | 82.3 |
| Sample | Young’s Modulus (MPa) | Compressive Strength (MPa) | Maximum Compressive Strain (%) | |||
|---|---|---|---|---|---|---|
| Dry | Wet | Dry | Wet | Dry | Wet | |
| CS1G0 | 11.2 ± 1.4 | 1.13 ± 0.44 | 1.0 ± 0.3 | 0.17 ± 0.02 | 12.2 ± 2.3 | 13.20 ± 3.40 |
| CS2G0 | 9.9 ± 2.9 | 0.30 ± 0.10 | 0.8 ± 0.2 | 0.07 ± 0.01 | 18.1 ± 3.8 | 13.47 ± 0.21. |
| CS3G0 | 1.9 ± 0.1 | 0.30 ± 0.07 | 16.1 ± 4.2 | 0.09 ± 0.05 | 66.4 ± 7.0 | 12.60 ± 1.71 |
| CS4G0 | 2.8 ± 0.6 | 0.75 ± 0.25 | 1.46 ± 0.1 | 0.11 ± 0.04 | 50.4 ± 5.5 | 9.18 ± 1.55 |
| CS5G2 | 21.1 ± 7.0 | 0.78 ± 0.07 | 5.3 ± 4.8 | 0.19 ± 0.05 | 36.5 ± 25.6 | 12.92 ± 1.30 |
| CS6G2 | 7.7 ± 1.5 | 0.40 ± 0.01 | 22.6 ± 15.1 | 0.16 ± 0.01 | 77.6 ± 16.3 | 21.21 ± 2.09 |
| CS7G2 | 6.7 ± 1.3 | 0.16 ± 0.02 | 77.7 ± 2.7 | 0.09 ± 0.03 | 77.7 ± 1.6 | 22.89 ± 9.47 |
| CS8G4 | 32.4 ± 3.8 | 0.77 ± 0.30 | 5.6 ± 3.1 | 0.21 ± 0.01 | 35.2 ± 10.7 | 9.41 ± 3.01 |
| CS9G4 | 8.7 ± 0.5 | 0.82 ± 0.17 | 5.5 ± 2.9 | 0.27 ± 0.04 | 38.0 ± 14.9 | 10.58 ± 0.86 |
| CS10G4 | 50.3 ± 7.0 | 0.27 ± 0.03 | 95.7 ± 6.9 | 0.26 ± 0.03 | 76.2 ± 9.5 | 21.74 ± 1.37 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Reyes-Peces, M.V.; Pérez-Moreno, A.; de-los-Santos, D.M.; Mesa-Díaz, M.d.M.; Pinaglia-Tobaruela, G.; Vilches-Pérez, J.I.; Fernández-Montesinos, R.; Salido, M.; de la Rosa-Fox, N.; Piñero, M. Chitosan-GPTMS-Silica Hybrid Mesoporous Aerogels for Bone Tissue Engineering. Polymers 2020, 12, 2723. https://doi.org/10.3390/polym12112723
Reyes-Peces MV, Pérez-Moreno A, de-los-Santos DM, Mesa-Díaz MdM, Pinaglia-Tobaruela G, Vilches-Pérez JI, Fernández-Montesinos R, Salido M, de la Rosa-Fox N, Piñero M. Chitosan-GPTMS-Silica Hybrid Mesoporous Aerogels for Bone Tissue Engineering. Polymers. 2020; 12(11):2723. https://doi.org/10.3390/polym12112723
Chicago/Turabian StyleReyes-Peces, María V., A. Pérez-Moreno, Deseada María de-los-Santos, María del Mar Mesa-Díaz, Gonzalo Pinaglia-Tobaruela, Jose Ignacio Vilches-Pérez, Rafael Fernández-Montesinos, Mercedes Salido, Nicolás de la Rosa-Fox, and Manuel Piñero. 2020. "Chitosan-GPTMS-Silica Hybrid Mesoporous Aerogels for Bone Tissue Engineering" Polymers 12, no. 11: 2723. https://doi.org/10.3390/polym12112723
APA StyleReyes-Peces, M. V., Pérez-Moreno, A., de-los-Santos, D. M., Mesa-Díaz, M. d. M., Pinaglia-Tobaruela, G., Vilches-Pérez, J. I., Fernández-Montesinos, R., Salido, M., de la Rosa-Fox, N., & Piñero, M. (2020). Chitosan-GPTMS-Silica Hybrid Mesoporous Aerogels for Bone Tissue Engineering. Polymers, 12(11), 2723. https://doi.org/10.3390/polym12112723