Predicting Antibody Neutralization Efficacy in Hypermutated Epitopes Using Monte Carlo Simulations


Abstract
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Data Collection
2.2. Modeling of Three-Dimensional gp120 Structures
2.3. Simulating the Binding Process with PELE
2.4. Population Analysis of PELE Simulations
3. Results
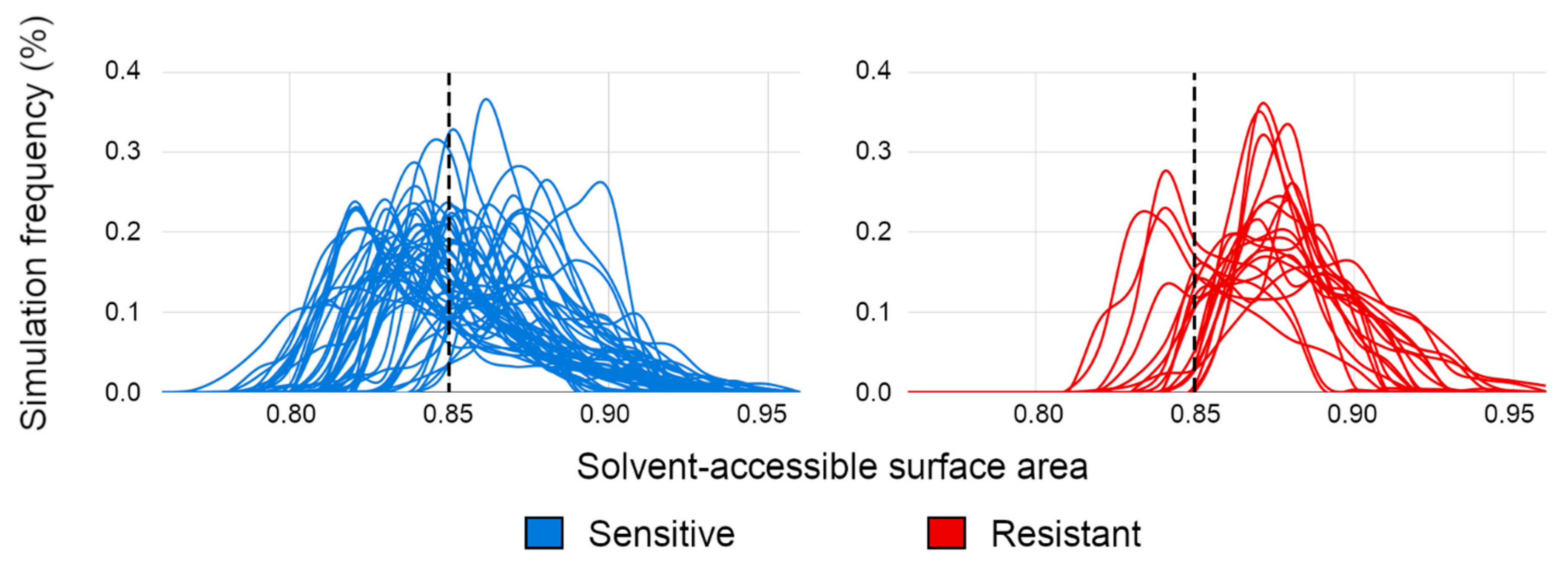
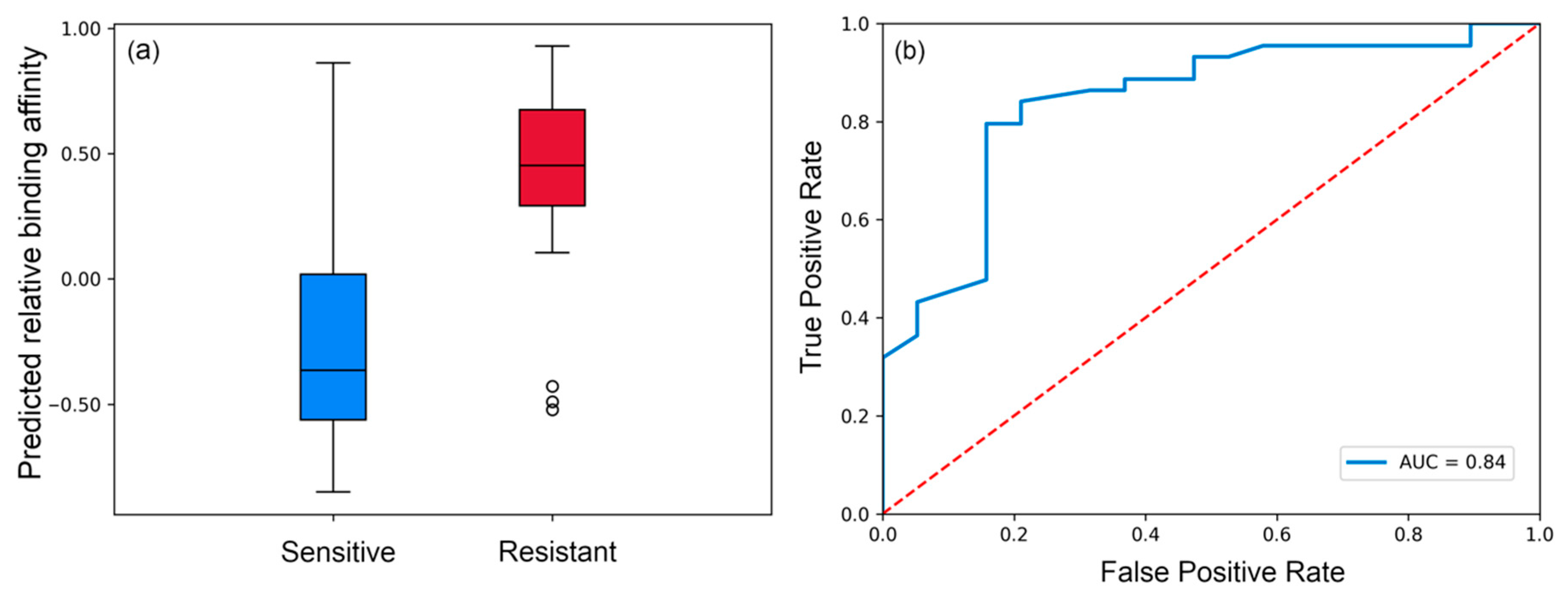
3.1. Evaluating Binding Profiles of Sensitive and Resistant Strains to VRC01
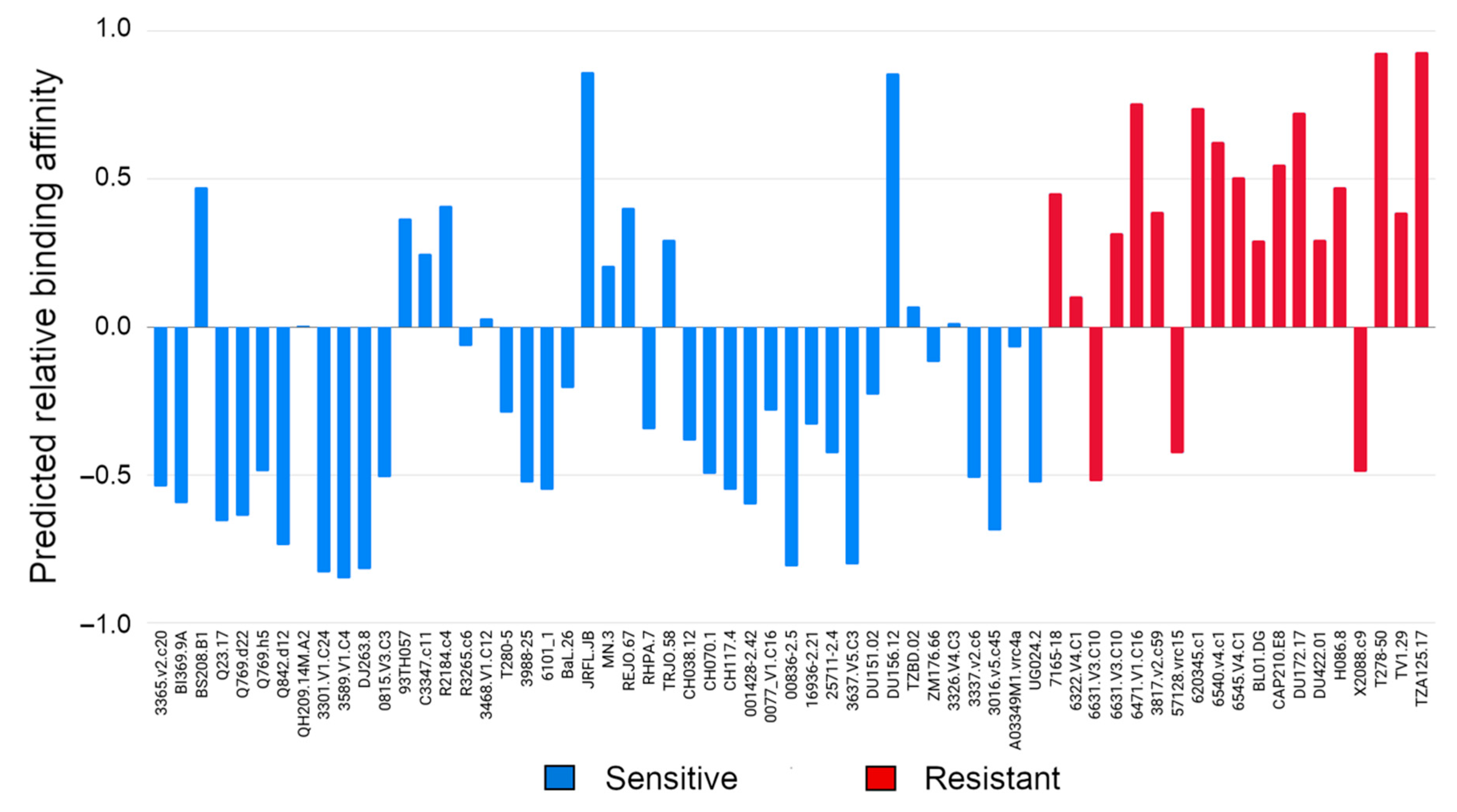
3.2. Evaluating Binding Affinity of VRC01, NIH45-46 and 3BNC117 to Multiple Viral Isolates
3.3. Structural Basis of Resistance Mechanisms
4. Discussion
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- HIV/AIDS. Available online: https://www.who.int/news-room/fact-sheets/detail/hiv-aids (accessed on 19 August 2020).
- Cuevas, J.M.; Geller, R.; Garijo, R.; López-Aldeguer, J.; Sanjuán, R. Extremely High Mutation Rate of HIV-1 In Vivo. PLoS Biol. 2015, 13, e1002251. [Google Scholar] [CrossRef] [PubMed]
- Zhuang, J.; Jetzt, A.E.; Sun, G.; Yu, H.; Klarmann, G.; Ron, Y.; Preston, B.D.; Dougherty, J.P. Human immunodeficiency virus type 1 recombination: Rate, fidelity, and putative hot spots. J. Virol. 2002, 76, 11273–11282. [Google Scholar] [CrossRef] [PubMed]
- Ramirez, B.C.; Simon-Loriere, E.; Galetto, R.; Negroni, M. Implications of recombination for HIV diversity. Virus Res. 2008, 134, 64–73. [Google Scholar] [CrossRef] [PubMed]
- Chun, T.W.; Finzi, D.; Margolick, J.; Chadwick, K.; Schwartz, D.; Siliciano, R.F. In vivo fate of HIV-1-infected T cells: Quantitative analysis of the transition to stable latency. Nat. Med. 1995, 1, 1284–1290. [Google Scholar] [CrossRef] [PubMed]
- Finzi, D.; Hermankova, M.; Pierson, T.; Carruth, L.M.; Buck, C.; Chaisson, R.E.; Quinn, T.C.; Chadwick, K.; Margolick, J.; Brookmeyer, R.; et al. Identification of a reservoir for HIV-1 in patients on highly active antiretroviral therapy. Science 1997, 278, 1295–1300. [Google Scholar] [CrossRef] [PubMed]
- Chun, T.W.; Carruth, L.; Finzi, D.; Shen, X.; DiGiuseppe, J.A.; Taylor, H.; Hermankova, M.; Chadwick, K.; Margolick, J.; Quinn, T.C.; et al. Quantification of latent tissue reservoirs and total body viral load in HIV-1 infection. Nature 1997, 387, 183–188. [Google Scholar] [CrossRef]
- Pitman, M.C.; Lau, J.S.Y.; McMahon, J.H.; Lewin, S.R. Barriers and strategies to achieve a cure for HIV. Lancet HIV 2018, 5, e317–e328. [Google Scholar] [CrossRef]
- Arts, E.J.; Hazuda, D.J. HIV-1 antiretroviral drug therapy. Cold Spring Harb. Perspect. Med. 2012, 2, a007161. [Google Scholar] [CrossRef]
- Atta, M.G.; De Seigneux, S.; Lucas, G.M. Clinical Pharmacology in HIV Therapy. Clin. J. Am. Soc. Nephrol. 2019, 14, 435–444. [Google Scholar] [CrossRef]
- Wandeler, G.; Johnson, L.F.; Egger, M. Trends in life expectancy of HIV-positive adults on antiretroviral therapy across the globe: Comparisons with general population. Curr. Opin. HIV AIDS 2016, 11, 492–500. [Google Scholar] [CrossRef]
- Oguntibeju, O.O. Quality of life of people living with HIV and AIDS and antiretroviral therapy. HIV AIDS 2012, 4, 117–124. [Google Scholar] [CrossRef]
- Davey, R.T.; Bhat, N.; Yoder, C.; Chun, T.-W.; Metcalf, J.A.; Dewar, R.; Natarajan, V.; Lempicki, R.A.; Adelsberger, J.W.; Miller, K.D.; et al. HIV-1 and T cell dynamics after interruption of highly active antiretroviral therapy (HAART) in patients with a history of sustained viral suppression. Proc. Natl. Acad. Sci. USA 1999, 96, 15109–15114. [Google Scholar] [CrossRef] [PubMed]
- Chun, T.W.; Davey, R.T., Jr.; Engel, D.; Lane, H.C.; Fauci, A.S. Re-emergence of HIV after stopping therapy. Nature 1999, 401, 874–875. [Google Scholar] [CrossRef] [PubMed]
- Pannus, P.; Rutsaert, S.; De Wit, S.; Allard, S.D.; Vanham, G.; Cole, B.; Nescoi, C.; Aerts, J.; De Spiegelaere, W.; Tsoumanis, A.; et al. Rapid viral rebound after analytical treatment interruption in patients with very small HIV reservoir and minimal on-going viral transcription. J. Int. AIDS Soc. 2020, 23, e25453. [Google Scholar] [CrossRef]
- Sok, D.; Burton, D.R. Recent progress in broadly neutralizing antibodies to HIV. Nat. Immunol. 2018, 19, 1179–1188. [Google Scholar] [CrossRef] [PubMed]
- Cohen, Y.Z.; Caskey, M. Broadly neutralizing antibodies for treatment and prevention of HIV-1 infection. Curr. Opin. HIV AIDS 2018, 13, 366. [Google Scholar] [CrossRef]
- Carrillo, J.; Clotet, B.; Blanco, J. Antibodies and Antibody Derivatives: New Partners in HIV Eradication Strategies. Front. Immunol. 2018, 9, 2429. [Google Scholar] [CrossRef]
- Gama, L.; Koup, R.A. New-Generation High-Potency and Designer Antibodies: Role in HIV-1 Treatment. Annu. Rev. Med. 2018, 69, 409–419. [Google Scholar] [CrossRef]
- Dhillon, A.K.; Donners, H.; Pantophlet, R.; Johnson, W.E.; Decker, J.M.; Shaw, G.M.; Lee, F.-H.; Richman, D.D.; Doms, R.W.; Vanham, G.; et al. Dissecting the neutralizing antibody specificities of broadly neutralizing sera from human immunodeficiency virus type 1-infected donors. J. Virol. 2007, 81, 6548–6562. [Google Scholar] [CrossRef][Green Version]
- Sather, D.N.; Armann, J.; Ching, L.K.; Mavrantoni, A.; Sellhorn, G.; Caldwell, Z.; Yu, X.; Wood, B.; Self, S.; Kalams, S.; et al. Factors associated with the development of cross-reactive neutralizing antibodies during human immunodeficiency virus type 1 infection. J. Virol. 2009, 83, 757–769. [Google Scholar] [CrossRef]
- Binley, J.M.; Lybarger, E.A.; Crooks, E.T.; Seaman, M.S.; Gray, E.; Davis, K.L.; Decker, J.M.; Wycuff, D.; Harris, L.; Hawkins, N.; et al. Profiling the Specificity of Neutralizing Antibodies in a Large Panel of Plasmas from Patients Chronically Infected with Human Immunodeficiency Virus Type 1 Subtypes B and C. J. Virol. 2008, 82, 11651–11668. [Google Scholar] [CrossRef] [PubMed]
- Gray, E.S.; Taylor, N.; Wycuff, D.; Moore, P.L.; Tomaras, G.D.; Wibmer, C.K.; Puren, A.; DeCamp, A.; Gilbert, P.B.; Wood, B.; et al. Antibody specificities associated with neutralization breadth in plasma from human immunodeficiency virus type 1 subtype C-infected blood donors. J. Virol. 2009, 83, 8925–8937. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Migueles, S.A.; Welcher, B.; Svehla, K.; Phogat, A.; Louder, M.K.; Wu, X.; Shaw, G.M.; Connors, M.; Wyatt, R.T.; et al. Broad HIV-1 neutralization mediated by CD4-binding site antibodies. Nat. Med. 2007, 13, 1032–1034. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Svehla, K.; Louder, M.K.; Wycuff, D.; Phogat, S.; Tang, M.; Migueles, S.A.; Wu, X.; Phogat, A.; Shaw, G.M.; et al. Analysis of neutralization specificities in polyclonal sera derived from human immunodeficiency virus type 1-infected individuals. J. Virol. 2009, 83, 1045–1059. [Google Scholar] [CrossRef] [PubMed]
- Scheid, J.F.; Horwitz, J.A.; Bar-On, Y.; Kreider, E.F.; Lu, C.-L.; Lorenzi, J.C.C.; Feldmann, A.; Braunschweig, M.; Nogueira, L.; Oliveira, T.; et al. HIV-1 antibody 3BNC117 suppresses viral rebound in humans during treatment interruption. Nature 2016, 535, 556–560. [Google Scholar] [CrossRef]
- Cohen, Y.Z.; Butler, A.L.; Millard, K.; Witmer-Pack, M.; Levin, R.; Unson-O’Brien, C.; Patel, R.; Shimeliovich, I.; Lorenzi, J.C.C.; Horowitz, J.; et al. Safety, pharmacokinetics, and immunogenicity of the combination of the broadly neutralizing anti-HIV-1 antibodies 3BNC117 and 10-1074 in healthy adults: A randomized, phase 1 study. PLoS ONE 2019, 14, e0219142. [Google Scholar] [CrossRef]
- Crowell, T.A.; Colby, D.J.; Pinyakorn, S.; Sacdalan, C.; Pagliuzza, A.; Intasan, J.; Benjapornpong, K.; Tangnaree, K.; Chomchey, N.; Kroon, E.; et al. Safety and efficacy of VRC01 broadly neutralising antibodies in adults with acutely treated HIV (RV397): A phase 2, randomised, double-blind, placebo-controlled trial. Lancet HIV 2019, 6, e297–e306. [Google Scholar] [CrossRef]
- Bar, K.J.; Sneller, M.C.; Harrison, L.J.; Justement, J.S.; Overton, E.T.; Petrone, M.E.; Salantes, D.B.; Seamon, C.A.; Scheinfeld, B.; Kwan, R.W.; et al. Effect of HIV Antibody VRC01 on Viral Rebound after Treatment Interruption. N. Engl. J. Med. 2016, 375, 2037–2050. [Google Scholar] [CrossRef]
- Riddler, S.A.; Zheng, L.; Durand, C.M.; Ritz, J.; Koup, R.A.; Ledgerwood, J.; Bailer, R.T.; Koletar, S.L.; Eron, J.J.; Keefer, M.C.; et al. Randomized Clinical Trial to Assess the Impact of the Broadly Neutralizing HIV-1 Monoclonal Antibody VRC01 on HIV-1 Persistence in Individuals on Effective ART. Open Forum Infect. Dis. 2018, 5, ofy242. [Google Scholar] [CrossRef]
- Caskey, M.; Schoofs, T.; Gruell, H.; Settler, A.; Karagounis, T.; Kreider, E.F.; Murrell, B.; Pfeifer, N.; Nogueira, L.; Oliveira, T.Y.; et al. Antibody 10-1074 suppresses viremia in HIV-1-infected individuals. Nat. Med. 2017, 23, 185–191. [Google Scholar] [CrossRef]
- Gaudinski, M.R.; Coates, E.E.; Houser, K.V.; Chen, G.L.; Yamshchikov, G.; Saunders, J.G.; Holman, L.A.; Gordon, I.; Plummer, S.; Hendel, C.S.; et al. Safety and pharmacokinetics of the Fc-modified HIV-1 human monoclonal antibody VRC01LS: A Phase 1 open-label clinical trial in healthy adults. PLoS Med. 2018, 15, e1002493. [Google Scholar] [CrossRef] [PubMed]
- Gaudinski, M.R.; Houser, K.V.; Doria-Rose, N.A.; Chen, G.L.; Rothwell, R.S.S.; Berkowitz, N.; Costner, P.; Holman, L.A.; Gordon, I.J.; Hendel, C.S.; et al. Safety and pharmacokinetics of broadly neutralising human monoclonal antibody VRC07-523LS in healthy adults: A phase 1 dose-escalation clinical trial. Lancet HIV 2019, 6, e667–e679. [Google Scholar] [CrossRef]
- Kong, R.; Louder, M.K.; Wagh, K.; Bailer, R.T.; deCamp, A.; Greene, K.; Gao, H.; Taft, J.D.; Gazumyan, A.; Liu, C.; et al. Improving neutralization potency and breadth by combining broadly reactive HIV-1 antibodies targeting major neutralization epitopes. J. Virol. 2015, 89, 2659–2671. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; O’Dell, S.; Walker, L.M.; Wu, X.; Guenaga, J.; Feng, Y.; Schmidt, S.D.; McKee, K.; Louder, M.K.; Ledgerwood, J.E.; et al. Mechanism of neutralization by the broadly neutralizing HIV-1 monoclonal antibody VRC01. J. Virol. 2011, 85, 8954–8967. [Google Scholar] [CrossRef]
- Huang, J.; Kang, B.H.; Ishida, E.; Zhou, T.; Griesman, T.; Sheng, Z.; Wu, F.; Doria-Rose, N.A.; Zhang, B.; McKee, K.; et al. Identification of a CD4-Binding-Site Antibody to HIV that Evolved Near-Pan Neutralization Breadth. Immunity 2016, 45, 1108–1121. [Google Scholar] [CrossRef]
- Scheid, J.F.; Mouquet, H.; Ueberheide, B.; Diskin, R.; Klein, F.; Oliveira, T.Y.K.; Pietzsch, J.; Fenyo, D.; Abadir, A.; Velinzon, K.; et al. Sequence and structural convergence of broad and potent HIV antibodies that mimic CD4 binding. Science 2011, 333, 1633–1637. [Google Scholar] [CrossRef]
- Barbas, C.F., 3rd; Björling, E.; Chiodi, F.; Dunlop, N.; Cababa, D.; Jones, T.M.; Zebedee, S.L.; Persson, M.A.; Nara, P.L.; Norrby, E. Recombinant human Fab fragments neutralize human type 1 immunodeficiency virus in vitro. Proc. Natl. Acad. Sci. USA 1992, 89, 9339–9343. [Google Scholar] [CrossRef]
- Walker, L.M.; Phogat, S.K.; Chan-Hui, P.-Y.; Wagner, D.; Phung, P.; Goss, J.L.; Wrin, T.; Simek, M.D.; Fling, S.; Mitcham, J.L.; et al. Broad and potent neutralizing antibodies from an African donor reveal a new HIV-1 vaccine target. Science 2009, 326, 285–289. [Google Scholar] [CrossRef]
- Sok, D.; van Gils, M.J.; Pauthner, M.; Julien, J.-P.; Saye-Francisco, K.L.; Hsueh, J.; Briney, B.; Lee, J.H.; Le, K.M.; Lee, P.S.; et al. Recombinant HIV envelope trimer selects for quaternary-dependent antibodies targeting the trimer apex. Proc. Natl. Acad. Sci. USA 2014, 111, 17624–17629. [Google Scholar] [CrossRef]
- Doria-Rose, N.A.; Bhiman, J.N.; Roark, R.S.; Schramm, C.A.; Gorman, J.; Chuang, G.-Y.; Pancera, M.; Cale, E.M.; Ernandes, M.J.; Louder, M.K.; et al. New Member of the V1V2-Directed CAP256-VRC26 Lineage That Shows Increased Breadth and Exceptional Potency. J. Virol. 2016, 90, 76–91. [Google Scholar] [CrossRef]
- Saunders, K.O.; Nicely, N.I.; Wiehe, K.; Bonsignori, M.; Meyerhoff, R.R.; Parks, R.; Walkowicz, W.E.; Aussedat, B.; Wu, N.R.; Cai, F.; et al. Vaccine Elicitation of High Mannose-Dependent Neutralizing Antibodies against the V3-Glycan Broadly Neutralizing Epitope in Nonhuman Primates. Cell Rep. 2017, 18, 2175–2188. [Google Scholar] [CrossRef] [PubMed]
- Mouquet, H.; Scharf, L.; Euler, Z.; Liu, Y.; Eden, C.; Scheid, J.F.; Halper-Stromberg, A.; Gnanapragasam, P.N.P.; Spencer, D.I.R.; Seaman, M.S.; et al. Complex-type N-glycan recognition by potent broadly neutralizing HIV antibodies. Proc. Natl. Acad. Sci. USA 2012, 109, E3268–E3277. [Google Scholar] [CrossRef] [PubMed]
- Julien, J.-P.; Sok, D.; Khayat, R.; Lee, J.H.; Doores, K.J.; Walker, L.M.; Ramos, A.; Diwanji, D.C.; Pejchal, R.; Cupo, A.; et al. Broadly neutralizing antibody PGT121 allosterically modulates CD4 binding via recognition of the HIV-1 gp120 V3 base and multiple surrounding glycans. PLoS Pathog. 2013, 9, e1003342. [Google Scholar] [CrossRef]
- Cardoso, R.M.F.; Zwick, M.B.; Stanfield, R.L.; Kunert, R.; Binley, J.M.; Katinger, H.; Burton, D.R.; Wilson, I.A. Broadly neutralizing anti-HIV antibody 4E10 recognizes a helical conformation of a highly conserved fusion-associated motif in gp41. Immunity 2005, 22, 163–173. [Google Scholar] [CrossRef]
- Huang, J.; Ofek, G.; Laub, L.; Louder, M.K.; Doria-Rose, N.A.; Longo, N.S.; Imamichi, H.; Bailer, R.T.; Chakrabarti, B.; Sharma, S.K.; et al. Broad and potent neutralization of HIV-1 by a gp41-specific human antibody. Nature 2012, 491, 406–412. [Google Scholar] [CrossRef]
- Molinos-Albert, L.M.; Clotet, B.; Blanco, J.; Carrillo, J. Immunologic Insights on the Membrane Proximal External Region: A Major Human Immunodeficiency Virus Type-1 Vaccine Target. Front. Immunol. 2017, 8, 1154. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Kang, B.H.; Pancera, M.; Lee, J.H.; Tong, T.; Feng, Y.; Imamichi, H.; Georgiev, I.S.; Chuang, G.-Y.; Druz, A.; et al. Broad and potent HIV-1 neutralization by a human antibody that binds the gp41-gp120 interface. Nature 2014, 515, 138–142. [Google Scholar] [CrossRef] [PubMed]
- Falkowska, E.; Le, K.M.; Ramos, A.; Doores, K.J.; Lee, J.H.; Blattner, C.; Ramirez, A.; Derking, R.; van Gils, M.J.; Liang, C.-H.; et al. Broadly Neutralizing HIV Antibodies Define a Glycan-Dependent Epitope on the Prefusion Conformation of gp41 on Cleaved Envelope Trimers. Immunity 2014, 40, 657–668. [Google Scholar] [CrossRef]
- Scharf, L.; Scheid, J.F.; Lee, J.H.; West, A.P., Jr.; Chen, C.; Gao, H.; Gnanapragasam, P.N.P.; Mares, R.; Seaman, M.S.; Ward, A.B.; et al. Antibody 8ANC195 reveals a site of broad vulnerability on the HIV-1 envelope spike. Cell Rep. 2014, 7, 785–795. [Google Scholar] [CrossRef]
- Schoofs, T.; Barnes, C.O.; Suh-Toma, N.; Golijanin, J.; Schommers, P.; Gruell, H.; West, A.P., Jr.; Bach, F.; Lee, Y.E.; Nogueira, L.; et al. Broad and Potent Neutralizing Antibodies Recognize the Silent Face of the HIV Envelope. Immunity 2019, 50, 1513–1529. [Google Scholar] [CrossRef]
- Crooks, E.T.; Osawa, K.; Tong, T.; Grimley, S.L.; Dai, Y.D.; Whalen, R.G.; Kulp, D.W.; Menis, S.; Schief, W.R.; Binley, J.M. Effects of partially dismantling the CD4 binding site glycan fence of HIV-1 Envelope glycoprotein trimers on neutralizing antibody induction. Virology 2017, 505, 193–209. [Google Scholar] [CrossRef]
- Seabright, G.E.; Doores, K.J.; Burton, D.R.; Crispin, M. Protein and Glycan Mimicry in HIV Vaccine Design. J. Mol. Biol. 2019, 431, 2223–2247. [Google Scholar] [CrossRef]
- Puigdomènech, I.; Massanella, M.; Cabrera, C.; Clotet, B.; Blanco, J. On the steps of cell-to-cell HIV transmission between CD4 T cells. Retrovirology 2009, 6, 89. [Google Scholar] [CrossRef] [PubMed]
- Bai, H.; Li, Y.; Michael, N.L.; Robb, M.L.; Rolland, M. The breadth of HIV-1 neutralizing antibodies depends on the conservation of key sites in their epitopes. PLoS Comput. Biol. 2019, 15, e1007056. [Google Scholar] [CrossRef] [PubMed]
- Rawi, R.; Mall, R.; Shen, C.-H.; Farney, S.K.; Shiakolas, A.; Zhou, J.; Bensmail, H.; Chun, T.-W.; Doria-Rose, N.A.; Lynch, R.M.; et al. Accurate Prediction for Antibody Resistance of Clinical HIV-1 Isolates. Sci. Rep. 2019, 9, 14696. [Google Scholar] [CrossRef]
- Buiu, C.; Putz, M.V.; Avram, S. Learning the Relationship between the Primary Structure of HIV Envelope Glycoproteins and Neutralization Activity of Particular Antibodies by Using Artificial Neural Networks. Int. J. Mol. Sci. 2016, 17, 1710. [Google Scholar] [CrossRef]
- Hepler, N.L.; Scheffler, K.; Weaver, S.; Murrell, B.; Richman, D.D.; Burton, D.R.; Poignard, P.; Smith, D.M.; Kosakovsky Pond, S.L. IDEPI: Rapid prediction of HIV-1 antibody epitopes and other phenotypic features from sequence data using a flexible machine learning platform. PLoS Comput. Biol. 2014, 10, e1003842. [Google Scholar] [CrossRef]
- Hake, A.; Pfeifer, N. Prediction of HIV-1 sensitivity to broadly neutralizing antibodies shows a trend towards resistance over time. PLOS Comput. Biol. 2017, 13, e1005789. [Google Scholar] [CrossRef] [PubMed]
- Borrelli, K.W.; Vitalis, A.; Alcantara, R.; Guallar, V. PELE: Protein Energy Landscape Exploration. A Novel Monte Carlo Based Technique. J. Chem. Theory Comput. 2005, 1, 1304–1311. [Google Scholar] [CrossRef]
- Hosseini, A.; Alibés, A.; Noguera-Julian, M.; Gil, V.; Paredes, R.; Soliva, R.; Orozco, M.; Guallar, V. Computational Prediction of HIV-1 Resistance to Protease Inhibitors. J. Chem. Inf. Model. 2016, 56, 915–923. [Google Scholar] [CrossRef] [PubMed]
- Jacobson, M.P.; Pincus, D.L.; Rapp, C.S.; Day, T.J.F.; Honig, B.; Shaw, D.E.; Friesner, R.A. A hierarchical approach to all-atom protein loop prediction. Proteins 2004, 55, 351–367. [Google Scholar] [CrossRef] [PubMed]
- Jacobson, M.P.; Friesner, R.A.; Xiang, Z.; Honig, B. On the Role of the Crystal Environment in Determining Protein Side-chain Conformations. J. Mol. Biol. 2002, 320, 597–608. [Google Scholar] [CrossRef]
- Zhou, T.; Georgiev, I.; Wu, X.; Yang, Z.-Y.; Dai, K.; Finzi, A.; Kwon, Y.D.; Scheid, J.F.; Shi, W.; Xu, L.; et al. Structural basis for broad and potent neutralization of HIV-1 by antibody VRC01. Science 2010, 329, 811–817. [Google Scholar] [CrossRef] [PubMed]
- Gallicchio, E.; Zhang, L.Y.; Levy, R.M. The SGB/NP hydration free energy model based on the surface generalized born solvent reaction field and novel nonpolar hydration free energy estimators. J. Comput. Chem. 2002, 23, 517–529. [Google Scholar] [CrossRef]
- Diskin, R.; Scheid, J.F.; Marcovecchio, P.M.; West, A.P., Jr.; Klein, F.; Gao, H.; Gnanapragasam, P.N.P.; Abadir, A.; Seaman, M.S.; Nussenzweig, M.C.; et al. Increasing the potency and breadth of an HIV antibody by using structure-based rational design. Science 2011, 334, 1289–1293. [Google Scholar] [CrossRef]
- Klein, F.; Diskin, R.; Scheid, J.F.; Gaebler, C.; Mouquet, H.; Georgiev, I.S.; Pancera, M.; Zhou, T.; Incesu, R.-B.; Fu, B.Z.; et al. Somatic mutations of the immunoglobulin framework are generally required for broad and potent HIV-1 neutralization. Cell 2013, 153, 126–138. [Google Scholar] [CrossRef]
- Lynch, R.M.; Wong, P.; Tran, L.; O’Dell, S.; Nason, M.C.; Li, Y.; Wu, X.; Mascola, J.R. HIV-1 fitness cost associated with escape from the VRC01 class of CD4 binding site neutralizing antibodies. J. Virol. 2015, 89, 4201–4213. [Google Scholar] [CrossRef]
- Gruell, H.; Klein, F. Antibody-mediated prevention and treatment of HIV-1 infection. Retrovirology 2018, 15, 1–11. [Google Scholar] [CrossRef]
- Santoro, M.M.; Perno, C.F. HIV-1 Genetic Variability and Clinical Implications. ISRN Microbiol. 2013, 2013, 481314. [Google Scholar] [CrossRef]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Amengual-Rigo, P.; Carrillo, J.; Blanco, J.; Guallar, V. Predicting Antibody Neutralization Efficacy in Hypermutated Epitopes Using Monte Carlo Simulations. Polymers 2020, 12, 2392. https://doi.org/10.3390/polym12102392
Amengual-Rigo P, Carrillo J, Blanco J, Guallar V. Predicting Antibody Neutralization Efficacy in Hypermutated Epitopes Using Monte Carlo Simulations. Polymers. 2020; 12(10):2392. https://doi.org/10.3390/polym12102392
Chicago/Turabian StyleAmengual-Rigo, Pep, Jorge Carrillo, Julià Blanco, and Victor Guallar. 2020. "Predicting Antibody Neutralization Efficacy in Hypermutated Epitopes Using Monte Carlo Simulations" Polymers 12, no. 10: 2392. https://doi.org/10.3390/polym12102392
APA StyleAmengual-Rigo, P., Carrillo, J., Blanco, J., & Guallar, V. (2020). Predicting Antibody Neutralization Efficacy in Hypermutated Epitopes Using Monte Carlo Simulations. Polymers, 12(10), 2392. https://doi.org/10.3390/polym12102392