Dependence of the Physical Properties and Molecular Dynamics of Thermotropic Liquid Crystalline Copolyesters on p-Hydroxybenzoic Acid Content


Abstract
1. Introduction
2. Materials and Methods
2.1. Materials
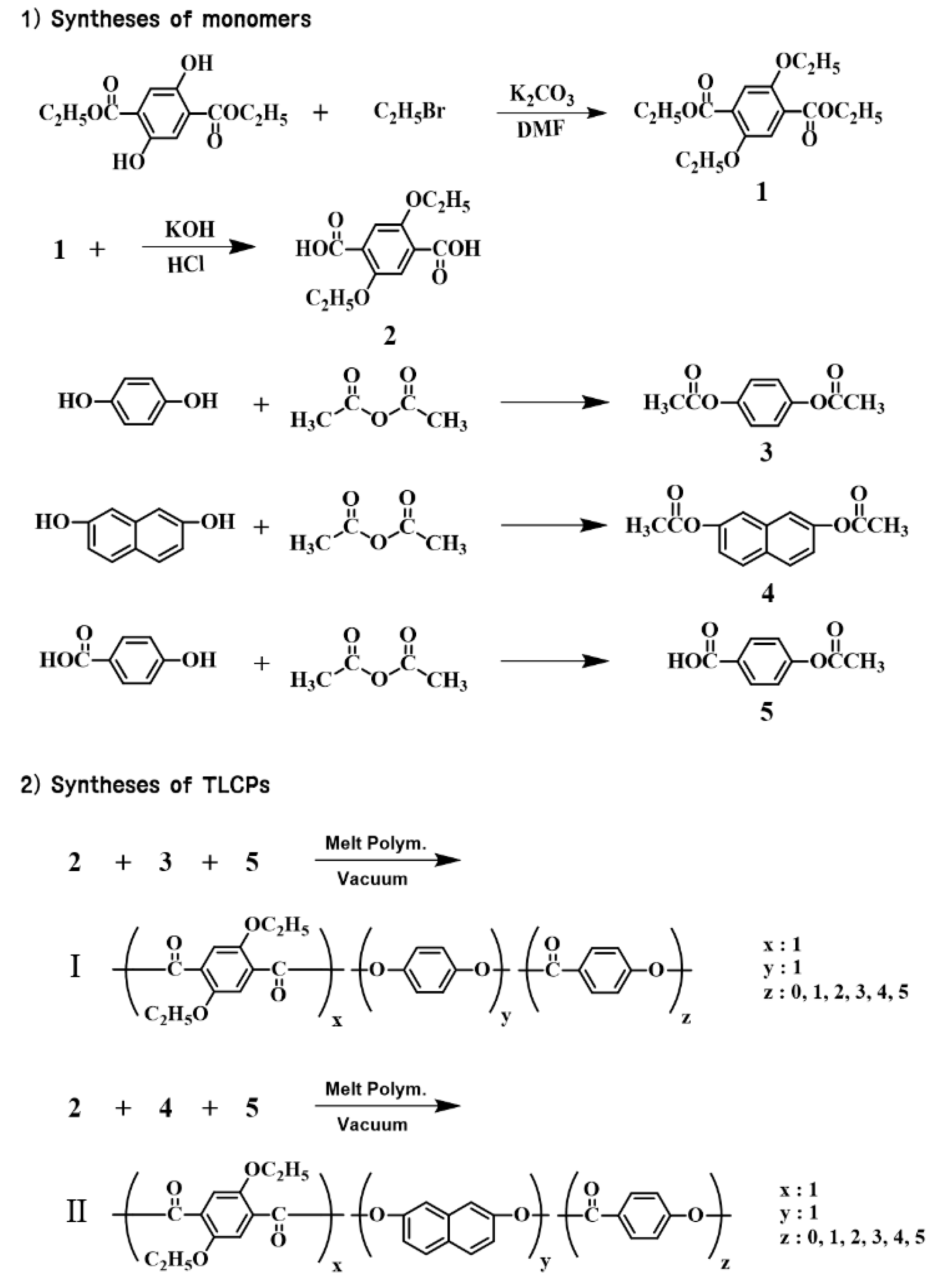
2.2. Syntheses of Monomers
2.3. Synthesis of TLCP-I
2.4. Synthesis of TLCP-II
2.5. NMR Spectroscopy
2.6. Characterization
3. Results and Discussion
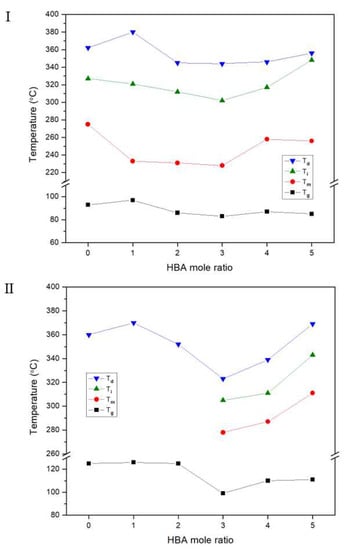
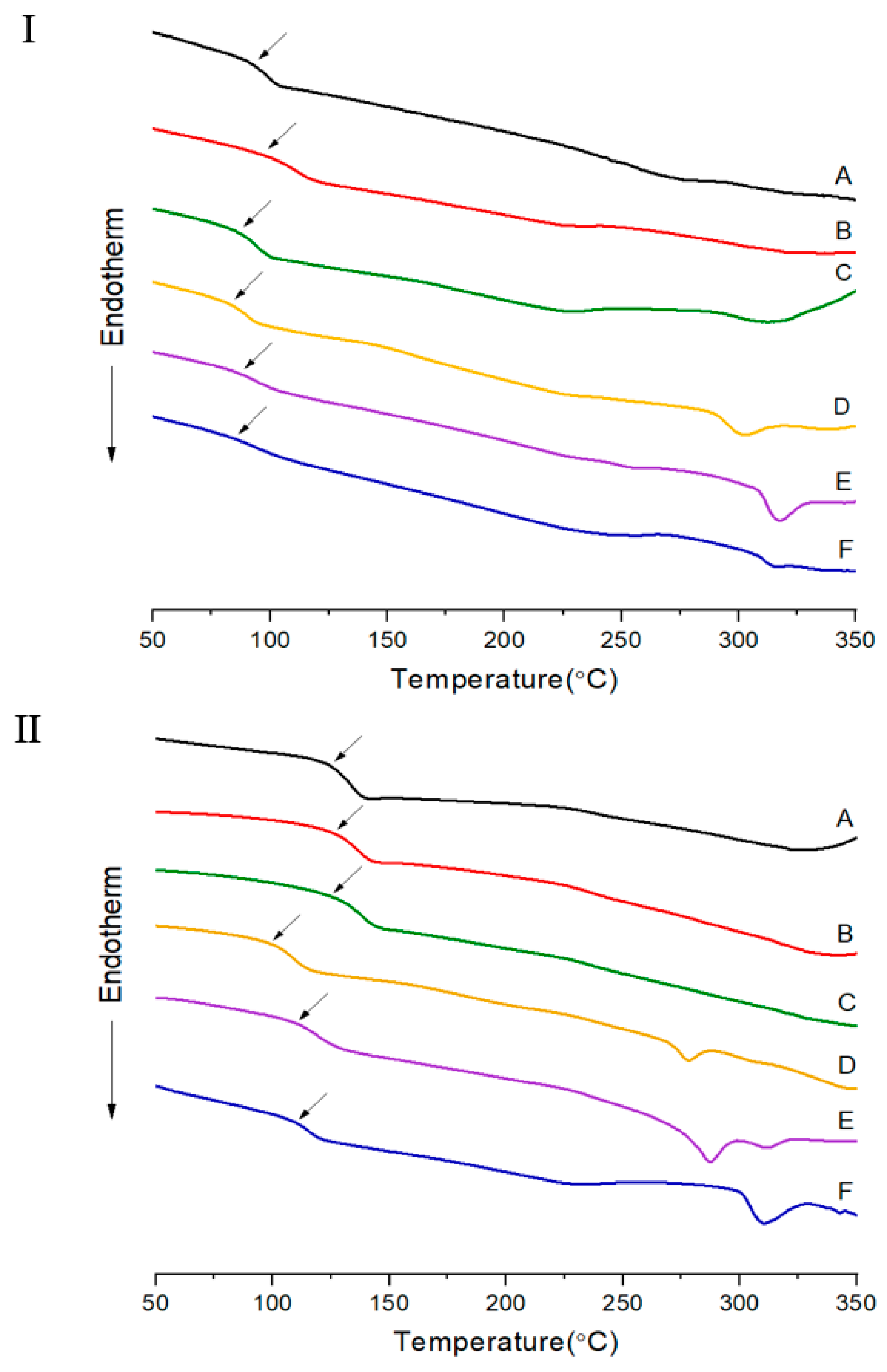
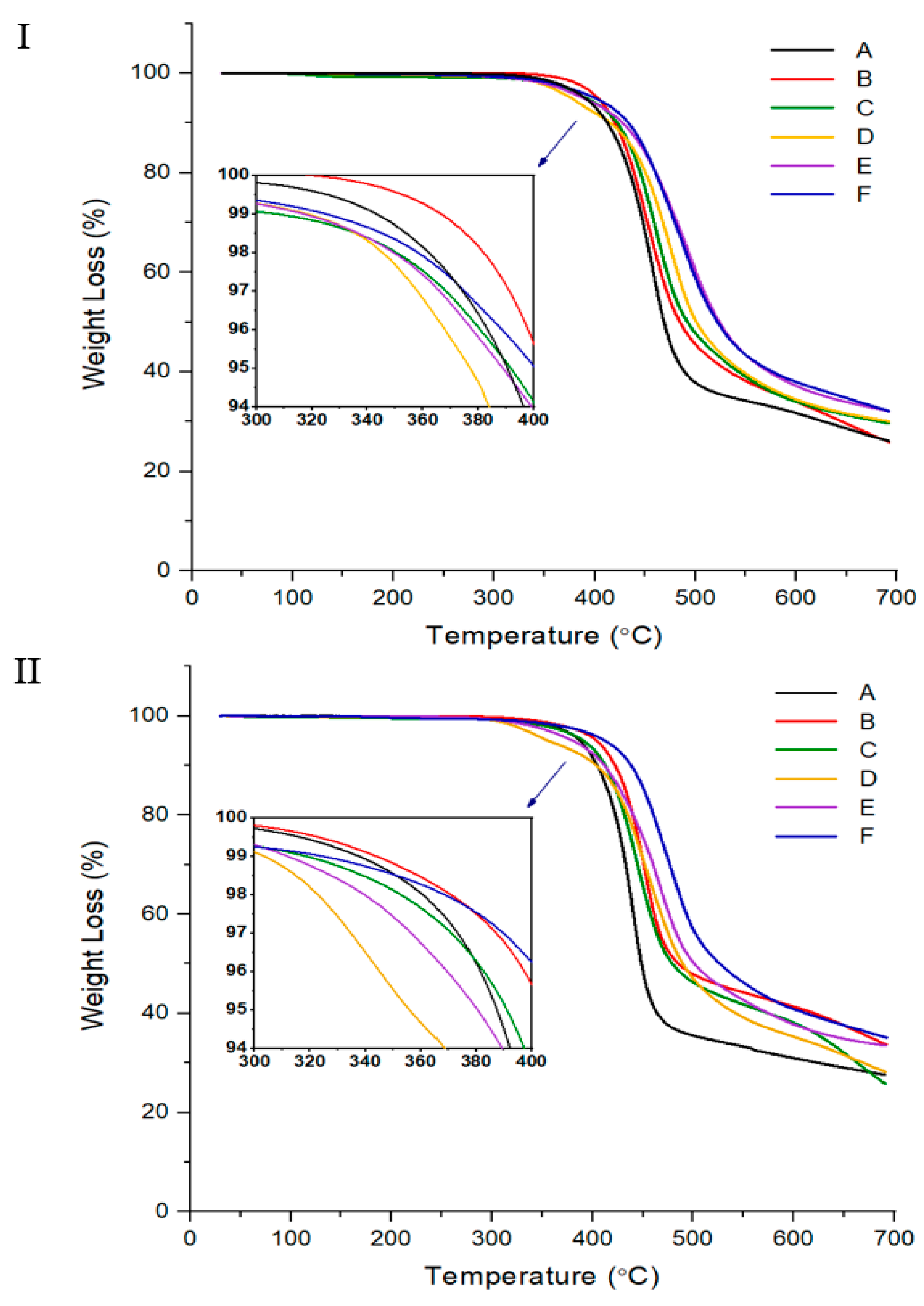
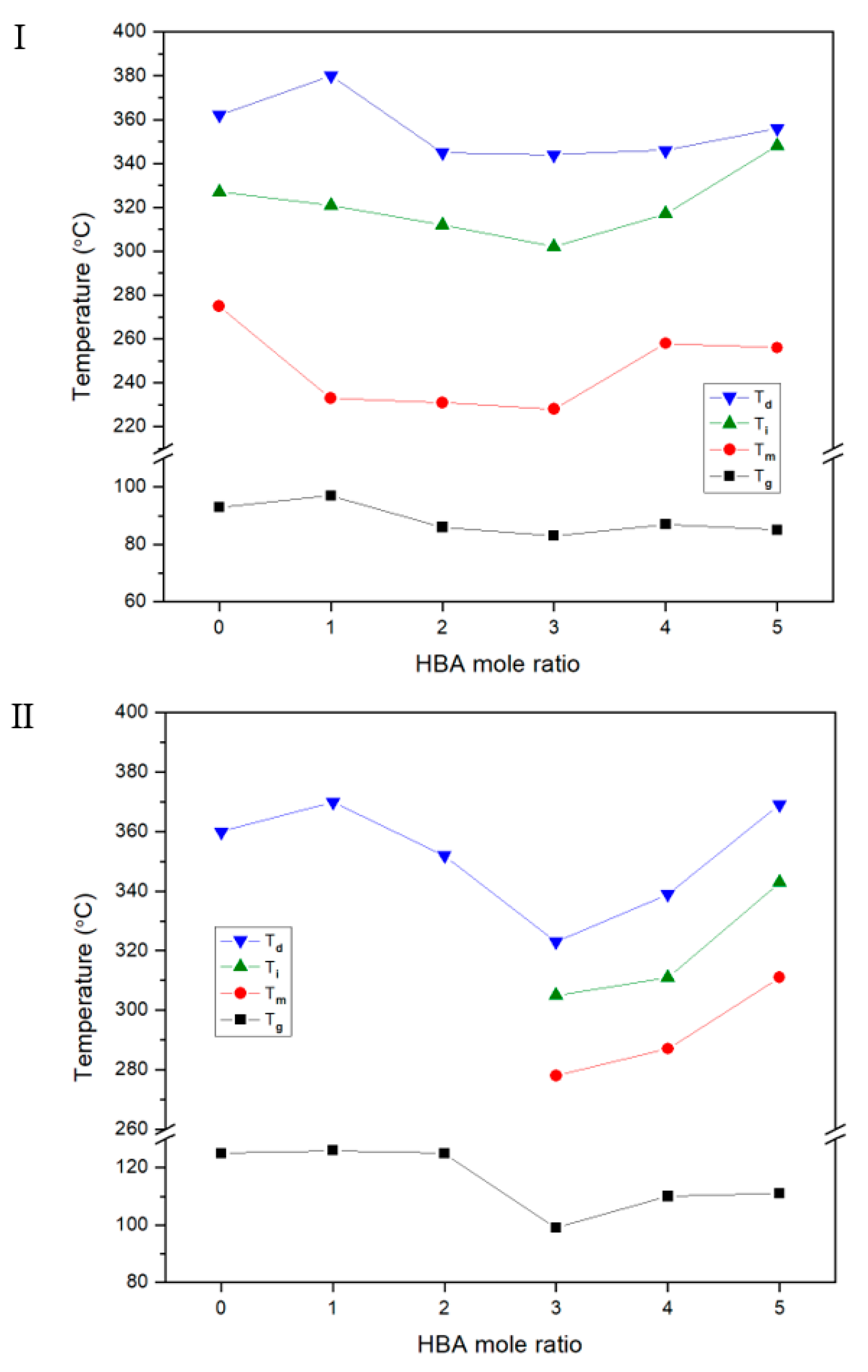
3.1. Thermal Behavior
3.2. Liquid Crystalline Mesophase
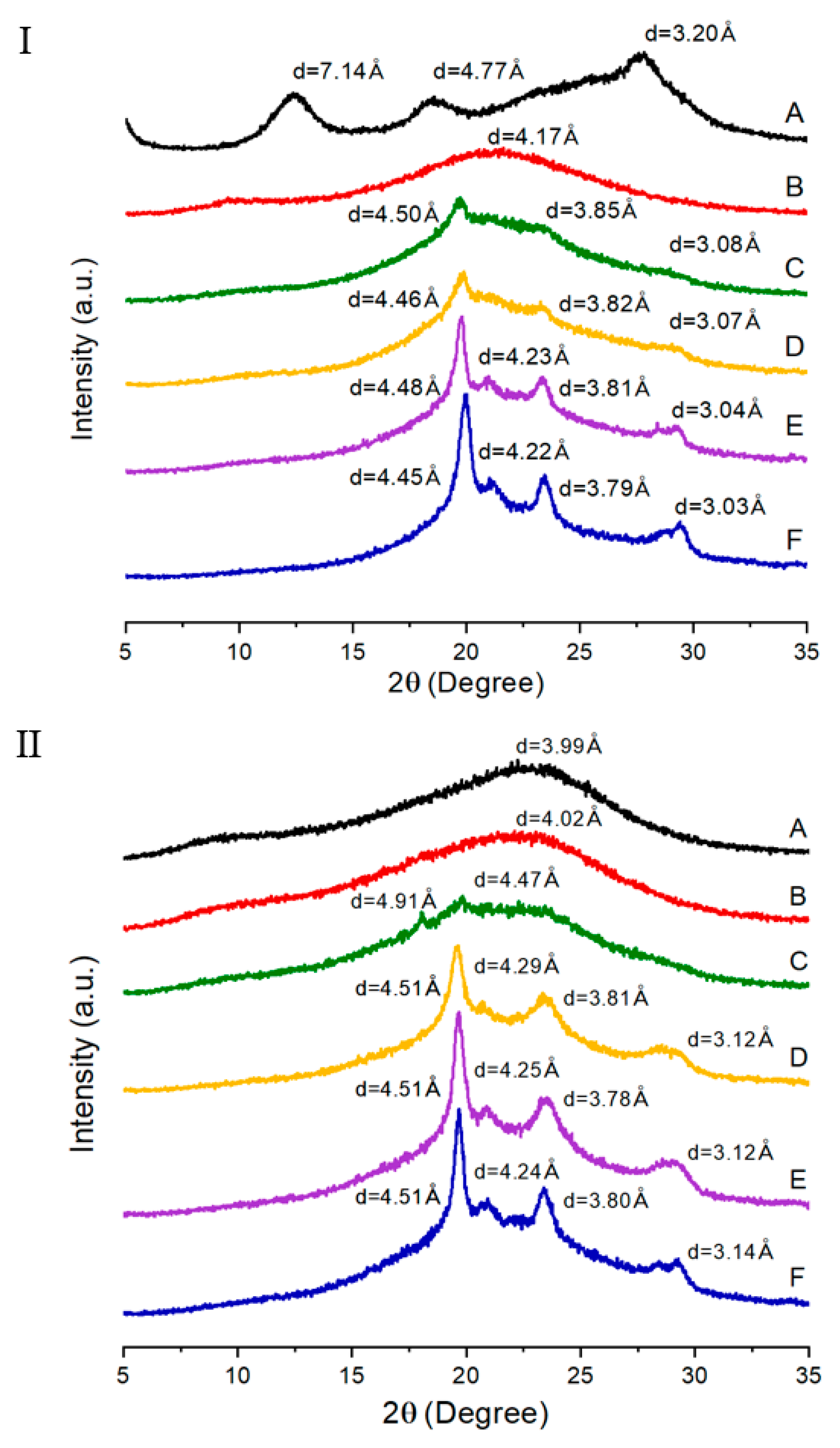
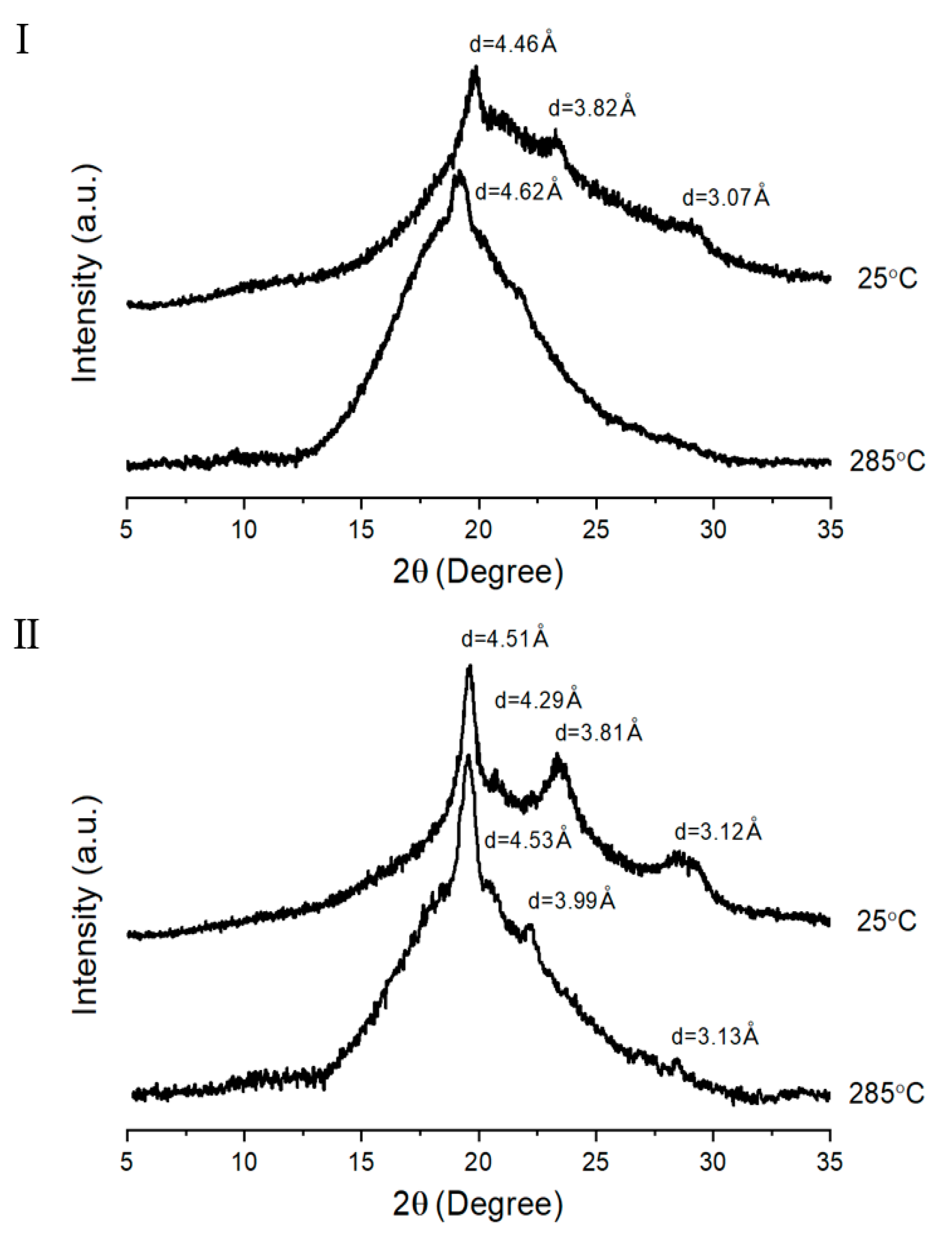
3.3. XRD
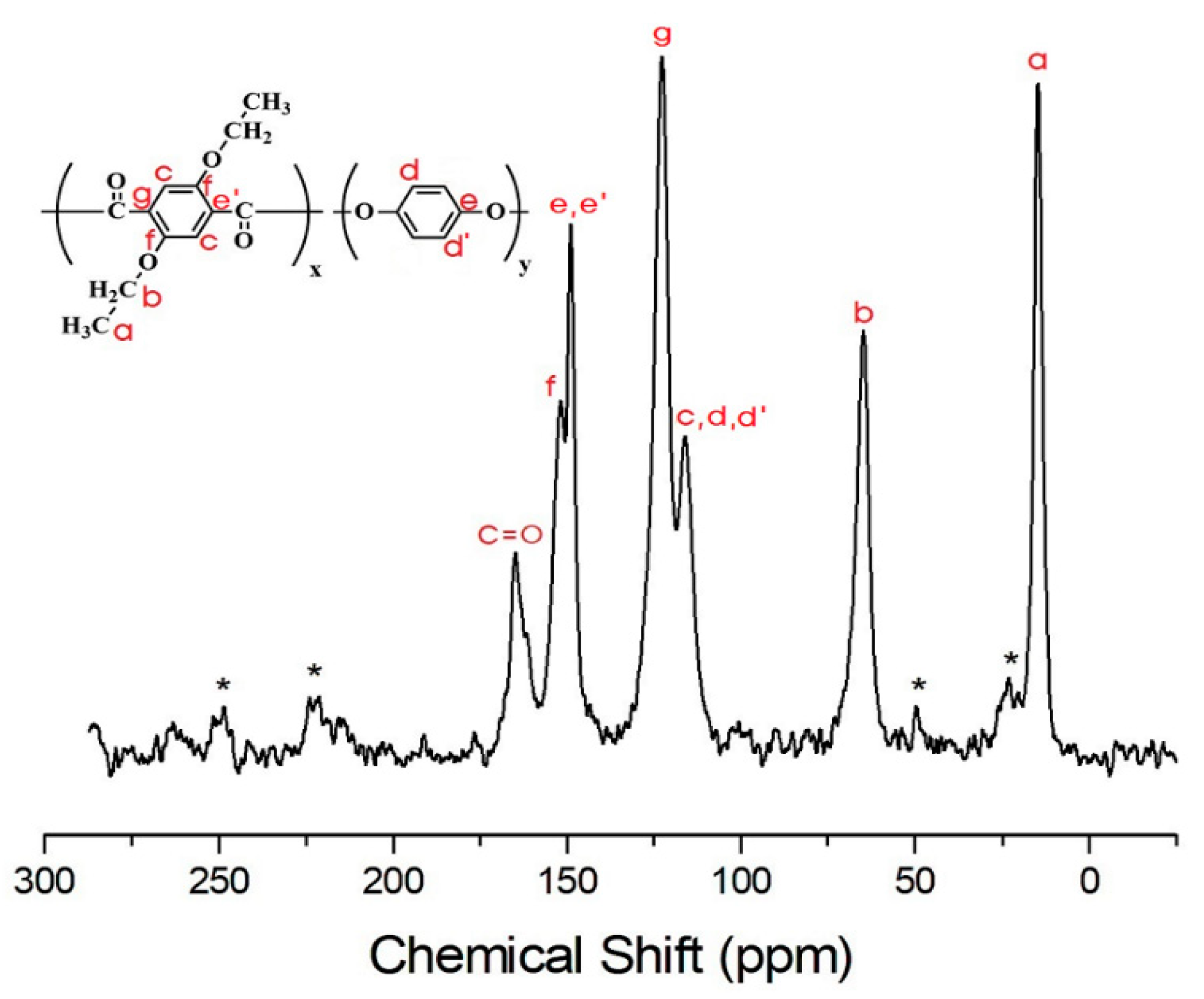
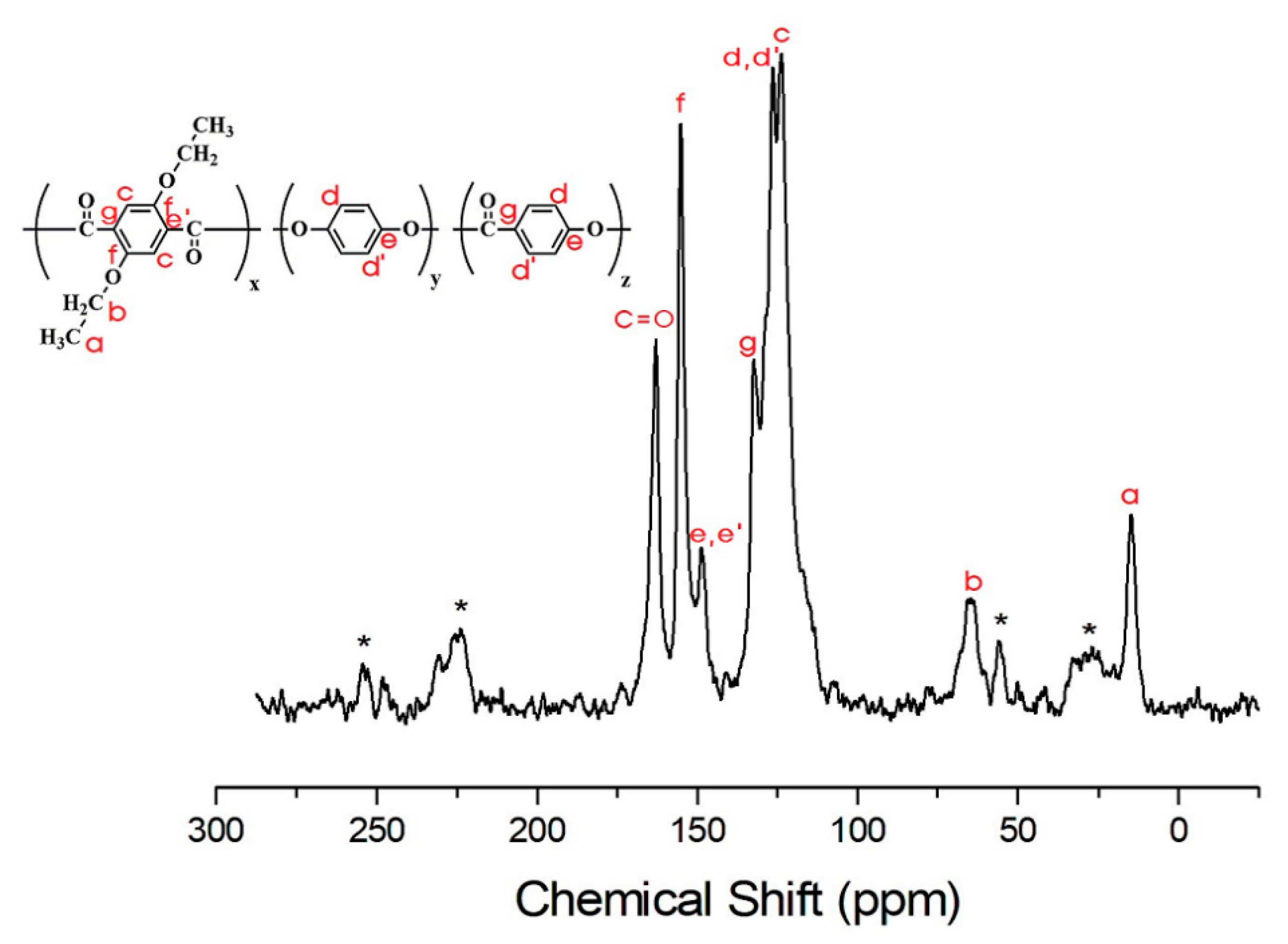
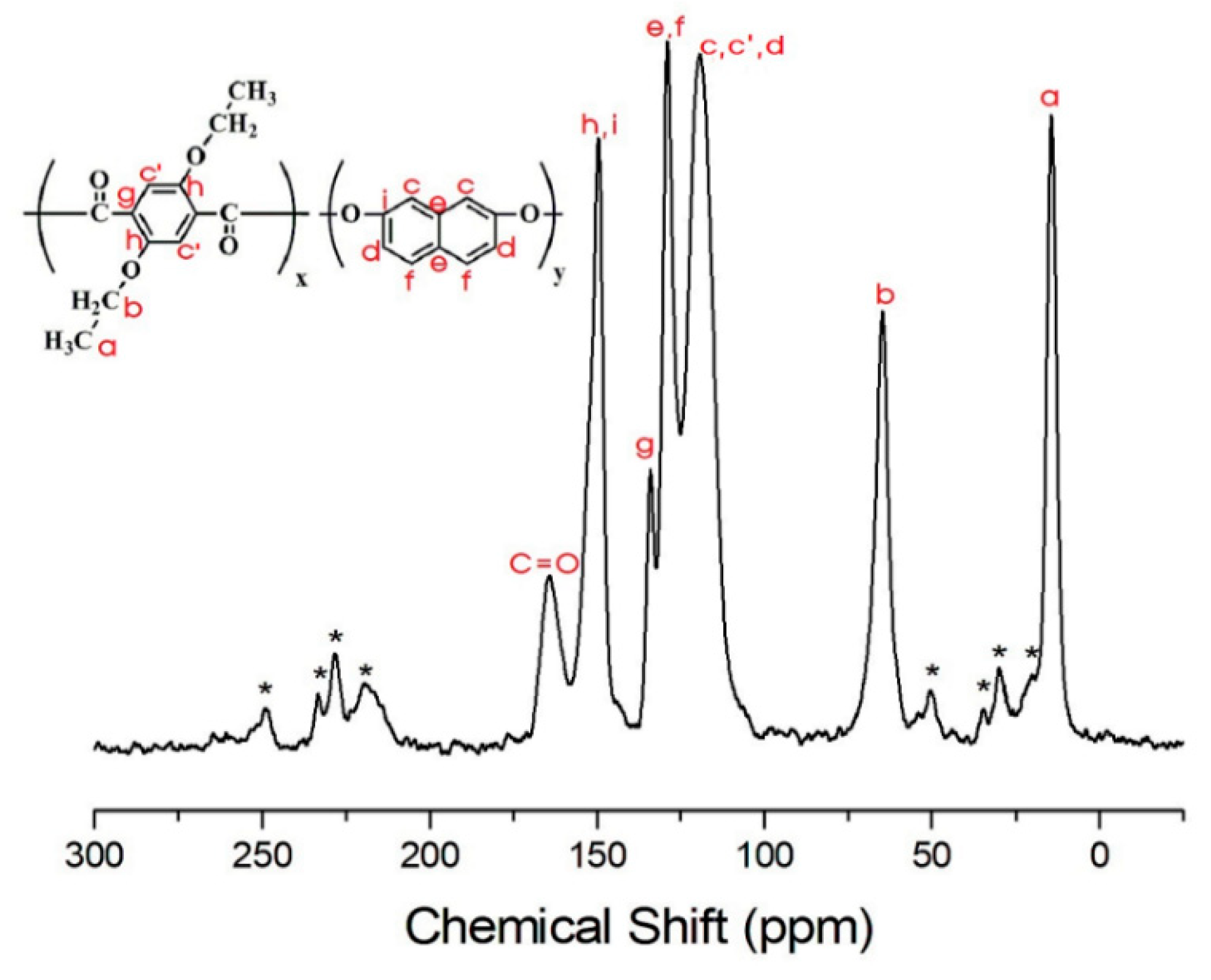
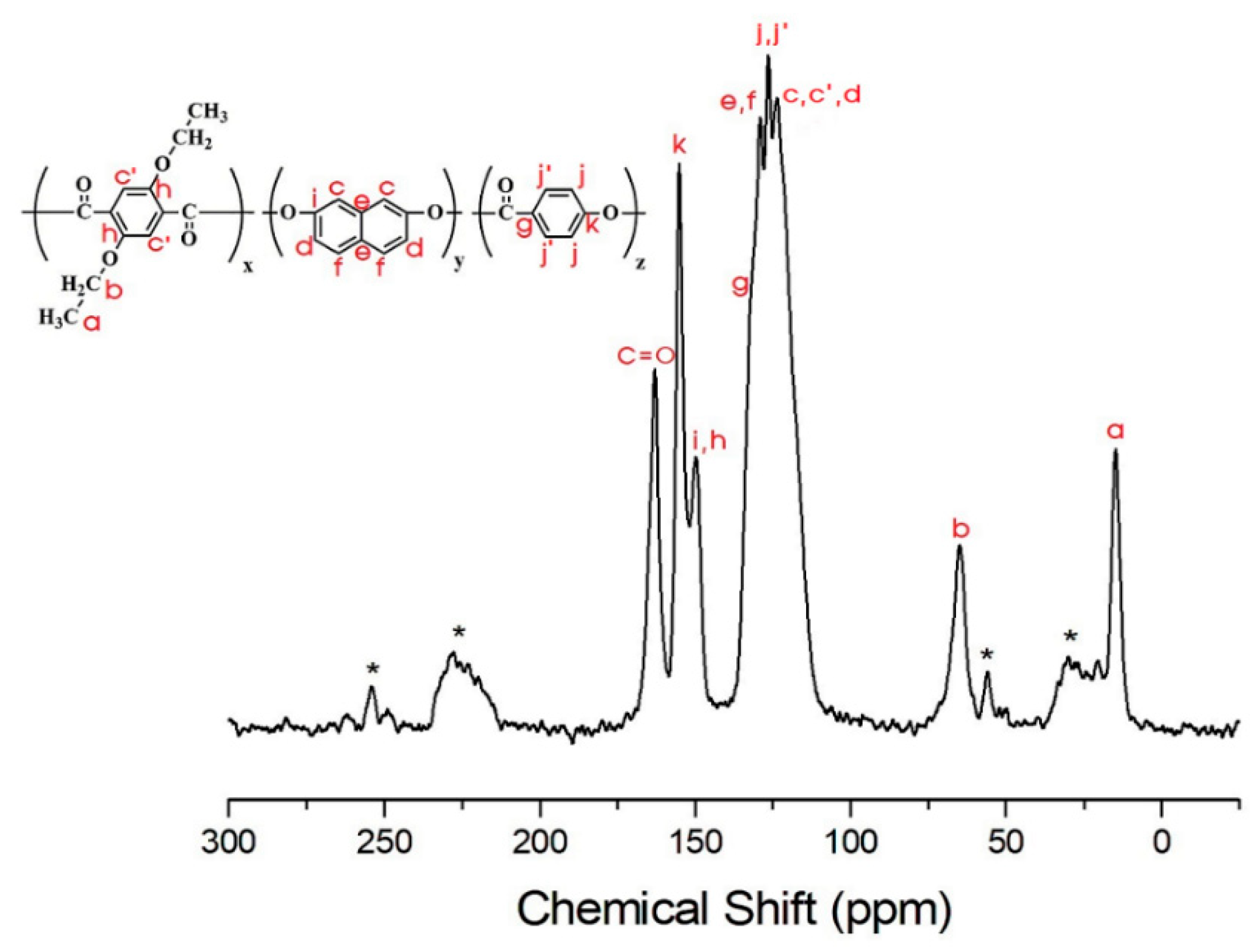
3.4. 13C Chemical Shifts and Relaxation Times
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Schaller, R.; Peijs, T.; Tervoort, T.A. High-performance liquid-crystalline polymer films for monolithic “composites”. Compos. Part A Appl. Sci. Manuf. 2016, 81, 296–304. [Google Scholar] [CrossRef]
- Beers, D.E.; Ramirez, J.E. Vectran High-performance Fibre. J. Text. Inst. 2008, 81, 561–574. [Google Scholar] [CrossRef]
- Kim, T.K.; Kim, S.O.; Chung, I.J. Synthesis and characterization of thermotropic liquid crystalline poly(ester imide)s. Polym. Adv. Technol. 1997, 8, 305–318. [Google Scholar] [CrossRef]
- Chen, Y.; Wombacher, R.; Wendorff, J.H.; Visjager, J.; Smith, P.; Greiner, A. Design, synthesis, and properties of new biodegradable aromatic/aliphatic liquid crystalline copolyesters. Biomacromolecules 2003, 4, 974–980. [Google Scholar] [CrossRef]
- Serbezeanu, D.; Vlad-Bubulac, T.; Hamciuc, C.; Aflori, M. Structure and properties of phosphorous-containing thermotropic liquid-crystalline aliphatic-aromatic copolyesters. Macromol. Chem. Phys. 2010, 211, 1460–1471. [Google Scholar] [CrossRef]
- Lefèvre, J.; Feldman, K.; Giesbrecht, J.; Smith, P.; Tervoort, T.A.; Meijer, H.E.H. “Foil spintrusion” of high-performance polymer films. J. Polym. Sci. Part B Polym. Phys. 2012, 50, 1713–1727. [Google Scholar] [CrossRef]
- Chae, H.G.; Kumar, S. Rigid-rod polymeric fibers. J. Appl. Polym. Sci. 2006, 100, 791–802. [Google Scholar] [CrossRef]
- Dingemans, T.; Knijnenberg, A.; Iqbal, M.; Weiser, E.; Stclair, T. All-aromatic liquid crystal thermosets: New high-performance materials for structural applications. Liq. Cryst. Today 2006, 15, 19–24. [Google Scholar] [CrossRef]
- Pramoda, K.P.; Chung, T.S. Crystallization and melting behavior of zenite thermotropic liquid crystalline polymers. Polym. Eng. Sci. 2002, 42, 439–451. [Google Scholar] [CrossRef]
- Pospiech, D.; Korwitz, A.; Eckstein, K.; Komber, H.; Jehnichen, D.; Suckow, M.; Lederer, A.; Arnhold, K.; Göbel, M.; Bremer, M.; et al. Fiber formation and properties of polyester/lignin blends. J. Appl. Polym. Sci. 2019, 136, 48257. [Google Scholar] [CrossRef]
- Wang, X.; Bu, H.; Luise, R.R. Synthesis and eutectic behavior of liquid crystalline copolyesters. J. Polym. Sci. Part B Polym. Phys. 2009, 47, 2171–2177. [Google Scholar] [CrossRef]
- Muhlebach, A.; Economy, J.; Johnson, R.D.; Karis, T.; Lyerla, J. Direct evidence for transesterification and randomization in a mixture of homopolyesters of poly(4-hydroxybenzoic acid) and poly(6-hydroxy-2-naphthoic acid) above 450 degree. Macromolecules 1990, 23, 1803–1809. [Google Scholar] [CrossRef]
- Bhowmik, P.K.; Han, H.; Garay, R.O. Fully aromatic thermotropic liquid crystalline homopolyesters of 3,4′-benzophenone dicarboxylic acid. J. Polym. Sci. Part A Polym. Chem. 1994, 32, 333–342. [Google Scholar] [CrossRef]
- Itoyama, K. Melt spinning of thermotropic liquid-crystal polyesters to form ultrahigh-modulus filaments. J. Polym. Sci. Part B Polym. Phys. 1988, 26, 1845–1863. [Google Scholar] [CrossRef]
- Nelson, A.M.; Fahs, G.B.; Moore, R.B.; Long, T.E. High-performance segmented liquid crystalline copolyesters. Macromol. Chem. Phys. 2015, 216, 1754–1763. [Google Scholar] [CrossRef]
- Wilsens, C.H.R.M.; Noordover, B.A.J.; Rastogi, S. Aromatic thermotropic polyesters based on 2,5-furandicarboxylic acid and vanillic acid. Polymer 2014, 55, 2432–2439. [Google Scholar] [CrossRef]
- Hirano, H.; Kadota, J.; Agari, Y.; Harada, T.; Tanaka, M.; Hasegawa, K. Linear polymers with sulfur in the main chain. IV. Synthesis of thermotropic liquid-crystalline polythioesters based on 4,4′-biphenyldithiol with excellent adhesive properties. Polym. Eng. Sci. 2007, 47, 262–269. [Google Scholar] [CrossRef]
- Young-Wan, K.; Hoon, C.D.; Jin, J.-I. Liquid crystalline aromatic polyesters. Polymer (Korea) 2005, 29, 523–535. [Google Scholar]
- Jackson, W.J. Liquid crystalline polymers. 5. Liquid crystalline polyesters containing naphthalene rings. Macromolecules 1983, 16, 1027–1033. [Google Scholar] [CrossRef]
- Blackwell, J.; Gutierrez, G. The structure of liquid crystalline copolyester fibers prepared from p-hydroxybenzoic acid, 2,6-dihydroxy naphthalene, and terephthalic acid. Polymer 1982, 23, 671–675. [Google Scholar] [CrossRef]
- Koenig, J.L. Spectroscopy of Polymers; Elsevier: Amsterdam, The Netherland, 1999. [Google Scholar]
- McBrierty, V.J.; Packer, K.J. Nuclear Magnetic Resonance in Solid Polymers; Cambridge University Press: Cambridge, UK, 2006. [Google Scholar]
- Lim, A.R. Effects of paramagnetic interactions by the partial replacement of Zn2+ ions with Cu2+ ions in lead-free zinc-based perovskite (MA)2ZnCl4 crystal by MAS NMR. AIP Adv. 2019, 9, 105115. [Google Scholar] [CrossRef]
- Damman, S.B.; Mercx, F.P.M.; Kootwijk-Damman, C.M. Liquid-crystalline main-chain polymers with a poly(p-phenylene terephthalate) backbone: 1. Synthesis, characterization and rheology of polyesters with alkoxy side chains. Polymer 1993, 34, 1891–1897. [Google Scholar] [CrossRef][Green Version]
- Chang, J.-H.; Ju, C.H.; Kim, S.H. Synthesis and characterization of a series of thermotropic liquid crystalline copolyester nanocomposites. J. Polym. Sci. Part B Polym. Phys. 2006, 44, 387–397. [Google Scholar] [CrossRef]
- Lim, A.R.; Chang, J.-H.; Kim, J.H.; Novak, B.M. Carbon-13 T1ρ investigation in two polycarbodiimides. Solid State Commun. 2000, 115, 23–28. [Google Scholar] [CrossRef]
- Bershtein, V.A.; Yegorov, V.M.; Yemel’yanov, Y.A. Relations between the main relaxation transitions in polymers and the length of segments and the character and degree of cooperation in molecular motion in the vicinity of Tg. Polym. Sci. U.S.S.R. 1985, 27, 2757–2764. [Google Scholar] [CrossRef]
- Chang, H.-S.; Wu, T.-Y.; Chen, Y. Synthesis and properties of TLCPs with 2,6-naphthalene-based mesogen, polymethylene spacer, and nonlinear 4,4′-thiodiphenyl links. J. Appl. Polym. Sci. 2002, 83, 1536–1546. [Google Scholar] [CrossRef]
- Sakurai, T.; Nojima, S. Significant increase in the melting temperature of poly(ɛ-caprolactone) blocks confined in the crystallized lamellar morphology of poly(ɛ-caprolactone)- block -polyethylene copolymers. Polym. J. 2011, 43, 370–377. [Google Scholar] [CrossRef]
- Jin, J.-I.; Kang, C.-S. Thermotropic main chain polyesters. Prog. Polym. Sci. 1997, 22, 937–973. [Google Scholar] [CrossRef]
- Langelaan, H.C.; de Boer, A.P. Crystallization of thermotropic liquid crystalline HBA/HNA copolymers. Polymer 1996, 37, 5667–5680. [Google Scholar] [CrossRef]
- Ballauff, M. Rigid rod polymers having flexible side chains, 1. Thermotropic poly(1,4-phenylene 2,5-dialkoxyterephthalate)s. Makromol. Chem., Rapid Commun. 1986, 7, 407–414. [Google Scholar] [CrossRef]
- Hakemi, H. On the miscibility of liquid crystalline polymers. Polymer 2000, 41, 6145–6150. [Google Scholar] [CrossRef]
- Wei, P.; Cakmak, M.; Chen, Y.; Wang, X.; Wang, Y.; Wang, Y. Aromatic liquid crystalline copolyesters with low Tm and high Tg: Synthesis, characterization, and properties. J. Appl. Polym. Sci. 2014, 131, 40487 (1–10). [Google Scholar] [CrossRef]
- Chen, B.-K.; Tsay, S.-Y.; Chen, J.-Y. Synthesis and properties of liquid crystalline polymers with low Tm and broad mesophase temperature ranges. Polymer 2005, 46, 8624–8633. [Google Scholar] [CrossRef]
- Elias, F.; Clarke, S.M.; Peck, R.; Terentjev, E.M. Equilibrium textures in main-chain liquid crystalline polymers. Europhys. Lett. 1999, 47, 442–448. [Google Scholar] [CrossRef][Green Version]
- Jin, J.-I.; Chang, J.-H.; Shim, H.K. Properties of aromatic copolyesters derived from p-hydroxybenzoic acid, terephthalic acid, and naphthalenediol isomers. Macromolecules 1989, 22, 93–99. [Google Scholar] [CrossRef]
- Shukla, U.; Rao, K.V.; Rakshit, A.K. Thermotropic liquid-crystalline polymers: Synthesis, characterization, and properties of poly(azomethine esters). J. Appl. Polym. Sci. 2003, 88, 153–160. [Google Scholar] [CrossRef]
- Babacan, V.; Aksoy, S.; Yerlikaya, Z.; Altinok, H. Thermal and morphological properties of thermotropic liquid-crystalline copolyesters containing poly(ethylene terephthalate), 4-hydroxyphenylacetic acid and main-chain rigid aromatic units. Polym. Int. 2010, 59, 749–755. [Google Scholar] [CrossRef]
- Lim, A.R.; Novak, B.M. Helix conformation in helical polycarbodiimides studied by solid state 13C NMR. Chem. Phys. 2001, 272, 199–212. [Google Scholar] [CrossRef]
- Pouchert, C.J. The Aldrich Library of 13C and 1H FT NMR Spectra; Aldrich Publication: New York, NY, USA, 1993. [Google Scholar]
- Jasinska-Walc, L.; Villani, M.; Dudenko, D.; van Asselen, O.; Klop, E.; Rastogi, S.; Hansen, M.R.; Koning, C.E. Local conformation and cocrystallization phenomena in renewable diaminoisoidide-based polyamides studied by FT-IR, solid state NMR, and WAXD, and WAXD. Macromolecules 2012, 45, 2796–2808. [Google Scholar] [CrossRef]
- Abragam, A. The Principles of Nuclear Magnetism; Oxford University Press: Oxford, UK, 1961. [Google Scholar]













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| TLCP | I a | II b | ||||
|---|---|---|---|---|---|---|
| ETA | HQ | HBA | ETA | DHN | HBA | |
| A | 1 | 1 | 0 | 1 | 1 | 0 |
| B | 1 | 1 | 1 | 1 | 1 | 1 |
| C | 1 | 1 | 2 | 1 | 1 | 2 |
| D | 1 | 1 | 3 | 1 | 1 | 3 |
| E | 1 | 1 | 4 | 1 | 1 | 4 |
| F | 1 | 1 | 5 | 1 | 1 | 5 |
| TLCP | I a | II b |
|---|---|---|
| Temperature (°C)/time (min)/pressure (Torr) | Temperature (°C)/time (min)/pressure (Torr) | |
| A | 240/120/760 → 260/120/760 → 280/60/760 → 290/60/760 → 300/40/760 → 310/40/300 → 320/30/1 | 250/60/760 → 270/50/760 → 280/30/760 → 290/50/760 → 290/30/240 → 290/50/1 |
| B | 240/120/760 → 260/80/760 → 280/60/760 → 300/45/760 → 300/30/300 → 310/60/1 | 250/40/760 → 270/120/760 → 270/60/240 → 270/40/1 |
| C | 240/150/760 → 260/60/760 → 280/90/760 → 300/75/760 → 310/35/300 → 310/40/1 | 250/30/760 → 270/30/760 → 280/30/760 → 290/80/760 → 290/30/240 → 290/50/1 |
| D | 240/120/760 → 260/60/760 → 280/60/760 → 300/60/760 → 305/45/300 → 310/30/1 | 245/120/760 → 250/30/760 → 265/50/760 → 270/50/240 → 285/40/1 |
| E | 240/120/760 → 260/60/760 → 280/60/760 → 300/60/760 → 305/35/300 → 310/40/1 | 250/120/760 → 265/50/760 → 280/60/760 → 285/50/240 → 290/30/1 |
| F | 245/120/760 → 260/60/760 → 280/60/760 → 300/60/760 → 310/30/300 → 310/30/1 | 260/60/760 → 270/60/760 → 280/60/760 → 295/30/760 → 295/30/240 → 295/30/1 |
| TLCP | Ⅰ a | Ⅱ b | |||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| IV c | Tg (°C) | Tm (°C) | Ti (°C) | ∆Hm (J/g) | ∆Hi (J/g) | TDi d (°C) | wtR600 e (%) | LC Phase | DC f (%) | IV | Tg (°C) | Tfg (°C) | Tm (°C) | Ti (°C) | ∆Hm (J/g) | ∆Hi (J/g) | TDi (°C) | wtR600 (%) | LC Phase | DC (%) | |
| A | Insol.h | 93 | 275 | 327 | 1.07 | 1.32 | 362 | 32 | Nematic | 39 | Insol. | 125 | 200 | 360 | 31 | No. | 0 | ||||
| B | Insol. | 96 | 233 | 321 | 1.28 | 1.42 | 380 | 34 | Nematic | 20 | Insol. | 126 | 200 | 370 | 41 | No. | 0 | ||||
| C | Insol. | 86 | 231 | 312 | 1.69 | 2.07 | 345 | 34 | Nematic | 20 | Insol. | 125 | 200 | 352 | 38 | No. | 3 | ||||
| D | Insol. | 83 | 228 | 302 | 1.61 | 2.50 | 344 | 34 | Nematic | 24 | Insol. | 99 | 278 | 305 | 2.66 | 1.04 | 323 | 36 | Nematic | 15 | |
| E | Insol. | 87 | 258 | 317 | 1.61 | 3.51 | 346 | 37 | Nematic | 26 | Insol. | 110 | 287 | 311 | 3.01 | 2.76 | 339 | 38 | Nematic | 16 | |
| F | Insol. | 86 | 256 | 348 | 2.74 | 1.47 | 356 | 38 | Nematic | 39 | Insol. | 111 | 311 | 343 | 3.42 | 1.01 | 369 | 41 | Nematic | 18 | |
| TLCP | d (Å) (2θ (degree)) | |||||||
|---|---|---|---|---|---|---|---|---|
| I a | II b | |||||||
| A | 7.14 (12.38) c | 4.77 (18.56) | 3.20 (27.82) | - | 3.99 (22.24) | - | - | - |
| B | 4.17 (21.28) | - | - | - | 4.02 (22.1) | - | - | - |
| C | 4.50 (19.72) | 3.85 (23.08) | 3.08 (28.96) | - | 4.91 (18.04) | 4.47 (19.84) | - | - |
| D | 4.46 (19.88) | 3.82 (23.26) | 3.07 (29.06) | - | 4.51 (19.64) | 4.29 (20.7) | 3.81 (23.34) | 3.12 (28.56) |
| E | 4.48 (19.78) | 4.23 (20.96) | 3.81 (23.32) | 3.04 (29.3) | 4.51 (19.68) | 4.25 (20.88) | 3.78 (23.52) | 3.12 (28.58) |
| F | 4.45 (19.94) | 4.22 (21.02) | 3.79 (23.44) | 3.03 (29.4) | 4.51 (19.68) | 4.24 (20.94) | 3.80 (23.4) | 3.14 (28.42) |
| TLCP | I-A | I-B | I-C | I-D | I-E | I-F |
|---|---|---|---|---|---|---|
| CH3-a | 39.2 | 39.7 | 23.8 | 24.4 | 25.4 | 25.7 |
| CH2-b | 4.4 | 3.5 | 4.1 | 3.4 | 5.1 | 2.9 |
| c | 12.2 | 17.4 | 11.9 | 14.7 | 14.1 | 14.0 |
| d | 41.9 | 46.0 | 42.9 | 42.9 | 47.3 | 45.2 |
| e | 125.9 | 95.9 | 90.8 | 80.4 | 72.9 | 66.6 |
| f | 121.0 | 81.5 | 83.1 | 70.4 | 77.8 | 87.8 |
| g | 12.2 | 8.4 | 6.7 | 6.1 | 5.8 | 6.7 |
| C=O | 92.0 | 62.2 | 56.0 | 59.1 | 65.8 | 65.8 |
| TLCP | II-A | II-B | II-C | II-D | II-E | II-F |
|---|---|---|---|---|---|---|
| CH3-a | 55.1 | 42.5 | 38.5 | 34.0 | 24.5 | 36.6 |
| CH2-b | 8.2 | 5.8 | 5.8 | 6.4 | 4.5 | 6.7 |
| c,d | 29.3 | 21.2 | 18.0 | 19.0 | 18.0 | 27.4 |
| e,f | 68.9 | 53.1 | 42.7 | 23.9 | 22.3 | 20.5 |
| g | 119.9 | 118.0 | ||||
| h,i | 191.7 | 144.1 | 136.5 | 130.1 | 106.5 | 108.1 |
| j | 80.5 | 75.2 | ||||
| k | 107.8 | 114.2 | 101.1 | 94.6 | ||
| C=O | 182.6 | 101.1 | 100.4 | 102.3 | 83.1 | 77.6 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Park, G.T.; Lee, W.J.; Chang, J.-H.; Lim, A.R. Dependence of the Physical Properties and Molecular Dynamics of Thermotropic Liquid Crystalline Copolyesters on p-Hydroxybenzoic Acid Content. Polymers 2020, 12, 198. https://doi.org/10.3390/polym12010198
Park GT, Lee WJ, Chang J-H, Lim AR. Dependence of the Physical Properties and Molecular Dynamics of Thermotropic Liquid Crystalline Copolyesters on p-Hydroxybenzoic Acid Content. Polymers. 2020; 12(1):198. https://doi.org/10.3390/polym12010198
Chicago/Turabian StylePark, Gi Tae, Won Jun Lee, Jin-Hae Chang, and Ae Ran Lim. 2020. "Dependence of the Physical Properties and Molecular Dynamics of Thermotropic Liquid Crystalline Copolyesters on p-Hydroxybenzoic Acid Content" Polymers 12, no. 1: 198. https://doi.org/10.3390/polym12010198
APA StylePark, G. T., Lee, W. J., Chang, J.-H., & Lim, A. R. (2020). Dependence of the Physical Properties and Molecular Dynamics of Thermotropic Liquid Crystalline Copolyesters on p-Hydroxybenzoic Acid Content. Polymers, 12(1), 198. https://doi.org/10.3390/polym12010198