Fabrication and Characterization of Carboxymethyl Starch/Poly(l-Lactide) Acid/β-Tricalcium Phosphate Composite Nanofibers via Electrospinning


 ,
,
Abstract

1. Introduction
2. Materials and Methods
2.1. Electrospinning
2.2. Characterizations
2.2.1. Chemical Interactions
2.2.2. Thermal Behavior
2.2.3. X-ray Diffractions (XRD)
2.2.4. Wettability
2.2.5. Mechanical Properties
3. Results and Discussion
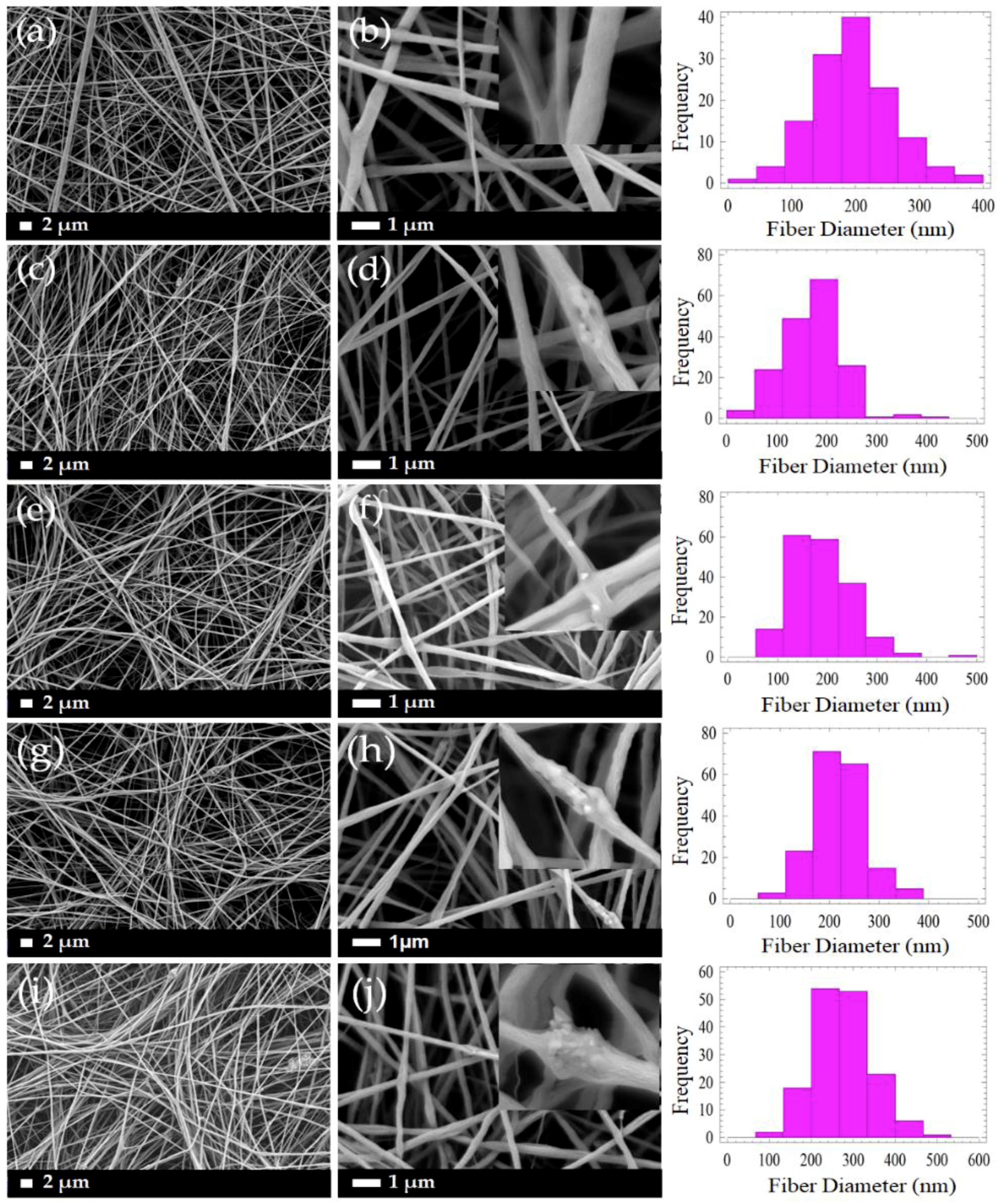
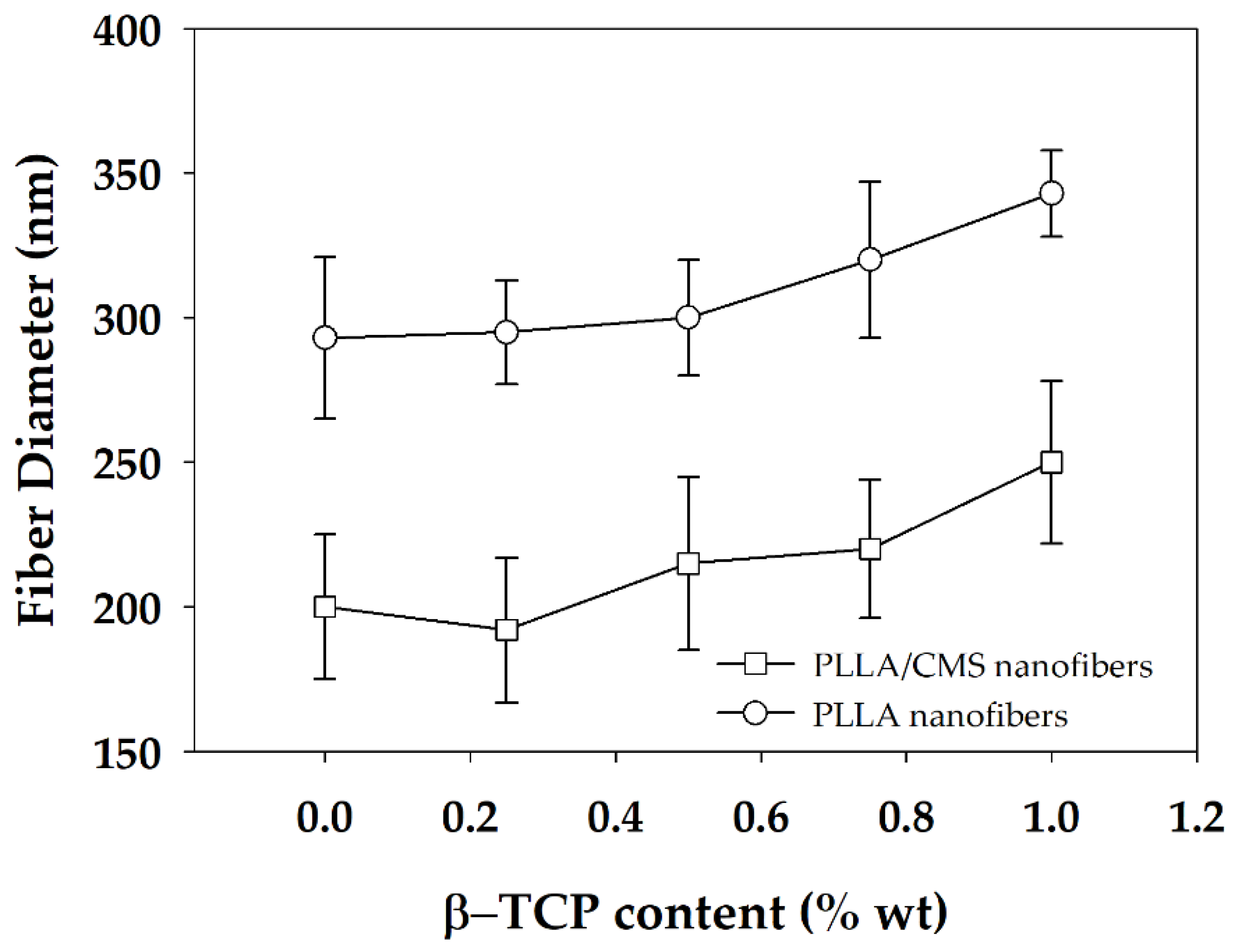
3.1. Morphology of PLLA/CMS/β-TCP Composite Nanofibers
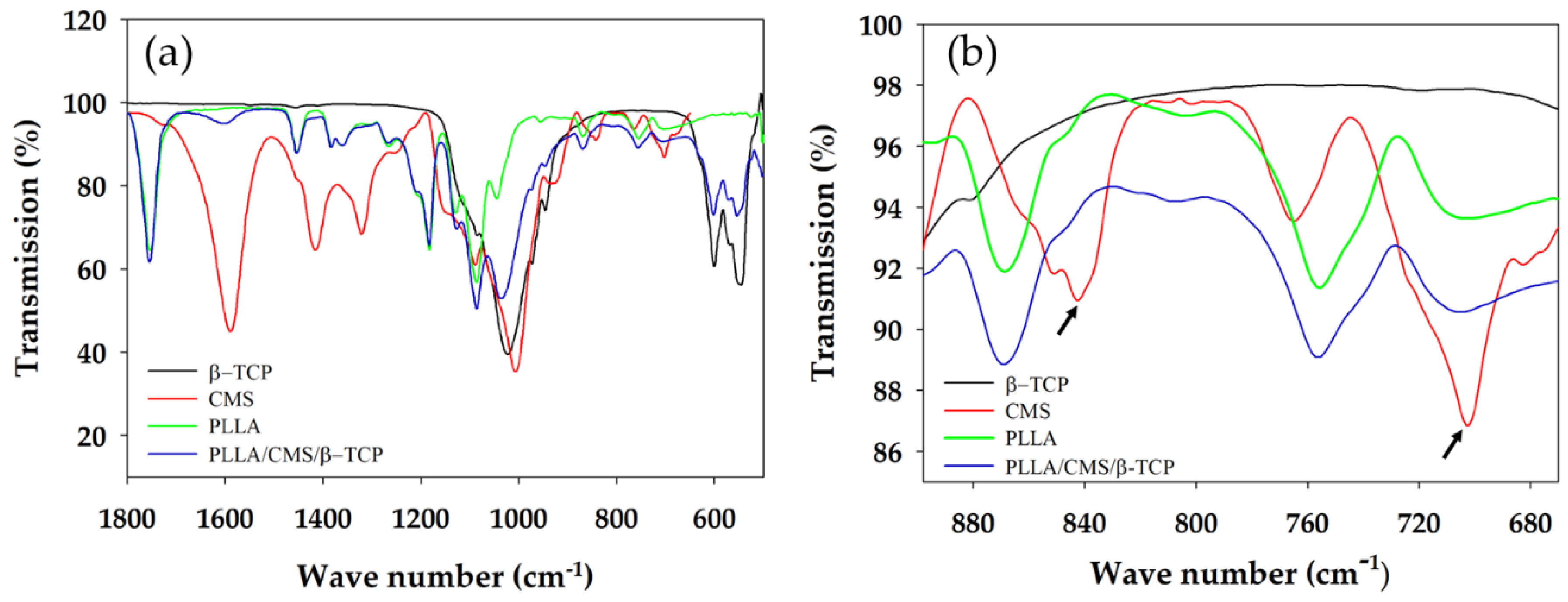
3.2. FTIR Analysis
3.3. Differential Scanning Calorimeter (DSC)
3.4. X-ray Diffraction Analysis (XRD)
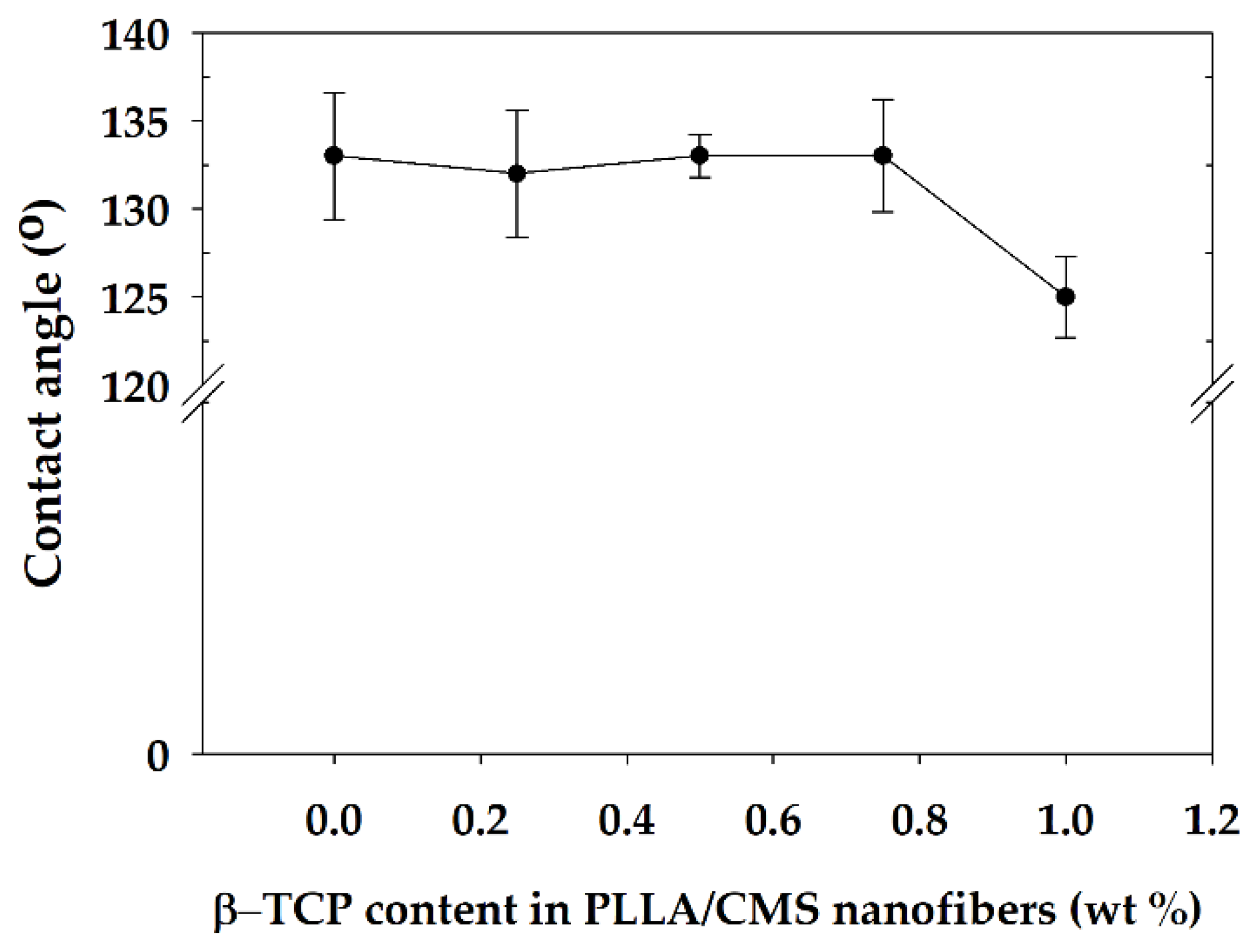
3.5. Wettability
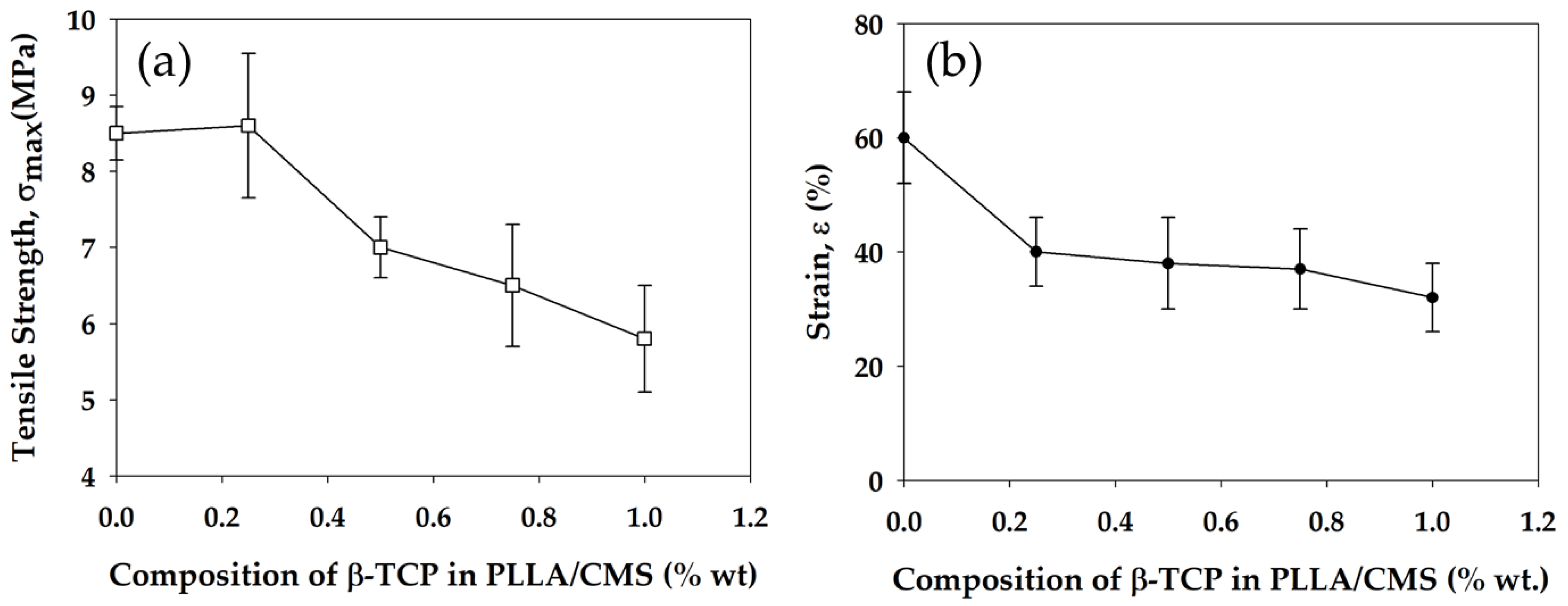
3.6. Mechanical Strength
4. Conclusions
Author Contributions
Funding
Acknowledgments
- Part of this work supported by Fundamental Research Grant Scheme (FRGS/1/2018/STG 07/UKM/02/03—Hybrid Nanofibers under Ionizing Radiation as a Guided Bone Regeneration Membrane, Universiti Kebangsaan Malaysia, Ministry of Higher Education.
- Part of this work was supported by the Ministry of Education, Youth and Sports of the Czech Republic and the European Union—European Structural and Investment Funds in the frames of Operational Programme Research, Development and Education—project Hybrid Materials for Hierarchical Structures (HyHi, Reg. No. CZ.02.1.01/0.0/0.0/16_019/0000843).
Conflicts of Interest
References
- Ramakrishna, S.; Fujihara, K.; Teo, W.E.; Lim, T.C.; Ma, Z. An Introduction to Electrospinning and Nanofibers; World Scientific: Singapore, 2005. [Google Scholar]
- Lv, D.; Wang, R.; Tang, G.; Mou, Z.; Lei, J.; Han, J. Ecofriendly Electrospun Membranes Loaded with Visible-Light-Responding Nanoparticles for Multifunctional Usages: Highly Efficient Air Filtration, Dye Scavenging, and Bactericidal Activity. ACS Appl. Mater. Inter. 2019, 11, 12880–12889. [Google Scholar] [CrossRef] [PubMed]
- Hanis, H.; Reusmaazran, Y.M.; Rashid, M.R.Z.; Rusymah, I.; Roy, C.S. Human Amniotic Membrane with Aligned Electrospun Fiber as Scaffold for Aligned Tissue Regeneration. Tissue Eng. Part C Methods 2018, 24, 368–378. [Google Scholar]
- Zhou, S.; Zhou, G.; Jiang, S.; Fan, P.; Hou, H. Flexible and refractory tantalum carbide-carbon electrospun nanofibers with high modulus and electric conductivity. Mater. Lett. 2017, 200, 97–100. [Google Scholar] [CrossRef]
- Sundarrajan, S.; Luck Tan, K.; Huat Lim, S.; Ramakrishna, S. Electrospun Nanofibers for Air Filtration Applications. Procedia Eng. 2014, 75, 159–163. [Google Scholar] [CrossRef]
- Yalcinkaya, F.; Siekierka, A.; Bryjak, M. Surface modification of electrospun nanofibrous membranes for oily wastewater separation. RSC Adv. 2017, 7, 56704–56712. [Google Scholar] [CrossRef]
- Lv, D.; Zhu, M.; Jiang, Z.; Jiang, S.; Zhang, Q.; Xiong, R. Green Electrospun Nanofibers and Their Application in Air Filtration. Macromol. Mater. Eng. 2018, 303, 1800336. [Google Scholar] [CrossRef]
- Jiang, S.; Chen, Y.; Duan, G.; Mei, C.; Greiner, A.; Agarwal, S. Electrospun nanofiber reinforced composites: A review. Polymer Chem. 2018, 9, 2685–2720. [Google Scholar] [CrossRef]
- Roche, R.; Yalcinkaya, F. Electrospun Polyacrylonitrile Nanofibrous Membranes for Point-of-Use Water and Air Cleaning. Chem. Open 2019, 8, 97–103. [Google Scholar] [CrossRef]
- Yalcinkaya, F.; Hruza, J. Effect of Laminating Pressure on Polymeric Multilayer Nanofibrous Membranes for Liquid Filtration. Nanomaterials 2018, 8, 272. [Google Scholar] [CrossRef]
- Yalcinkaya, F. A review on advanced nanofiber technology for membrane distillation. J. Eng. Fiber. Fabr. 2019, 14, 1–12. [Google Scholar] [CrossRef]
- Sill, T.J.; Von Recum, H.A. Electrospinning: Applications in drug delivery and tissue engineering. Biomaterials 2008, 29, 1989–2006. [Google Scholar] [CrossRef] [PubMed]
- Hu, X.; Liu, S.; Zhou, G.; Huang, Y.; Xie, Z.; Jing, X. Electrospinning of polymeric nanofibers for drug delivery applications. J. Control. Release 2014, 185, 12–21. [Google Scholar] [CrossRef] [PubMed]
- Pillay, V.; Dott, C.; Choonara, Y.E.; Tyagi, C.; Tomar, L.; Kumar, P.; du Toit, L.C.; Ndesendo, V.M.K. A Review of the Effect of Processing Variables on the Fabrication of Electrospun Nanofibers for Drug Delivery Applications. J. Nanomater. 2013, 2013, 1–22. [Google Scholar] [CrossRef]
- Sundarrajan, S.; Ramakrishna, S. The use of nanomaterials in smart protective clothing. In Smart Textiles for Protection; Woodhead Publishing Limited: Sawston, Cambridge, UK, 2012; pp. 127–147. [Google Scholar]
- Mirjalili, M.; Zohoori, S. Review for application of electrospinning and electrospun nanofibers technology in textile industry. J. Nanostructure Chem. 2016, 6, 207–213. [Google Scholar] [CrossRef]
- Lou, T.; Wang, X.; Song, G.; Gu, Z.; Yang, Z. Fabrication of PLLA/β-TCP nanocomposite scaffolds with hierarchical porosity for bone tissue engineering. Int. J. Biol. Macromol. 2014, 69, 464–470. [Google Scholar] [CrossRef]
- Thomas, S.; Grohens, Y.; Ninan, N. Nanotechnology Applications for Tissue Engineering; Elsevier: Amsterdam, The Netherlands, 2015. [Google Scholar]
- Arahira, T.; Maruta, M.; Matsuya, S.; Todo, M. Development and characterization of a novel porous β-TCP scaffold with a three-dimensional PLLA network structure for use in bone tissue engineering. Mater. Lett. 2015, 152, 148–150. [Google Scholar] [CrossRef]
- Balagangadharan, K.; Dhivya, S.; Selvamurugan, N. Chitosan based nanofibers in bone tissue engineering. Int. J. Biol. Macromol. 2017, 104, 1372–1382. [Google Scholar] [CrossRef]
- Yalcinkaya, F.; Komarek, M.; Lubasova, D.; Sanetrnik, F.; Maryska, J. Preparation of Antibacterial Nanofibre/Nanoparticle Covered Composite Yarns. J. Nanomater. 2016, 2016, 1–7. [Google Scholar] [CrossRef]
- Spasova, M.; Stoilova, O.; Manolova, N.; Rashkov, I.; Altankov, G. Preparation of PLLA/PEG nanofibers by electrospinning and potential applications. J. Bioact. Compat. Polym. 2007, 22, 62–76. [Google Scholar] [CrossRef]
- Xu, X.; Zhong, W.; Zhou, S.; Trajtman, A.; Alfa, M. Electrospun PEG-PLA nanofibrous membrane for sustained Release of hydrophilic antibiotics. J. Appl. Polym. Sci. 2010, 118, 588–595. [Google Scholar] [CrossRef]
- Cooper, A.; Bhattarai, N.; Zhang, M. Fabrication and cellular compatibility of aligned chitosan-PCL fibers for nerve tissue regeneration. Carbohydr. Polym. 2011, 85, 149–156. [Google Scholar] [CrossRef]
- Nguyen, T.T.T.; Chung, O.H.; Park, J.S. Coaxial electrospun poly(lactic acid)/chitosan (core/shell) composite nanofibers and their antibacterial activity. Carbohydr. Polym. 2011, 86, 1799–1806. [Google Scholar] [CrossRef]
- Frone, A.N.; Berlioz, S.; Chailan, J.F.; Panaitescu, D.M. Morphology and thermal properties of PLA-cellulose nanofibers composites. Carbohydr. Polym. 2013, 91, 377–384. [Google Scholar] [CrossRef]
- Lemma, S.M.; Bossard, F.; Rinaudo, M. Preparation of pure and stable chitosan nanofibers by electrospinning in the presence of poly(ethylene oxide). Int. J. Mol. Sci. 2016, 17, 1790. [Google Scholar] [CrossRef] [PubMed]
- Zhao, M.L.; Sui, G.; Deng, X.L.; Lu, J.G.; Ryu, S.K.; Yang, X.P. PLLA/HA Electrospin Hybrid Nanofiber Scaffolds: Morphology, In Vitro Degradation and Cell Culture Potential. Adv. Mater. Res. 2006, 11–12, 243–246. [Google Scholar] [CrossRef]
- Tang, Y.; Chen, L.; Zhao, K.; Wu, Z.; Wang, Y.; Tan, Q. Fabrication of PLGA/HA (core)-collagen/amoxicillin (shell) nanofiber membranes through coaxial electrospinning for guided tissue regeneration. Compos. Sci. Technol. 2016, 125, 100–107. [Google Scholar] [CrossRef]
- Ma, Z.; Chen, F.; Zhu, Y.J.; Cui, T.; Liu, X.Y. Amorphous calcium phosphate/poly(d,l-lactic acid) composite nanofibers: Electrospinning preparation and biomineralization. J. Colloid Interface Sci. 2011, 359, 371–379. [Google Scholar] [CrossRef]
- Keivani, F.; Shokrollahi, P.; Zandi, M.; Irani, S.; Shokrolahi, F.; Khorasani, S.C. Engineered electrospun poly(caprolactone)/polycaprolactone-g-hydroxyapatite nano-fibrous scaffold promotes human fibroblasts adhesion and proliferation. Mater. Sci. Eng. C 2016, 68, 78–88. [Google Scholar] [CrossRef]
- Sharma, P.R.; Zheng, B.; Sharma, S.K.; Zhan, C.; Wang, R.; Bhatia, S.R. High Aspect Ratio Carboxycellulose Nanofibers Prepared by Nitro-Oxidation Method and Their Nanopaper Properties. ACS Appl. Nano Mater. 2018, 1, 3969–3980. [Google Scholar] [CrossRef]
- Sharma, P.R.; Chattopadhyay, A.; Sharma, S.K.; Hsiao, B.S. Efficient Removal of UO22+ from Water Using Carboxycellulose Nanofibers Prepared by the Nitro-Oxidation Method. Ind. Eng. Chem. Res. 2017, 56, 13885–13893. [Google Scholar] [CrossRef]
- Sharma, P.R.; Chattopadhyay, A.; Sharma, S.K.; Geng, L.; Amiralian, N.; Martin, D. Nanocellulose from Spinifex as an Effective Adsorbent to Remove Cadmium(II) from Water. ACS Sustain. Chem. Eng. 2018, 6, 3279–3290. [Google Scholar] [CrossRef]
- He, M.; Zhang, B.; Dou, Y.; Yin, G.; Cui, Y.; Chen, X. Fabrication and characterization of electrospun feather keratin/poly(vinyl alcohol) composite nanofibers. RSC Adv. 2017, 7, 9854–9861. [Google Scholar] [CrossRef]
- Ahmed, F.E.; Lalia, B.S.; Hashaikeh, R. A review on electrospinning for membrane fabrication: Challenges and applications. Desalination 2015, 356, 15–30. [Google Scholar] [CrossRef]
- Zulkifli, F.H.; Jahir Hussain, F.S.; Abdull Rasad, M.S.B.; Mohd Yusoff, M. In vitro degradation study of novel HEC/PVA/collagen nanofibrous scaffold for skin tissue engineering applications. Polym. Degrad. Stab. 2014, 110, 473–481. [Google Scholar] [CrossRef]
- Esmaeili, A.; Haseli, M. Electrospinning of thermoplastic carboxymethyl cellulose/poly(ethylene oxide) nanofibers for use in drug-release systems. Mater. Sci. Eng. C 2017, 77, 1117–1127. [Google Scholar] [CrossRef] [PubMed]
- Ren, K.; Wang, Y.; Sun, T.; Yue, W.; Zhang, H. Electrospun PCL/gelatin composite nanofiber structures for effective guided bone regeneration membranes. Mater. Sci. Eng. C 2017, 78, 324–332. [Google Scholar] [CrossRef] [PubMed]
- Elgali, I.; Turri, W.; Xia, B.; Norlindh, A.; Johansson, C. Guided bone regeneration using resorbable membrane and different bone substitutes: Early histological and molecular events. Acta Biomater. 2012, 125, 315–337. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.W.; Song, J.H.; Kim, H.E. Nanofiber generation of gelatin-hydroxyapatite biomimetics for guided tissue regeneration. Adv. Funct. Mater. 2005, 15, 1988–1994. [Google Scholar] [CrossRef]
- Song, X.; Ling, F.; Ma, L.; Yang, C.; Chen, X. Electrospun hydroxyapatite grafted poly(l-lactide)/poly(lactic-co-glycolic acid) nanofibers for guided bone regeneration membrane. Compos. Sci. Technol. 2013, 79, 8–14. [Google Scholar] [CrossRef]
- Yusof, M.R.; Shamsudin, R.; Abdullah, Y.; Yalcinkaya, F.; Yaacob, N. Electrospinning of carboxymethyl starch/poly(l-lactide acid) composite nanofiber. Polym. Adv. Technol. 2018, 29, 1843–1851. [Google Scholar] [CrossRef]
- Yaacob, B.; Cairul, M.; Amin, I.M.; Kamaruddin, H.; Bakar, B.A. Optimization of Reaction Conditions for Carboxymethylated Sago Starch. Iran. Polym. J. 2011, 20, 10–15. [Google Scholar]
- Siqueira, L.; Passador, F.R.; Costa, M.M.; Lobo, A.O.; Sousa, E. Influence of the addition of β-TCP on the morphology, thermal properties and cell viability of poly(lactic acid) fibers obtained by electrospinning. Mater. Sci. Eng. C 2015, 52, 135–143. [Google Scholar] [CrossRef] [PubMed]
- Yener, F.; Jirsak, O. Comparison between the Needle and Roller Electrospinning of Polyvinylbutyral. J. Nanomater. 2012, 2012, 1–6. [Google Scholar] [CrossRef]
- Bow, J.S.; Liou, S.C.; Chen, S.Y. Structural characterization of room-temperature synthesized nano-sized β-tricalcium phosphate. Biomaterials 2004, 25, 3155–3161. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Liu, S.; Wang, G. Kinetics and adsorption behavior of carboxymethyl starch on α-alumina in aqueous medium. J. Colloid Interface Sci. 2006, 303, 380–387. [Google Scholar] [CrossRef]
- Gay, S.; Arostegui, S.; Lemaitre, J. Preparation and characterization of dense nanohydroxyapatite/PLLA composites. Mater. Sci. Eng. C 2009, 29, 172–177. [Google Scholar] [CrossRef]
- Ferri, J.M.; Gisbert, I.; García-Sanoguera, D.; Reig, M.J.; Balart, R. The effect of beta-tricalcium phosphate on mechanical and thermal performances of poly(lactic acid). J. Compos. Mater. 2016, 50, 4189–4198. [Google Scholar] [CrossRef]
- Vert, M.; Li, S.M.; Spenlehauer, G.; Guerin, P. Bioresorbability and biocompatibility of aliphatic polyesters. J. Mater. Sci. Mater. Med. 1992, 3, 432–446. [Google Scholar] [CrossRef]
- Ma, P.X. Scaffolds for tissue fabrication. Mater. Today 2004, 7, 30–40. [Google Scholar] [CrossRef]
- Hu, H.T.; Lee, S.Y.; Chen, C.C.; Yang, Y.C.; Yang, J.C. Processing and properties of hydrophilic electrospun polylactic acid/beta-tricalcium phosphate membrane for dental applications. Polym. Eng. Sci. 2013, 53, 833–842. [Google Scholar] [CrossRef]
- Deplaine, H.; Ribelles, J.L.L.G.; Ferrer, G.G. Effect of the content of hydroxyapatite nanoparticles on the pr4operties and bioactivity of poly(l-lactide)—Hybrid membranes. Compos. Sci. Technol. 2010, 70, 1805–1812. [Google Scholar] [CrossRef]
- Tammaro, L.; Vittoria, V.; Wyrwa, R.; Weisser, J.; Beer, B.; Thein, S.; Schnabelrauch, M. Fabrication and characterization of electrospun polylactide/β-tricalcium phosphate hybrid meshes for potential applications in hard tissue repair. BioNanoMaterials 2014, 15, 9–20. [Google Scholar] [CrossRef]
- Sui, G.; Yang, X.; Mei, F.; Hu, X.; Chen, G.; Deng, X.; Ryu, S. Poly-l-lactic acid/hydroxyapatite hybrid membrane for bone tissue regeneration. J. Biomed. Mater. Res. Part A 2007, 82, 445–454. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Lim, J.Y.; Donahue, H.J.; Dhurjati, R.; Mastro, A.M.; Vogler, E.A. Influence of substratum surface chemistry/energy and topography on the human fetal osteoblastic cell line hFOB 1.19: Phenotypic and genotypic responses observed in vitro. Biomaterials 2007, 28, 4535–4550. [Google Scholar] [CrossRef] [PubMed]
- Ma, F.; Chen, S.; Liu, P.; Geng, F.; Li, W.; Liu, X.; He, D.; Pan, D. Improvement of β-TCP/PLLA biodegradable material by surface modification with stearic acid. Mater. Sci. Eng. C 2016, 62, 407–413. [Google Scholar] [CrossRef]
- Yang, M.; Di, Z.; Lee, J.K. Facile control of surface wettability in TiO2/poly(methyl methacrylate) composite films. J. Colloid Interface Sci. 2012, 368, 603–607. [Google Scholar] [CrossRef]
- Ngiam, M.; Liao, S.; Patil, A.J.; Cheng, Z.; Chan, C.K.; Ramakrishna, S. The fabrication of nano-hydroxyapatite on PLGA and PLGA/collagen nanofibrous composite scaffolds and their effects in osteoblastic behavior for bone tissue engineering. Bone 2009, 45, 4–16. [Google Scholar] [CrossRef]
- McCullen, S.D.; Zhu, Y.; Bernacki, S.H.; Narayan, R.J.; Pourdeyhimi, B.; Gorga, R.E.; Loboa, E.G. Electrospun composite poly(l-lactic acid)/tricalcium phosphate scaffolds induce proliferation and osteogenic differentiation of human adipose-derived stem cells. Biomed. Mater. 2009, 4, 035002. [Google Scholar] [CrossRef]
- Cai, N.; Dai, Q.; Wang, Z.; Luo, X.; Xue, Y.; Yu, F. Toughening of electrospun poly(l-lactic acid) nanofiber scaffolds with unidirectionally aligned halloysite nanotubes. J. Mater. Sci. 2015, 50, 1435–1445. [Google Scholar] [CrossRef]
- Heydary, H.A.; Karamian, E.; Poorazizi, E.; Heydaripour, J.; Khandan, A. Electrospun of polymer/bioceramic nanocomposite as a new soft tissue for biomedical applications. J. Asian Ceram. Soc. 2015, 3, 417–425. [Google Scholar] [CrossRef]
- Ba Linh, N.T.; Lee, K.H.; Lee, B.T. Functional nanofiber mat of polyvinyl alcohol/gelatin containing nanoparticles of biphasic calcium phosphate for bone regeneration in rat calvaria defects. J. Biomed. Mater. Res. Part A 2013, 101 A, 2412–2423. [Google Scholar] [CrossRef]














{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Samples | Tg (°C) | Tc (°C) | Tm (°C) | * ∆Hc (J/g) | * ∆Hm (J/g) | * Xc (%) |
|---|---|---|---|---|---|---|
| PLLA/CMS | 53.6 | 69.0 | 170.0 | 9.5 | 45.8 | 38.2 |
| 0.25% β-TCP | 56.6 | 70.8 | 171.7 | 8.7 | 42.3 | 35.3 |
| 1 % β-TCP | 55.0 | 67.5 | 166.1 | 14.1 | 35.0 | 22.0 |
| Sample (%β-TCP) | Peaks Intensity (cps) | FWHM (radian) | Crystallite Size (nm) |
|---|---|---|---|
| 0 0.25 0.5 0.75 1.0 | 138.5 57.4 45.9 37.2 106.8 | 0.050 0.0261 0.0346 0.0376 0.0510 | 0.506 0.976 0.736 0.692 No data obtained |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yusof, M.R.; Shamsudin, R.; Zakaria, S.; Abdul Hamid, M.A.; Yalcinkaya, F.; Abdullah, Y.; Yacob, N. Fabrication and Characterization of Carboxymethyl Starch/Poly(l-Lactide) Acid/β-Tricalcium Phosphate Composite Nanofibers via Electrospinning. Polymers 2019, 11, 1468. https://doi.org/10.3390/polym11091468
Yusof MR, Shamsudin R, Zakaria S, Abdul Hamid MA, Yalcinkaya F, Abdullah Y, Yacob N. Fabrication and Characterization of Carboxymethyl Starch/Poly(l-Lactide) Acid/β-Tricalcium Phosphate Composite Nanofibers via Electrospinning. Polymers. 2019; 11(9):1468. https://doi.org/10.3390/polym11091468
Chicago/Turabian StyleYusof, Mohd Reusmaazran, Roslinda Shamsudin, Sarani Zakaria, Muhammad Azmi Abdul Hamid, Fatma Yalcinkaya, Yusof Abdullah, and Norzita Yacob. 2019. "Fabrication and Characterization of Carboxymethyl Starch/Poly(l-Lactide) Acid/β-Tricalcium Phosphate Composite Nanofibers via Electrospinning" Polymers 11, no. 9: 1468. https://doi.org/10.3390/polym11091468
APA StyleYusof, M. R., Shamsudin, R., Zakaria, S., Abdul Hamid, M. A., Yalcinkaya, F., Abdullah, Y., & Yacob, N. (2019). Fabrication and Characterization of Carboxymethyl Starch/Poly(l-Lactide) Acid/β-Tricalcium Phosphate Composite Nanofibers via Electrospinning. Polymers, 11(9), 1468. https://doi.org/10.3390/polym11091468