The Effects of Magnetic Field Alignment on Lithium Ion Transport in a Polymer Electrolyte Membrane with Lamellar Morphology


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
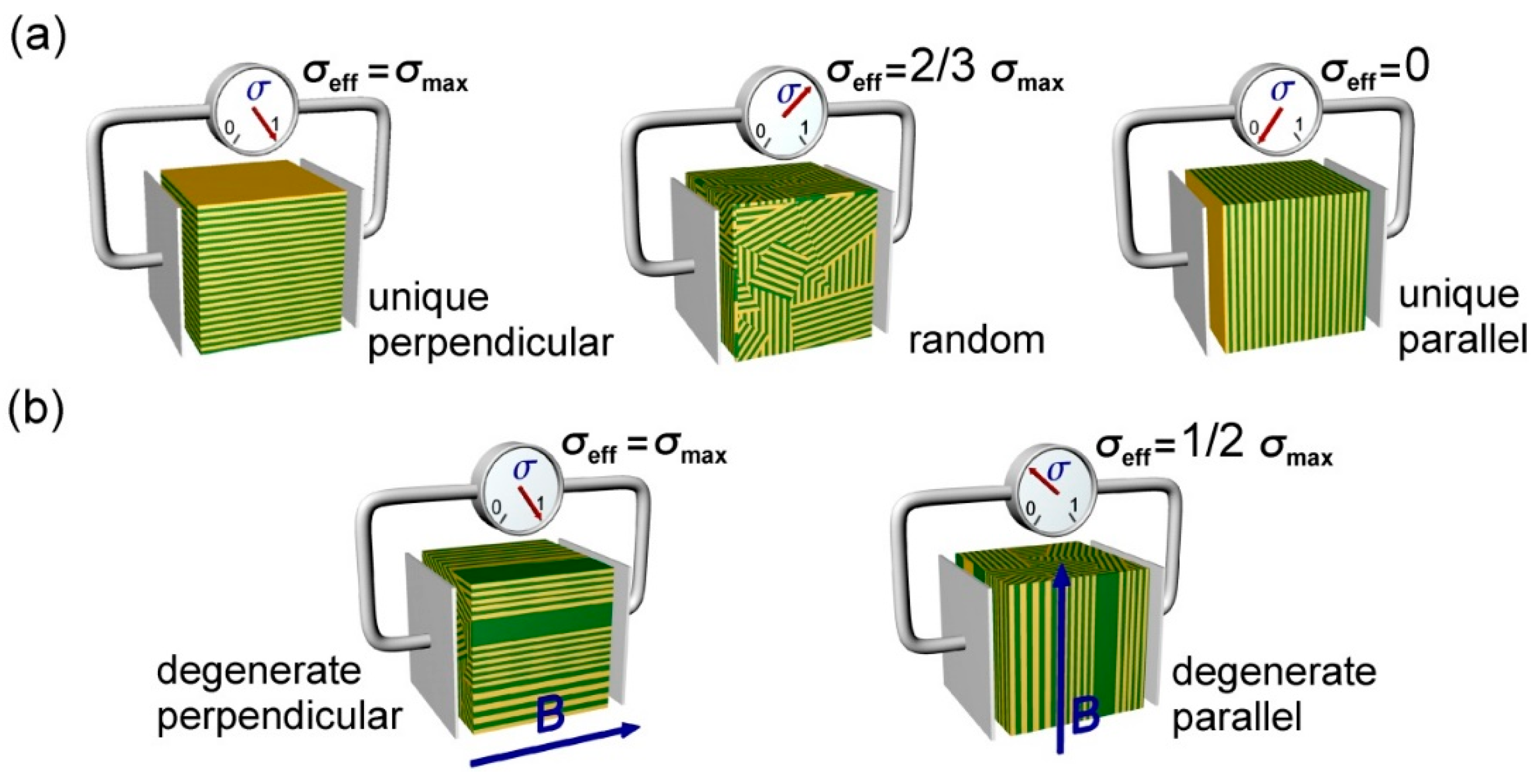
1.1. Microstructural Conductivity Model
1.2. Magnetic Alignment of Ion-Conducting Block Copolymers
2. Materials and Methods
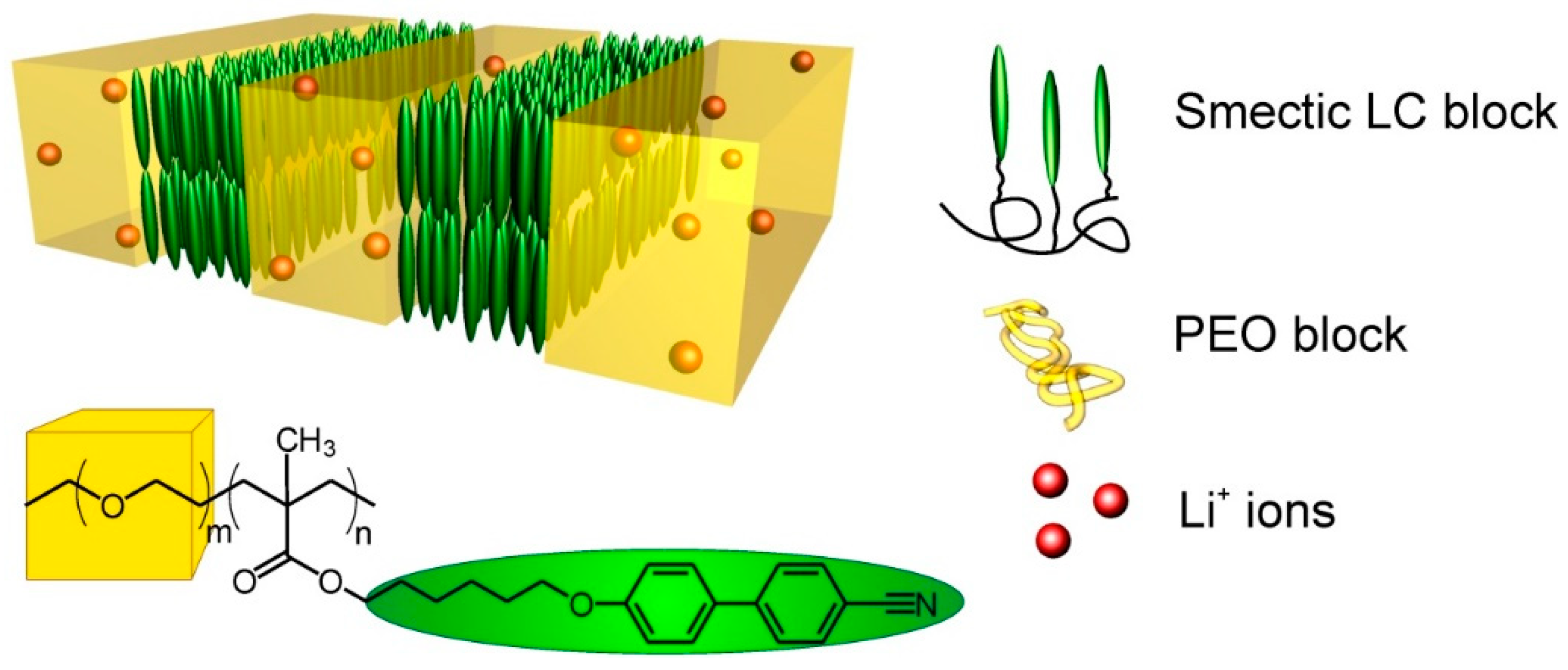
2.1. Materials
2.2. Preparation of LiClO4 Doped Polymer Samples
2.3. Assembly of Conductivity and SAXS/WAXS Cells
2.4. Magnetic Alignment
2.5. Small Angle X-ray Scattering
2.6. Differential Scanning Calorimetry
2.7. Conductivity Measurements
2.8. Atomic Force Microscopy (AFM)
3. Results and Discussion
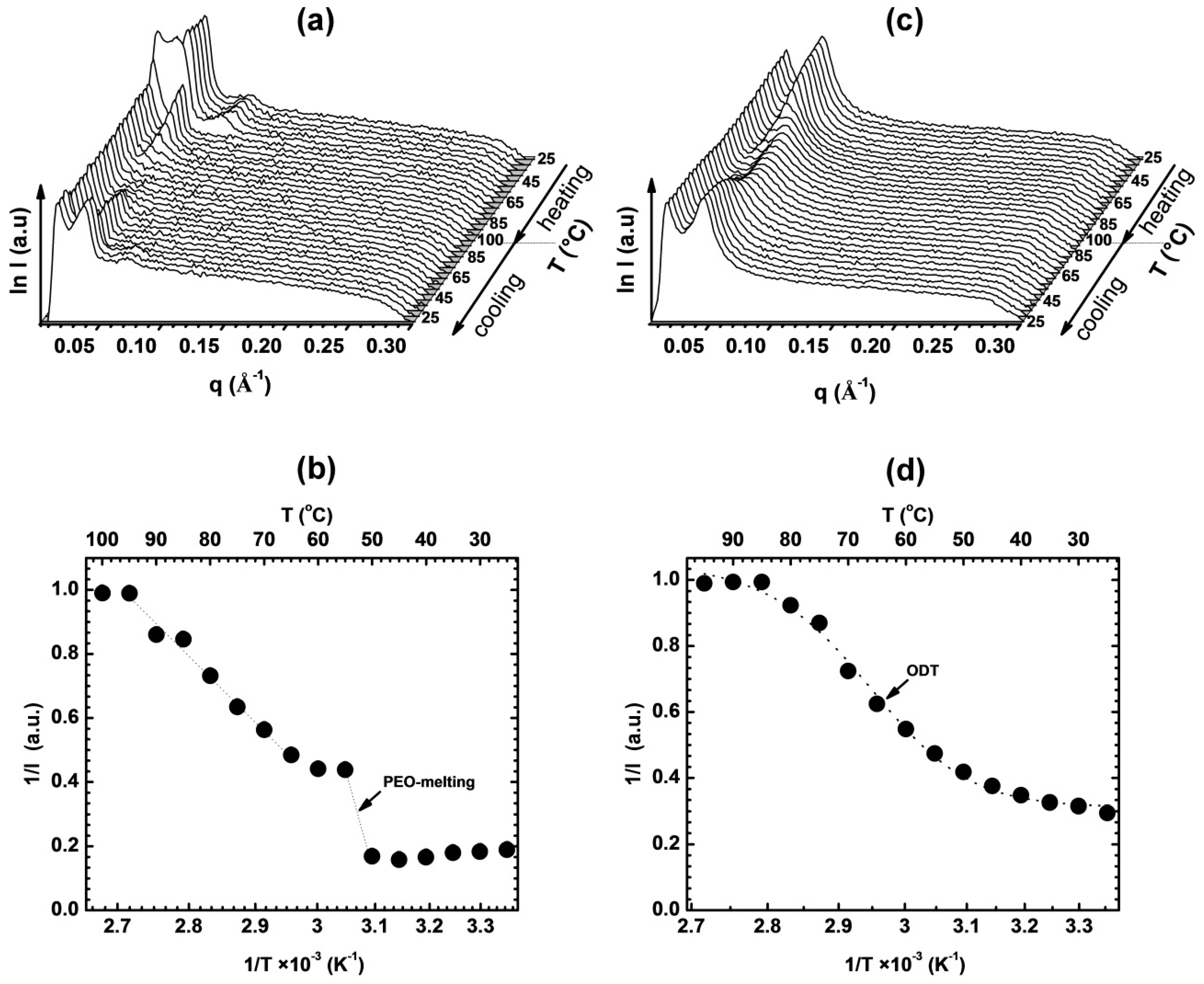
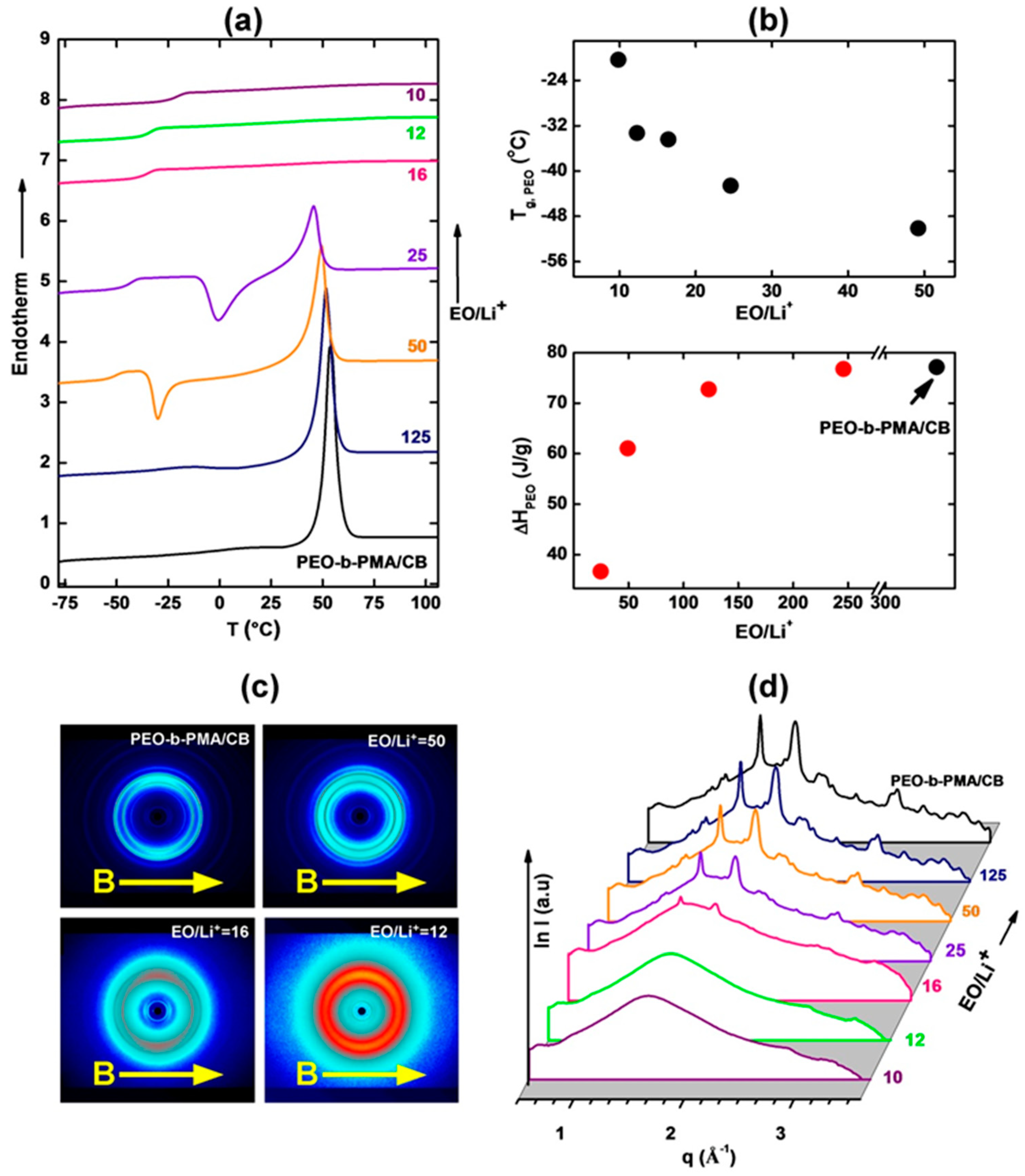
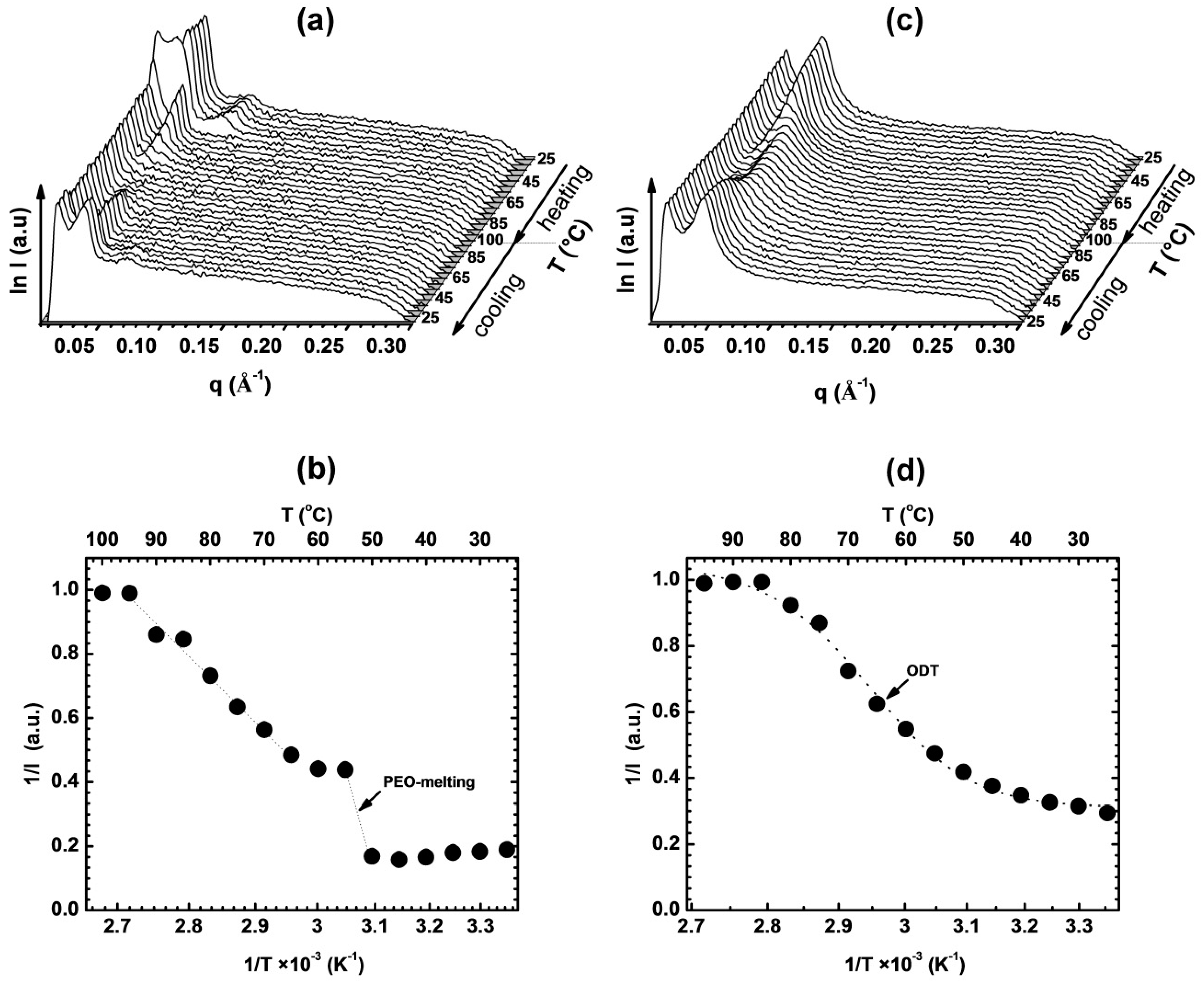
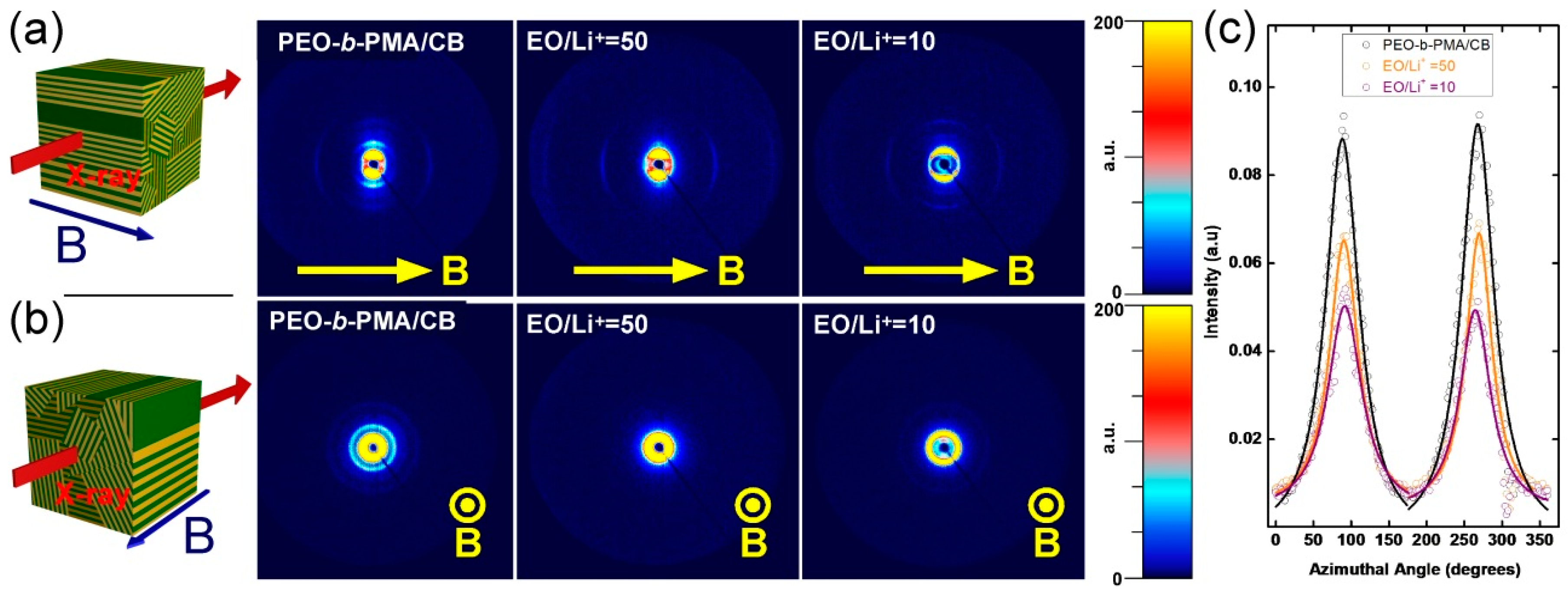
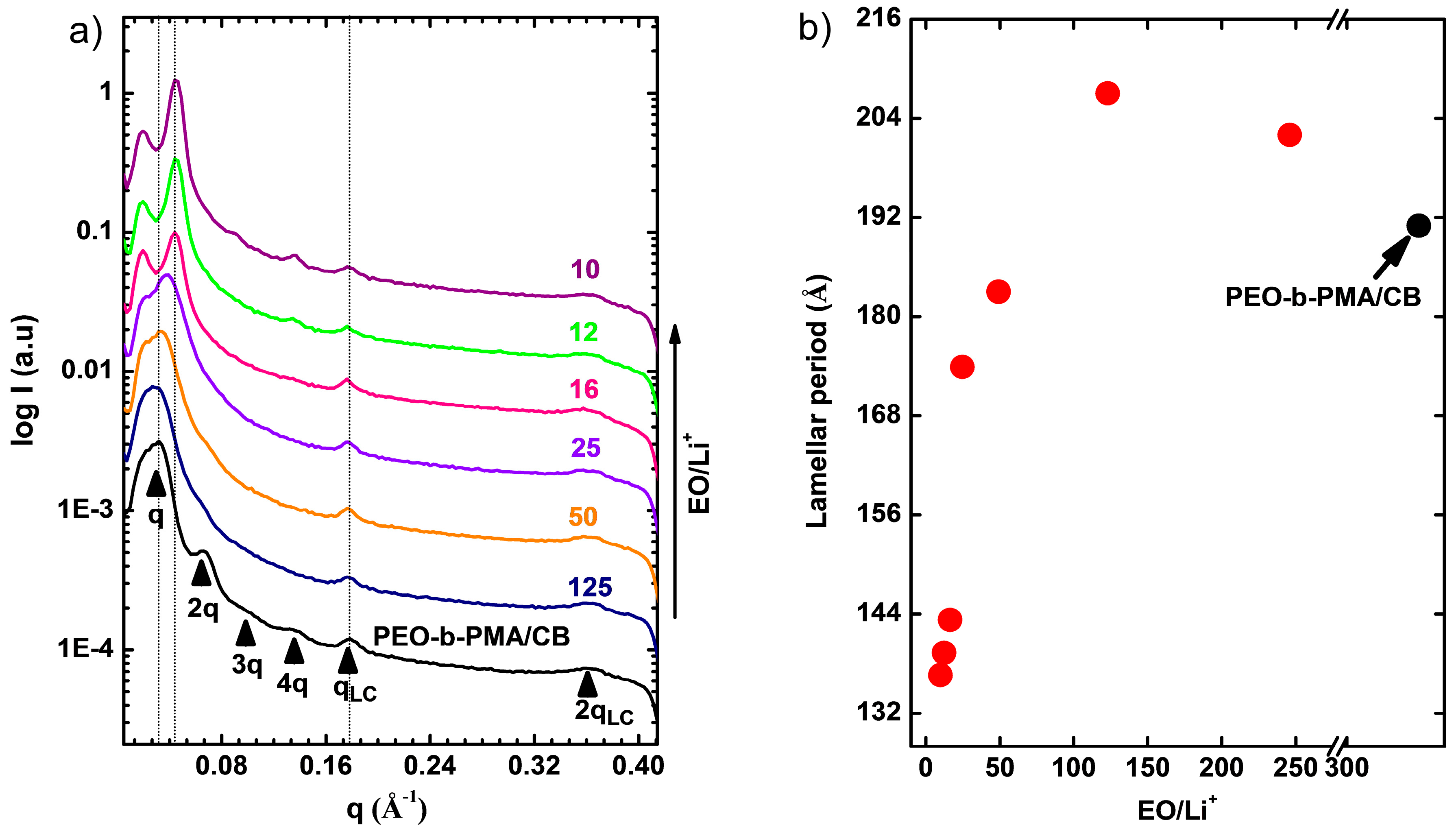
3.1. Thermal and Morphological Characterization
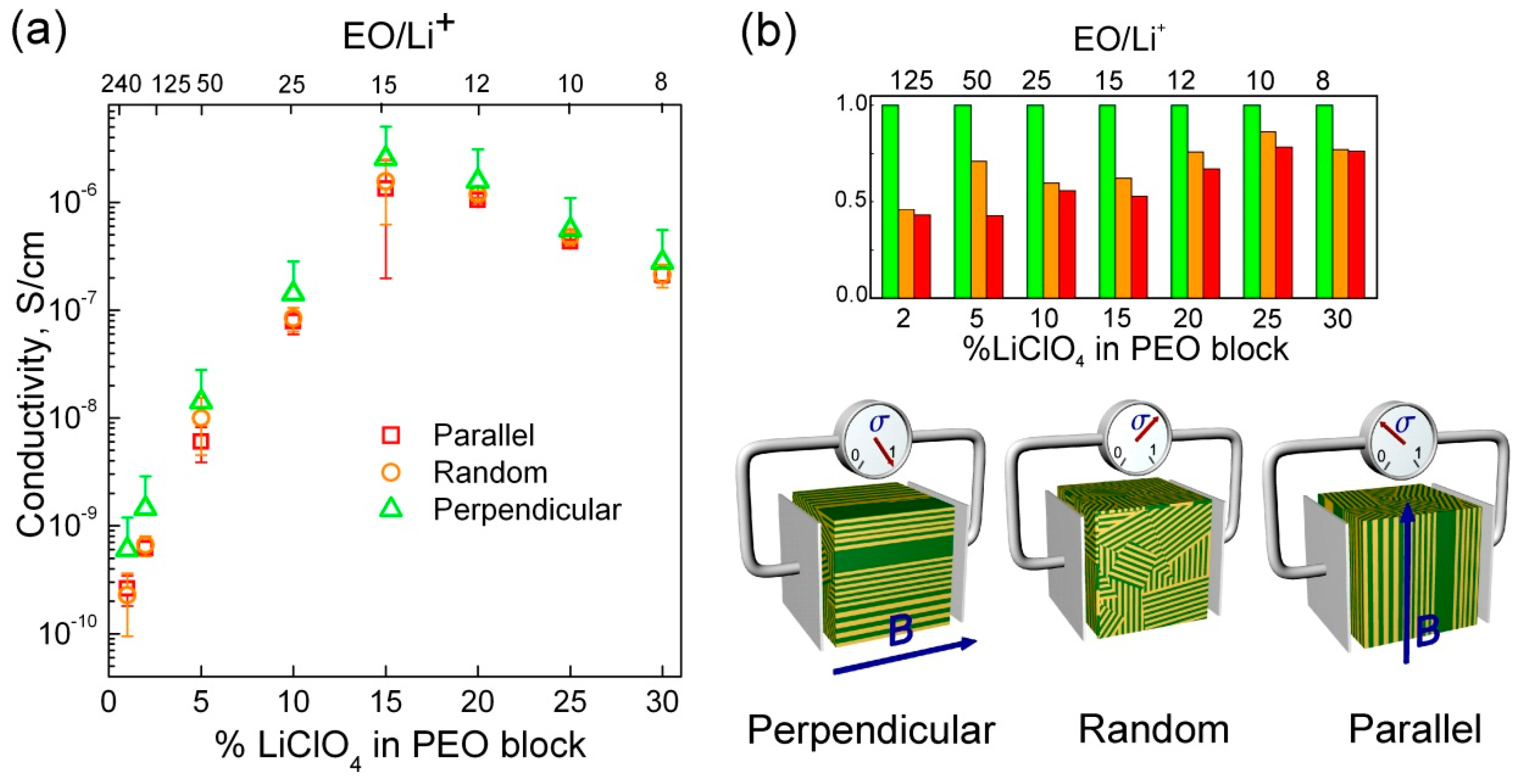
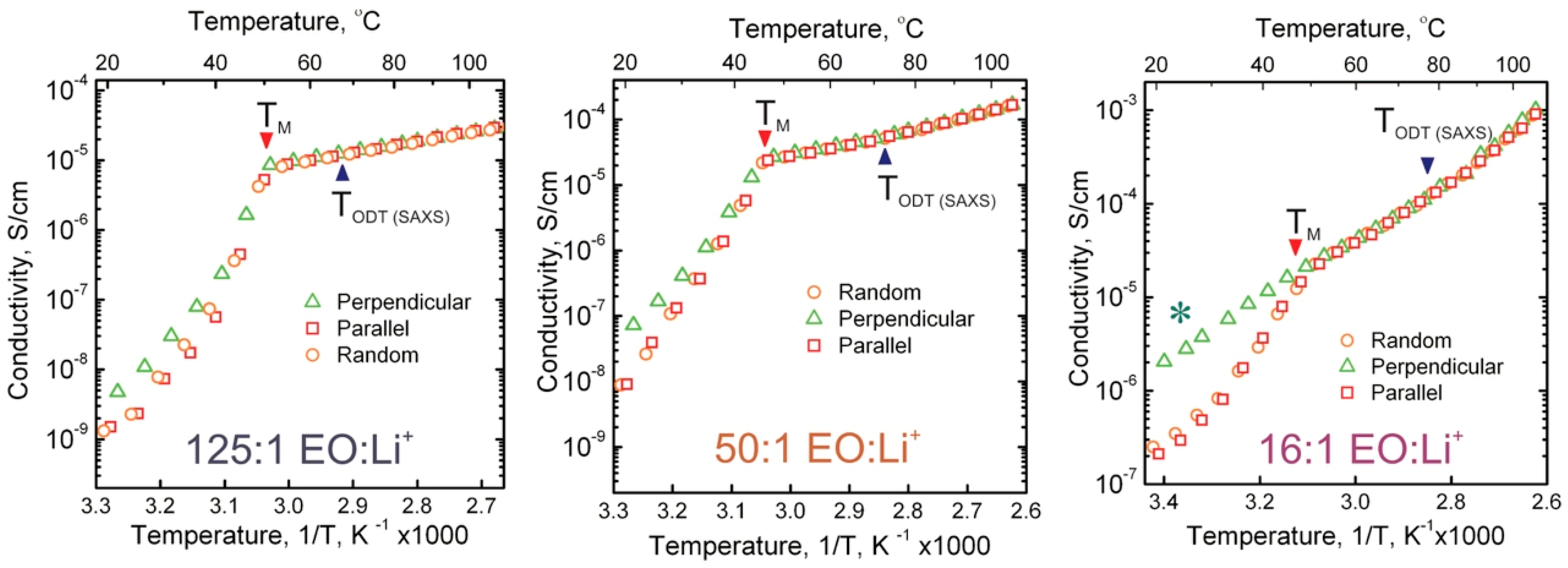
3.2. Ionic Conductivity
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Bates, C.M.; Bates, F.S. 50th anniversary perspective: Block polymers pure potential. Macromolecules 2016, 50, 3–22. [Google Scholar] [CrossRef]
- Park, M.J.; Balsara, N.P. Anisotropic proton conduction in aligned block copolymer electrolyte membranes at equilibrium with humid air. Macromolecules 2009, 43, 292–298. [Google Scholar] [CrossRef]
- Young, W.-S.; Epps Iii, T.H. Salt doping in peo-containing block copolymers: Counterion and concentration effects. Macromolecules 2009, 42, 2672–2678. [Google Scholar] [CrossRef]
- Panday, A.; Mullin, S.; Gomez, E.D.; Wanakule, N.; Chen, V.L.; Hexemer, A.; Pople, J.; Balsara, N.P. Effect of molecular weight and salt concentration on conductivity of block copolymer electrolytes. Macromolecules 2009, 42, 4632–4637. [Google Scholar] [CrossRef]
- Gopinadhan, M.; Majewski, P.W.; Osuji, C.O. Facile alignment of amorphous poly(ethylene oxide) microdomains in a liquid crystalline block copolymer using magnetic fields: Toward ordered electrolyte membranes. Macromolecules 2010, 43, 3286–3293. [Google Scholar] [CrossRef]
- Digiano, F.A.; Roudman, A.; Arnold, M.; Freeman, B. Novel block copolymers as nanofiltration materials. Environ. Eng. Sci. 2002, 19, 497–511. [Google Scholar] [CrossRef]
- Jha, A.K.; Chen, L.; Offeman, R.D.; Balsara, N.P. Effect of nanoscale morphology on selective ethanol transport through block copolymer membranes. J. Membr. Sci. 2011, 373, 112–120. [Google Scholar] [CrossRef]
- Phillip, W.A.; O’Neill, B.; Rodwogin, M.; Hillmyer, M.A.; Cussler, E.L. Self-assembled block copolymer thin films as water filtration membranes. ACS Appl. Mater. Interfaces 2010, 2, 847–853. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Mulvenna, R.A.; Qu, S.; Boudouris, B.W.; Phillip, W.A. Block polymer membranes functionalized with nanoconfined polyelectrolyte brushes achieve sub-nanometer selectivity. ACS Macro Lett. 2017, 6, 726–732. [Google Scholar]
- Zhang, Y.; Sargent, J.L.; Boudouris, B.W.; Phillip, W.A. Nanoporous membranes generated from self-assembled block polymer precursors: Q uo v adis? J. Appl. Polym. Sci. 2015, 132. [Google Scholar]
- Yu, H.; Qiu, X.; Moreno, N.; Ma, Z.; Calo, V.M.; Nunes, S.P.; Peinemann, K.V. Self-assembled asymmetric block copolymer membranes: Bridging the gap from ultra-to nanofiltration. Angew. Chem. Int. Ed. 2015, 54, 13937–13941. [Google Scholar] [CrossRef] [PubMed]
- Young, W.S.; Kuan, W.F.; Epps Iii, T.H. Block copolymer electrolytes for rechargeable lithium batteries. J. Polym. Sci. Pol. Phys. 2014, 52, 1–16. [Google Scholar] [CrossRef]
- Ruzette, A.-V.G.; Soo, P.P.; Sadoway, D.R.; Mayes, A.M. Melt-formable block copolymer electrolytes for lithium rechargeable batteries. J. Electrochem. Soc. 2001, 148, A537–A543. [Google Scholar] [CrossRef]
- Stone, G.M.; Mullin, S.A.; Teran, A.A.; Hallinan, D.T.; Minor, A.M.; Hexemer, A.; Balsara, N.P. Resolution of the modulus versus adhesion dilemma in solid polymer electrolytes for rechargeable lithium metal batteries. J. Electrochem. Soc. 2012, 159, A222–A227. [Google Scholar] [CrossRef]
- Khurana, R.; Schaefer, J.L.; Archer, L.A.; Coates, G.W. Suppression of lithium dendrite growth using cross-linked polyethylene/poly(ethylene oxide) electrolytes: A new approach for practical lithium-metal polymer batteries. J. Am. Chem. Soc. 2014, 136, 7395–7402. [Google Scholar] [CrossRef]
- Maxwell, J.C. Conduction through heterogeneous media. A Treatise On Electricity And Magnetism 1891, 1, 435–441. [Google Scholar]
- Rayleigh, L. Lvi. On the influence of obstacles arranged in rectangular order upon the properties of a medium. London Edinburgh Dublin Philos. Mag. J. Sci. 1892, 34, 481–502. [Google Scholar] [CrossRef]
- Sax, J.; Ottino, J.M. Modeling of transport of small molecules in polymer blends: Application of effective medium theory. Polym. Eng. Sci. 1983, 23, 165–176. [Google Scholar] [CrossRef]
- Chintapalli, M.; Chen, X.C.; Thelen, J.L.; Teran, A.A.; Wang, X.; Garetz, B.A.; Balsara, N.P. Effect of grain size on the ionic conductivity of a block copolymer electrolyte. Macromolecules 2014, 47, 5424–5431. [Google Scholar] [CrossRef]
- Shen, K.-H.; Brown, J.R.; Hall, L.M. Diffusion in lamellae, cylinders, and double gyroid block copolymer nanostructures. ACS Macro Lett. 2018, 7, 1092–1098. [Google Scholar] [CrossRef]
- Kambe, Y.; Arges, C.G.; Patel, S.; Stoykovish, M.P.; Nealey, P.F. Ion conduction in microphase-separated block copolymer electrolytes. Electrochem. Soc. Interface 2017, 26, 61–67. [Google Scholar] [CrossRef]
- Darling, S.B. Directing the self-assembly of block copolymers. Prog. Polym. Sci. 2007, 32, 1152–1204. [Google Scholar] [CrossRef]
- Hu, H.; Gopinadhan, M.; Osuji, C.O. Directed self-assembly of block copolymers: A tutorial review of strategies for enabling nanotechnology with soft matter. Soft Matter 2014, 10, 3867–3889. [Google Scholar] [CrossRef]
- Majewski, P.W.; Osuji, C.O. Non-degenerate magnetic alignment of self-assembled mesophases. Soft Matter 2009, 5, 3417–3421. [Google Scholar] [CrossRef]
- Majewski, P.W.; Gopinadhan, M.; Jang, W.-S.; Lutkenhaus, J.L.; Osuji, C.O. Anisotropic ionic conductivity in block copolymer membranes by magnetic field alignment. J. Am. Chem. Soc. 2010, 132, 17516–17522. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Hsu, S.L. An analysis of the crystallization behavior of poly(ethylene oxide)/poly(methyl methacrylate) blends by spectroscopic and calorimetric techniques. J. Polym. Sci. Polym. Phys. Ed. 1984, 22, 1331–1342. [Google Scholar] [CrossRef]
- Richardson, P.H.; Richards, R.W.; Blundell, D.J.; MacDonald, W.A.; Mills, P. Differential scanning calorimetry and optical microscopy investigations of the isothermal crystallization of a poly(ethylene oxide)-poly(methyl methacrylate) block copolymer. Polymer 1995, 36, 3059–3069. [Google Scholar] [CrossRef]
- Chan, L.L.; Wong, K.H.; Smid, J. Complexation of lithium, sodium, and potassium carbanion pairs with polyglycol dimethyl ethers (glymes). Effect of chain length and temperature. J. Am. Chem. Soc. 1970, 92, 1955–1963. [Google Scholar] [CrossRef]
- Gunkel, I.; Thurn-Albrecht, T. Thermodynamic and structural changes in ion-containing symmetric diblock copolymers: A small-angle x-ray scattering study. Macromolecules 2011, 45, 283–291. [Google Scholar] [CrossRef]
- Naidu, S.; Ahn, H.; Gong, J.; Kim, B.; Ryu, D.Y. Phase behavior and ionic conductivity of lithium perchlorate-doped polystyrene-b-poly(2-vinylpyridine) copolymer. Macromolecules 2011, 44, 6085–6093. [Google Scholar] [CrossRef]
- Wang, J.-Y.; Chen, W.; Russell, T.P. Ion-complexation-induced changes in the interaction parameter and the chain conformation of ps-b-pmma copolymers. Macromolecules 2008, 41, 4904–4907. [Google Scholar] [CrossRef]
- Hong, S.; Yang, L.; MacKnight, W.J.; Gido, S.P. Morphology of a crystalline/amorphous diblock copolymer: Poly((ethylene oxide)-b-butadiene). Macromolecules 2001, 34, 7009–7016. [Google Scholar] [CrossRef]
- Burns, A.B.; Register, R.A. Large, reversible, and coherent domain spacing dilation driven by crystallization under soft lamellar confinement. Macromolecules 2017, 50, 8106–8116. [Google Scholar] [CrossRef]
- Loo, Y.-L.; Register, R.A.; Ryan, A.J. Modes of crystallization in block copolymer microdomains: Breakout, templated, and confined. Macromolecules 2002, 35, 2365–2374. [Google Scholar] [CrossRef]
- Sadoway, D.R. Block and graft copolymer electrolytes for high-performance, solid-state, lithium batteries. J. Power Sources 2004, 129, 1–3. [Google Scholar] [CrossRef]
- Ryu, S.-W.; Trapa, P.E.; Olugebefola, S.C.; Gonzalez-Leon, J.A.; Sadoway, D.R.; Mayes, A.M. Effect of counter ion placement on conductivity in single-ion conducting block copolymer electrolytes. J. Electrochem. Soc. 2005, 152, A158–A163. [Google Scholar] [CrossRef]
- Bakker, A.; Lindgren, J.; Hermansson, K. Polymer electrolytes based on triblock-copoly(oxyethylene/oxypropylene/oxyethylene) systems. Polymer 1996, 37, 1871–1878. [Google Scholar] [CrossRef]
- Ratner, M.A.; Shriver, D.F. Ion transport in solvent-free polymers. Chem. Rev. 1988, 88, 109–124. [Google Scholar] [CrossRef]
- Hoffmann, J.F.; Pulst, M.; Kressler, J. Enhanced ion conductivity of poly(ethylene oxide)-based single ion conductors with lithium 1,2,3-triazolate end groups. J. Appl. Polym. Sci. 2019, 136, 46949. [Google Scholar] [CrossRef]
- Grigorova, T.; Pispas, S.; Hadjichristidis, N.; Thurn-Albrecht, T. Magnetic field induced orientation in diblock copolymers with one crystallizable block. Macromolecules 2005, 38, 7430–7433. [Google Scholar] [CrossRef]

© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Majewski, P.W.; Gopinadhan, M.; Osuji, C.O. The Effects of Magnetic Field Alignment on Lithium Ion Transport in a Polymer Electrolyte Membrane with Lamellar Morphology. Polymers 2019, 11, 887. https://doi.org/10.3390/polym11050887
Majewski PW, Gopinadhan M, Osuji CO. The Effects of Magnetic Field Alignment on Lithium Ion Transport in a Polymer Electrolyte Membrane with Lamellar Morphology. Polymers. 2019; 11(5):887. https://doi.org/10.3390/polym11050887
Chicago/Turabian StyleMajewski, Pawel W., Manesh Gopinadhan, and Chinedum O. Osuji. 2019. "The Effects of Magnetic Field Alignment on Lithium Ion Transport in a Polymer Electrolyte Membrane with Lamellar Morphology" Polymers 11, no. 5: 887. https://doi.org/10.3390/polym11050887
APA StyleMajewski, P. W., Gopinadhan, M., & Osuji, C. O. (2019). The Effects of Magnetic Field Alignment on Lithium Ion Transport in a Polymer Electrolyte Membrane with Lamellar Morphology. Polymers, 11(5), 887. https://doi.org/10.3390/polym11050887