3.1. Synthesis and Characterization of PEGF Copolyester
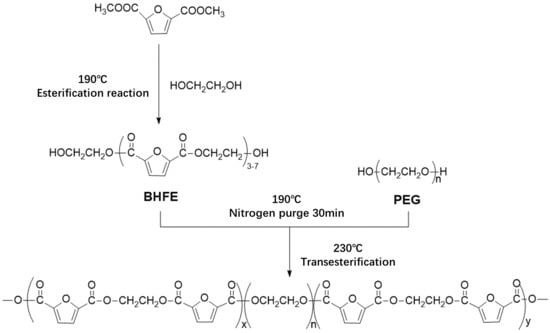
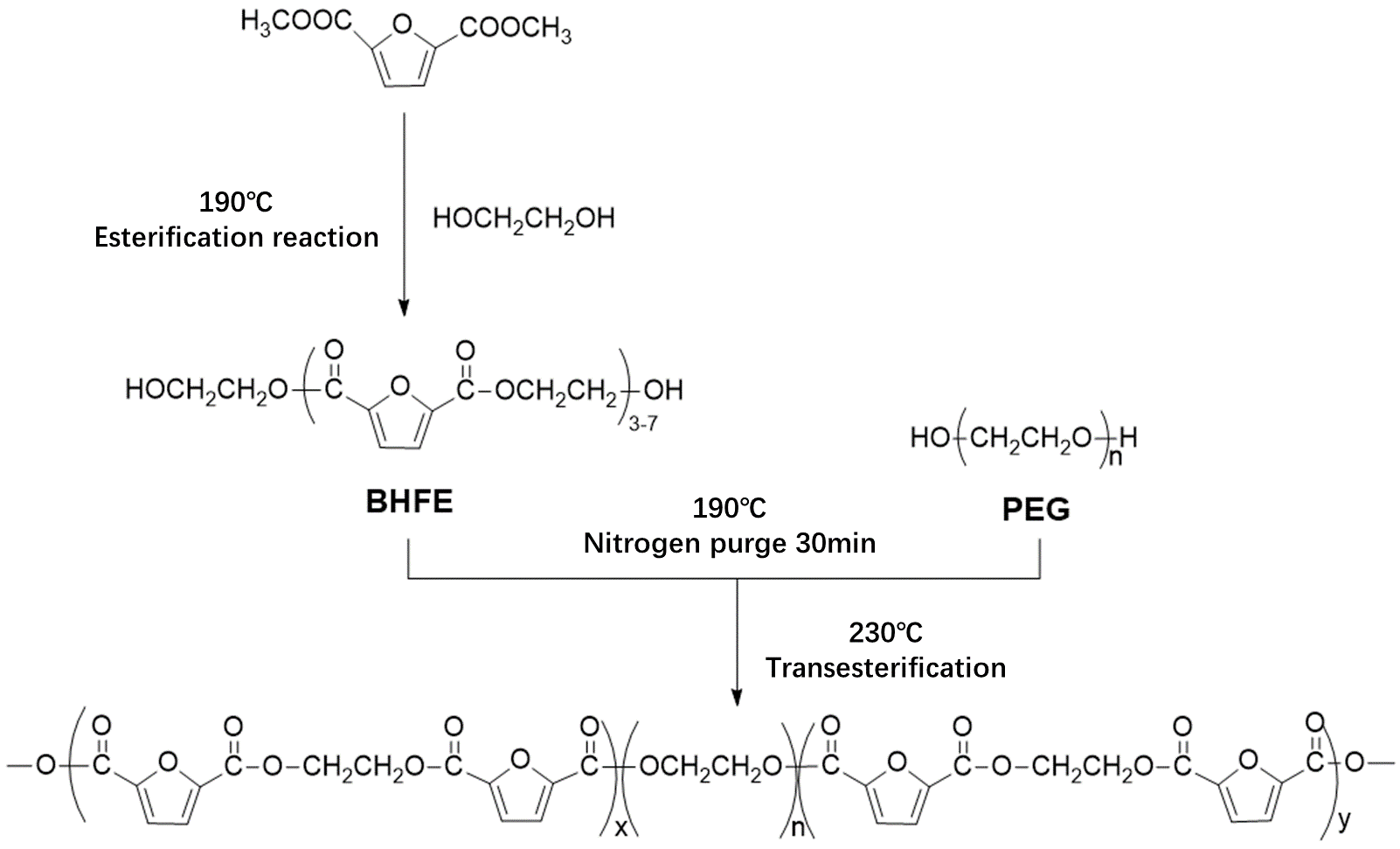
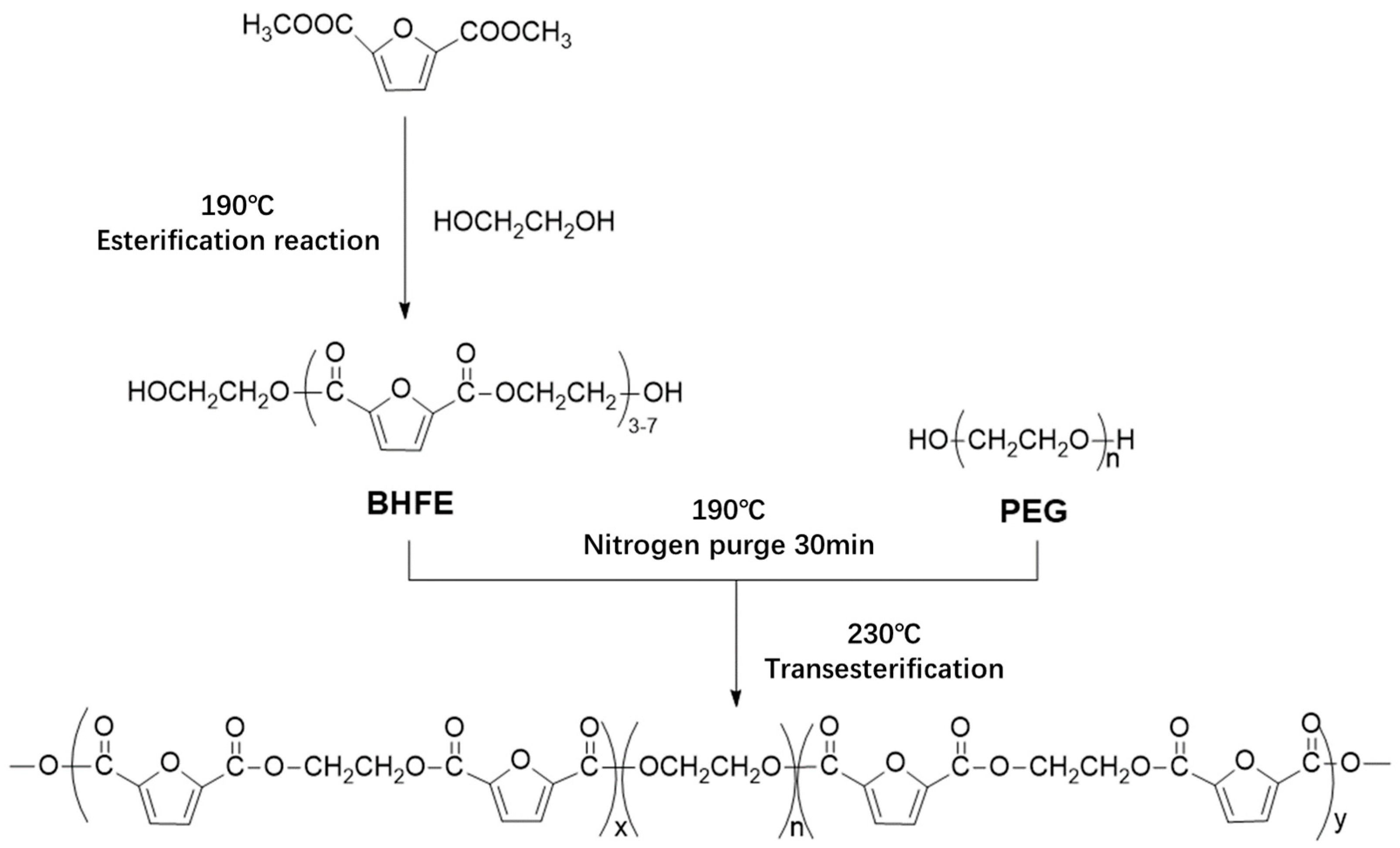
In this study, bio-based 2,5-DMFD, PEG and EG were used as reaction materials and the polymerization scheme is shown in
Figure 2. The reaction used different molecular weights of different PEGs (number average molecular weights of 600, 2000, 6000, 10,000 g/mol) and different PEG contents (10, 20, 40, 60 wt % relative to DMFD) of PEGF copolyester. The esterification rate during polymerization is shown in Table 2.
From the data in
Table 1, when the esterification temperature is 190 °C, the esterification rate of each sample is above 90%. The intrinsic viscosity of PEGFs is shown in
Table 1. The intrinsic viscosity of PEGF copolyester is between 0.62–0.99 dL/g, which is higher than the intrinsic viscosity of PEF (0.61 dL/g). When the content or molecular weight of PEG is increased, the intrinsic viscosity of PEGFs is increased, mainly because of the existence of flexible long-chain PEG, which increases the chain length and flexibility of PEGFs. These changes make part of the copolyester molecular chain tangle in the capillary, resulting in prolonged residence time of the solution in the capillary and indicating that the introduction of flexible PEG achieves a thickening effect on PEF.
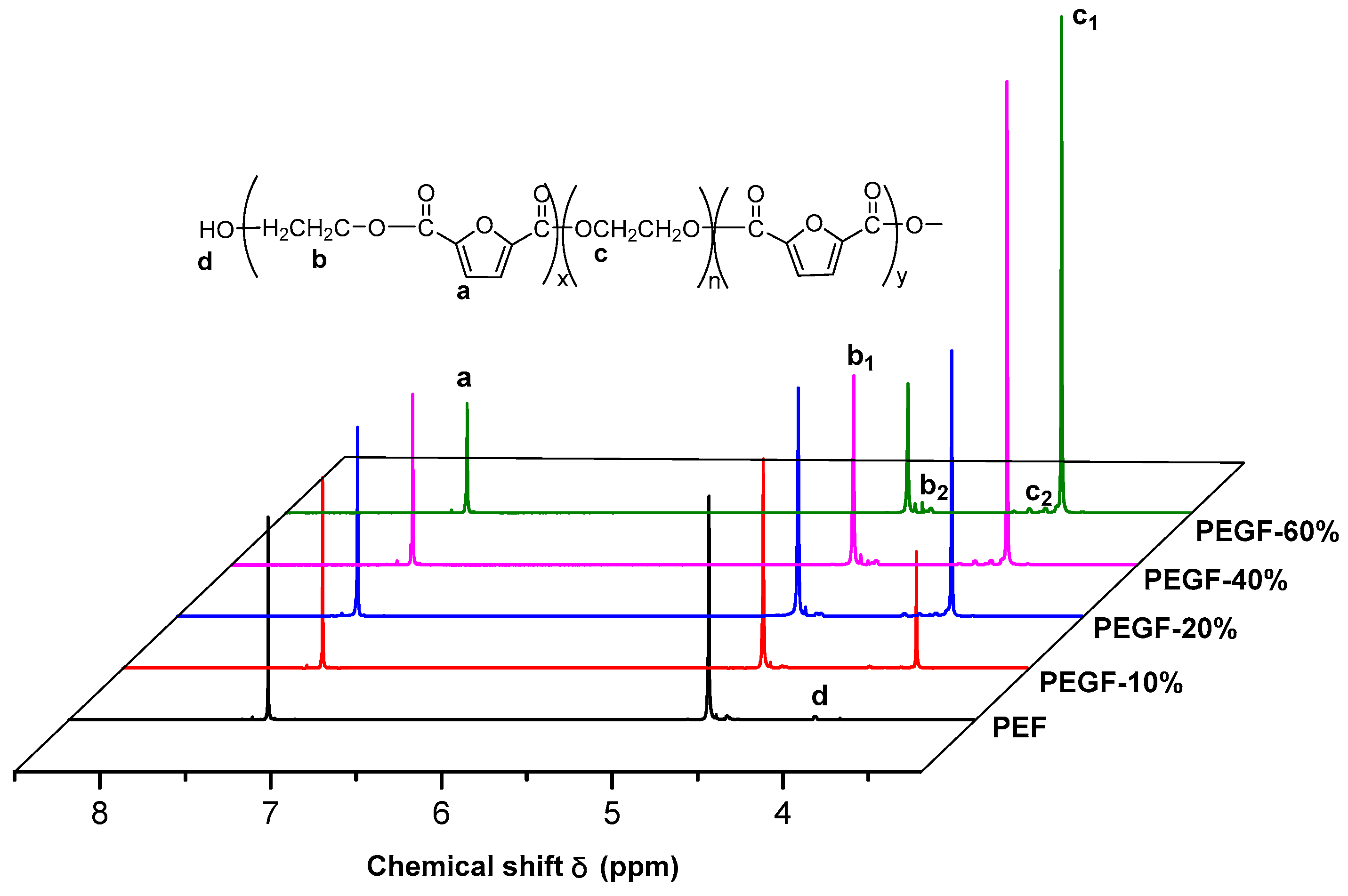
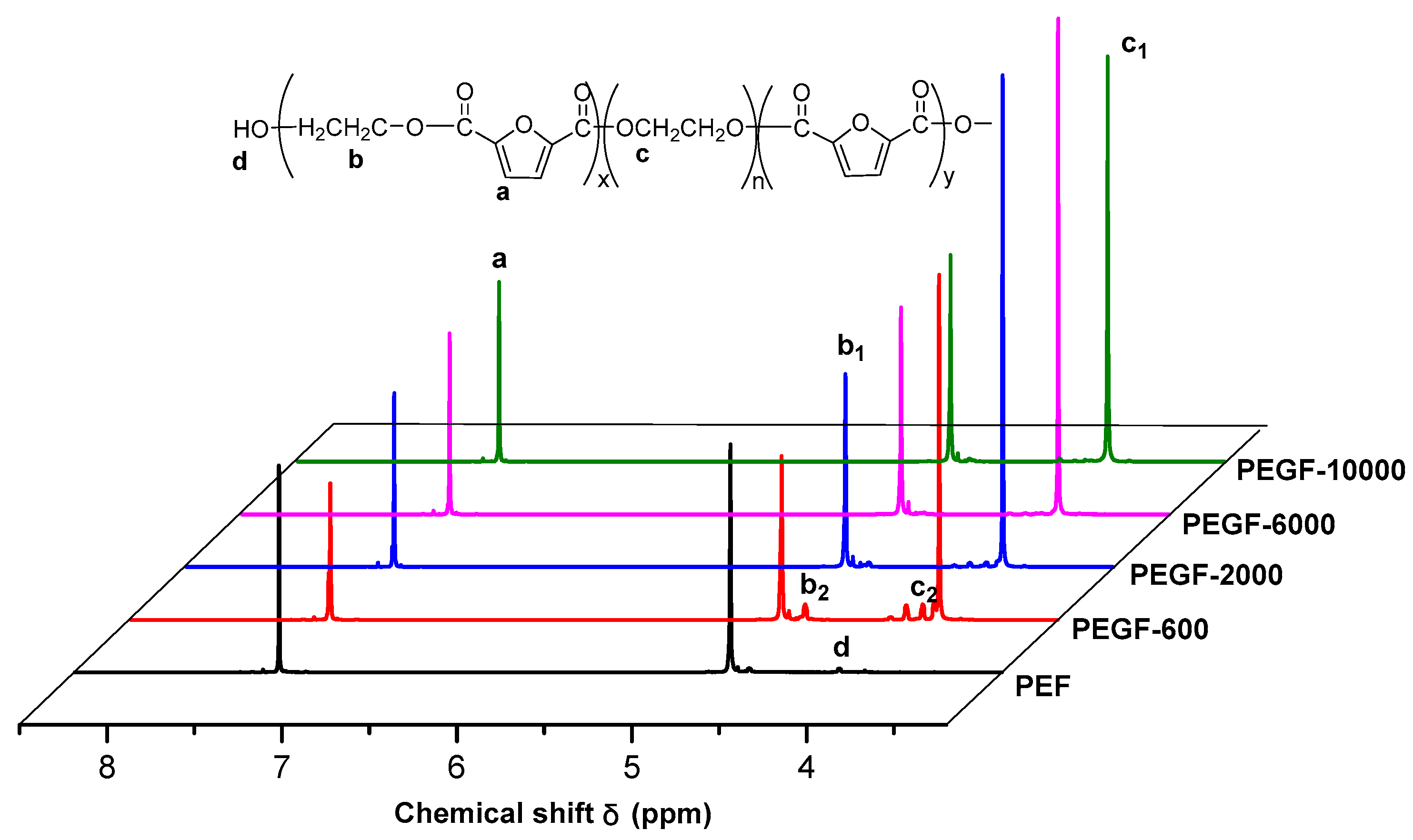
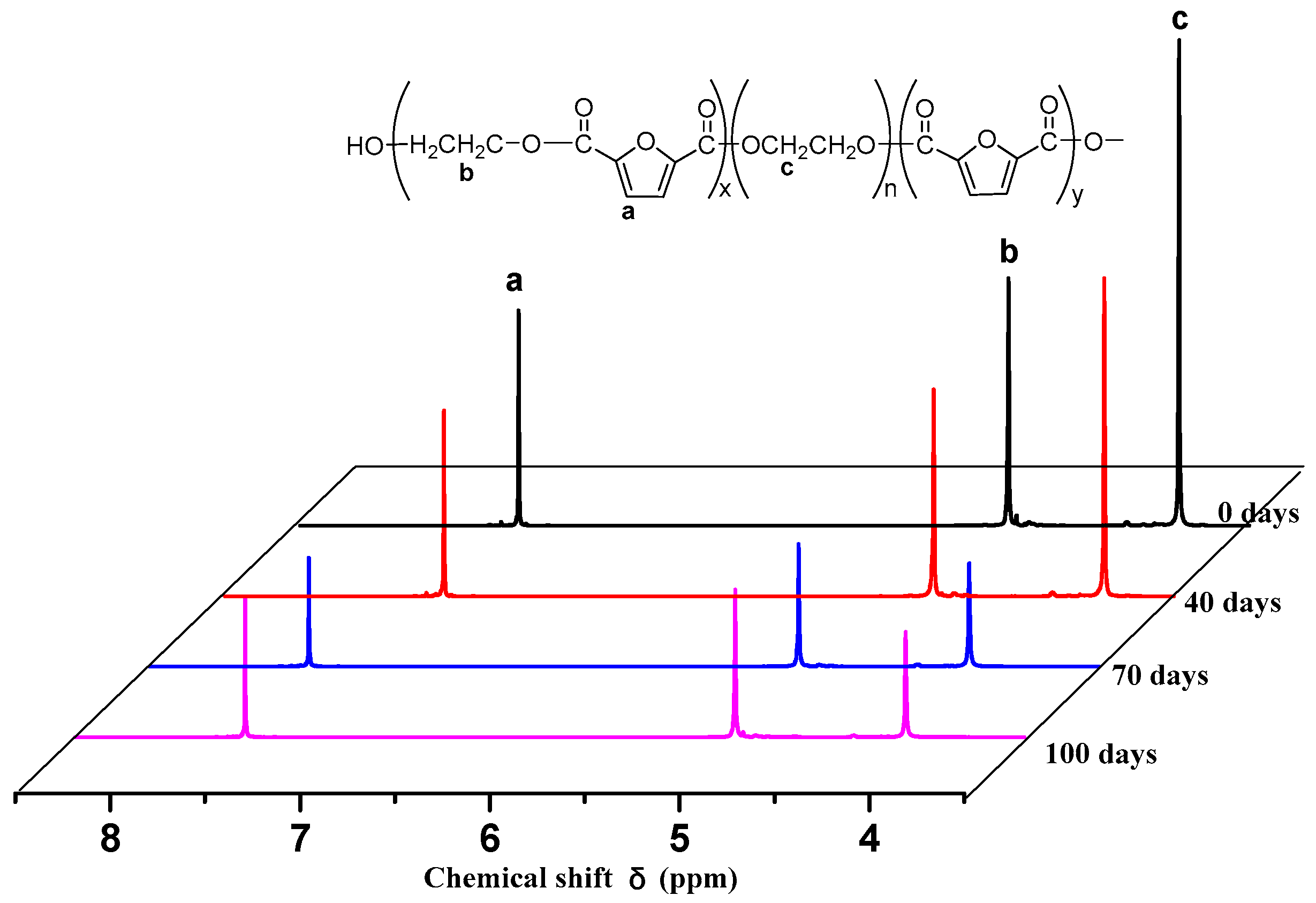
The
1H NMR spectra of PEGFs are presented. The structure of PEGFs with different PEG contents when the number average molecular weight of PEG is 2000 g/mol is shown in
Figure 3. The structure of PEGFs with different molecular weights when the PEG mass content is 40 wt % is shown in
Figure 4. As seen in the figure, each copolyester contains a total of four different types of H atoms with different chemical environments, where
δa = 7.32 ppm corresponds to the H atom on the furan ring,
δb = 4.73 ppm corresponds to the H atom of the methylene group in the hard segment of the polyester,
δc = 3.84 ppm corresponds to the H atom of the methylene group on the segment of PEG, and
δd = 3.81 ppm corresponds to the H atom in the terminal hydroxyl group of each copolyester. Due to the different positions of –CH
2– in the copolyester segment, the inductive effects of the surrounding atoms are different. The chemical shift of –CH
2– at the chain end will shift. Therefore, there are small proton peaks near the peaks of b and c, where b2 is the proton peak of –CH
2– in the PEF chain end, and c2 is the proton peak of –CH
2– in the PEG segment. According to the NMR results, when the molecular weight of PEG is 2000 g/mol, the PEG content increases, and the area of the c peak gradually increases. The above information indicates that the copolyester obtained by the polymerization is the target product.
The content of PEG in the copolyester was calculated according to Equations (3) and (4), and the results are shown in
Table 1. According to the results of the calculation, the actual added PEG content is consistent with the nuclear magnetic calculation results, indicating that the actually added PEG is fully involved in the reaction.
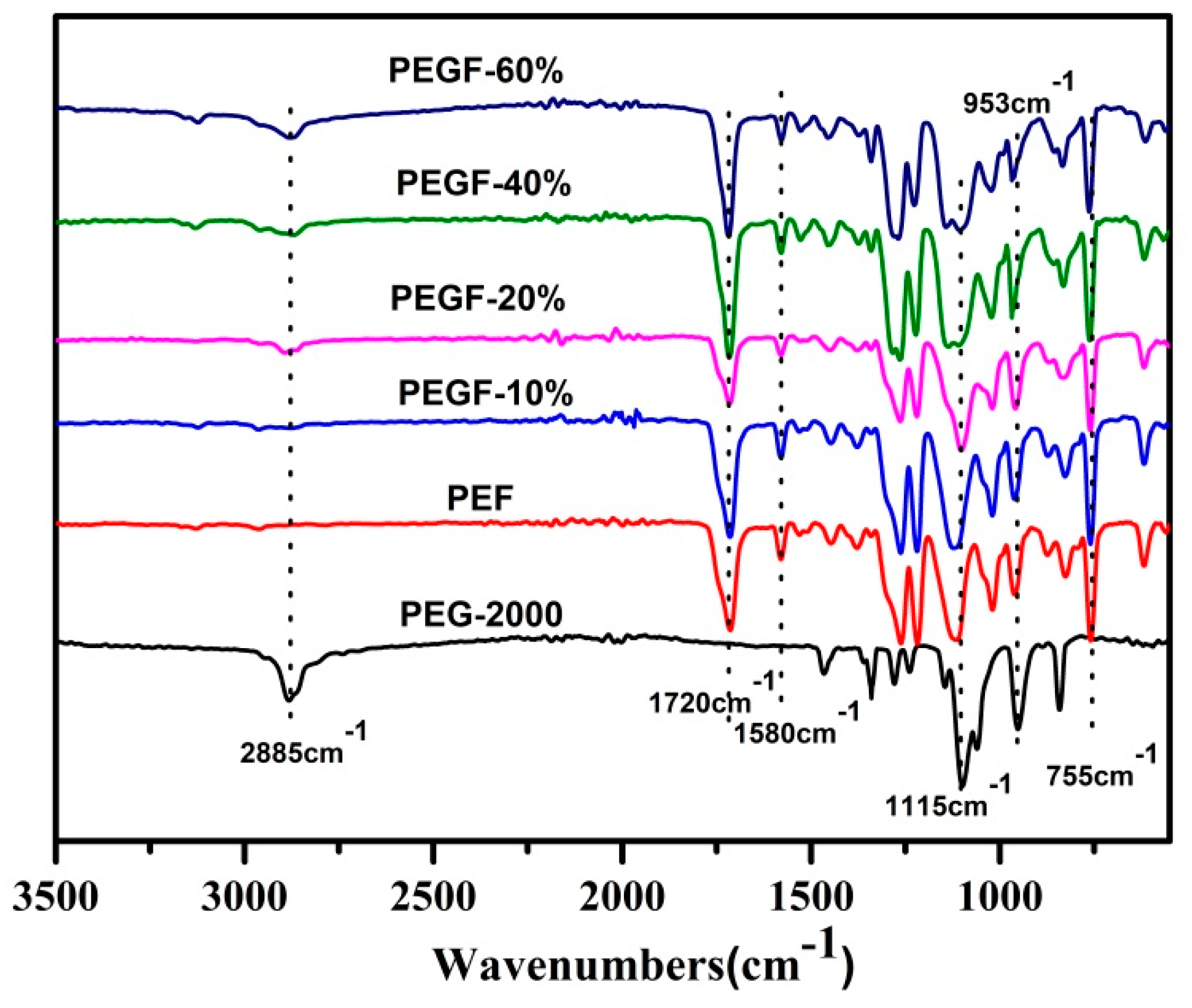
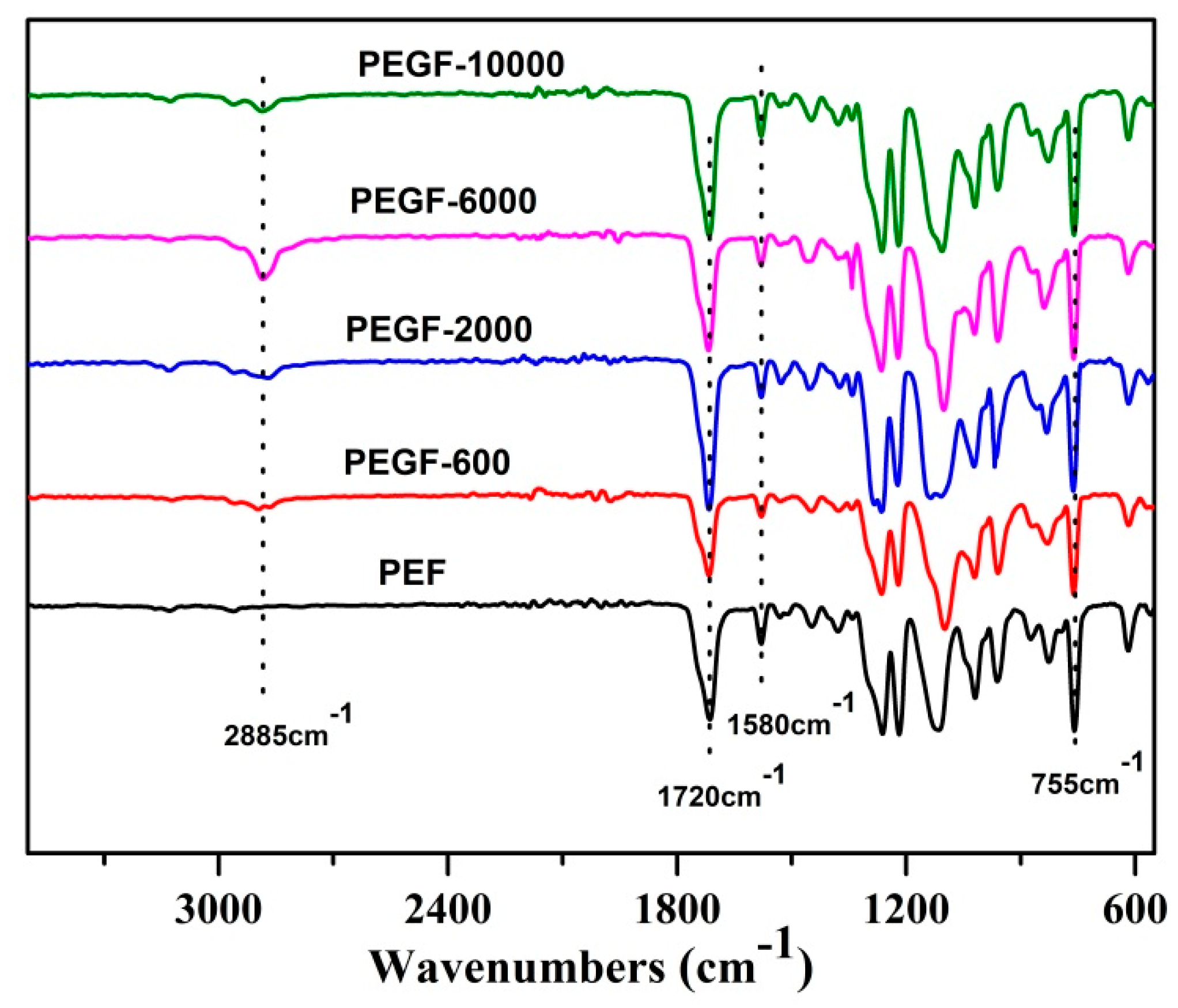
The chemical structure of the PEGFs was confirmed by FTIR. The infrared spectrum of the copolyester of different content of PEG when the number average molecular weight of PEG is 2000 g/mol is shown in
Figure 5; the infrared spectrum of the copolyester of different PEG molecular weights when the content of PEG is 40 wt % is shown in
Figure 6. The peaks at 2885 and 1115 cm
−1 are attributed to the –CH
2– and C–O–C vibration modes of PEG, respectively. In the copolyester, not only the characteristic peak of PEG, but also the characteristic peak of PEF appeared. The peak at 1720 cm
−1 is attributed to the C=O stretching vibration. The stretching vibration peaks of C–O–C appear at 1260 cm
−1. The stretching vibration peak of –CH
2– is observed at 2962 cm
−1. The C–H and C=C stretching vibration peaks on the furan are observed at 3120 and 1580 cm
−1, respectively, and the bending vibration peaks of the furan ring appeared at 960, 828, and 760 cm
−1. According to the
1H NMR and FTIR results, the positions of the characteristic peaks of the synthesized products are consistent with the positions of the characteristic peaks of each group in PEF and PEG. After the addition of PEG, the C–O–C peak of the product at 1115 cm
−1 appeared to move to the low wavelength region. When the number of average molecular weight of PEG was 2000 g/mol, the peak width at that point increased with the increasing PEG content. When the molecular weight of PEG gradually increases, the peak widths change a little, indicating that when the molecular weight of PEG is 2000 g/mol, the obtained product is a PEGF copolyester. When the molecular weight of PEG is higher than 2000 g/mol, the resulting product may be a blend of PEGF copolyester and PEG, which remains to be further characterized.
3.2. Thermal Properties of PEGF Copolyesters
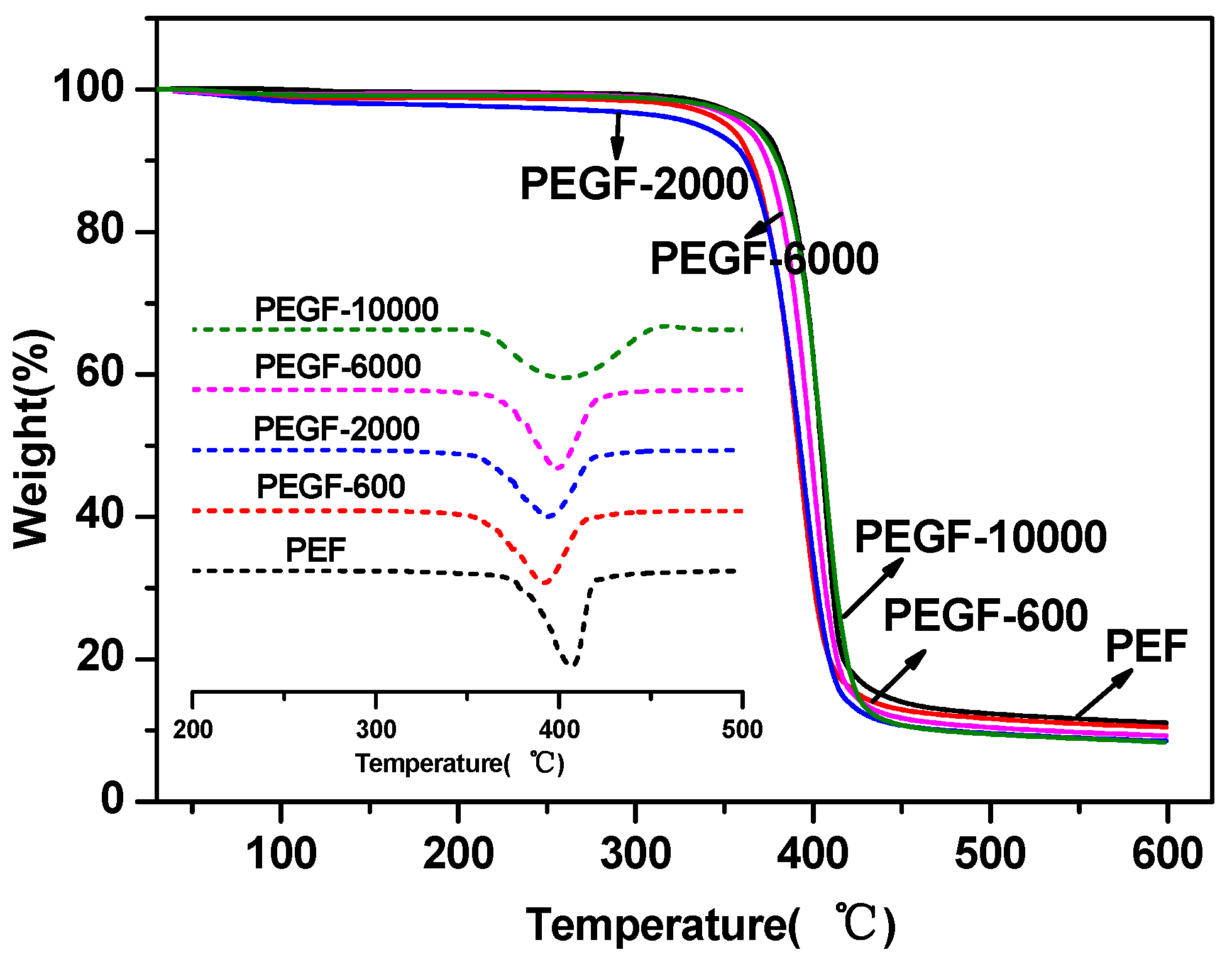
The thermal stability of the PEGFs was characterized by TGA. The results are shown in
Figure 7 and
Figure 8. From
Table 2, the thermal decomposition temperature of each copolyester is approximately 350 °C, the temperature at the fastest thermal decomposition rate is approximately 395 °C, and the remaining copolyester approximately 9% after the temperature is raised to 600 °C. Compared with that of PEF, the thermal stability of PEGFs is slightly decreased, but it is still much higher than the spinning temperature of conventional polyesters. Therefore, the addition of PEG does not affect its practical application in terms of thermal stability.
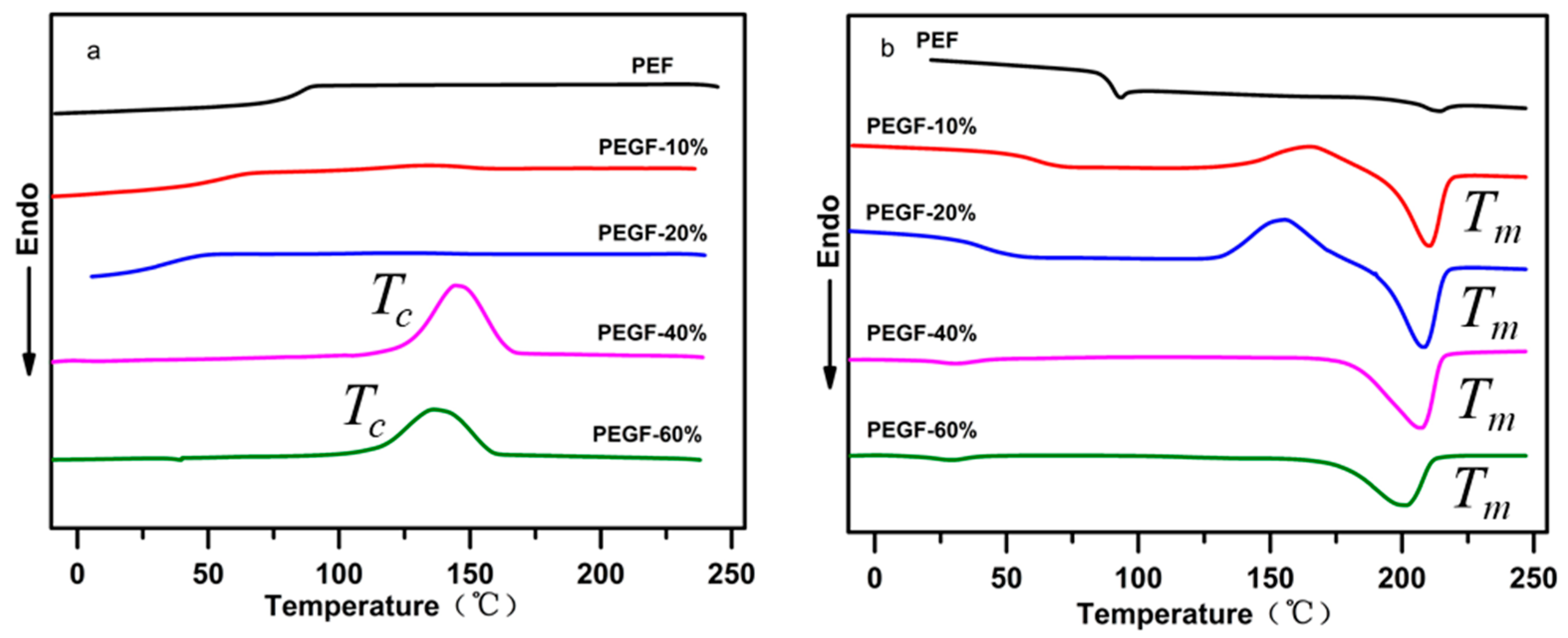
The crystallization behavior and melting performance of PEGFs were investigated by DSC. The crystallization and melting curves of PEGFs with different PEG contents (PEG
Mn = 2000 g/mol) are shown in
Figure 9a,b. Compared with PEF, when the PEG content increases gradually, the melting peak area of PEGFs increases gradually. When the PEG content reaches 40 and 60 wt %, the copolyester exhibits an obvious crystallization peak during cooling, indicating that the increased PEG content can promote the crystallization of PEGFs and increase the crystallization rate. This is mainly because the copolyester uses a part of the shorter PEF segment as the crystal nucleus during the cooling process; thus the PEG segment with strong kinetic ability will be rapidly arranged on the crystal nucleus formed by the PEF, the crystal will gradually grow and the crystallization rate will be increased. As seen from
Figure 9b, when the PEG content increases, the melting point of PEGFs gradually decreases. When the PEG content is 10 and 20 wt %, the crystallization peak appears before the melting of the PEGFs, indicating that when the PEG content in PEGFs is low, the crystallization rate of PEGFs is limited. In the cooling process, the PEGFs are not completely crystallized. During the heating process, when the temperature reaches the crystallization temperature again, the secondary crystals of PEGFs will appear, and imperfect crystallization peaks appear. When the PEG content is 40 and 60 wt %, the imperfect crystallization peak does not appear before the melting of the PEGFs, indicating that the crystallization rate of the PEGFs is faster with the increased PEG content, and that the crystallization is more perfect during the cooling process than before.
Figure 10a shows the cooling crystallization curves of PEGFs with PEG of different molecular weights (mass content: 40%). Compared with PEF, the addition of PEG promotes crystallization of the copolyester and increases its crystallization rate. When the molecular weight of PEG is 2000 g/mol, there is only one obvious crystallization peak during the cooling process of PEGFs. When the PEG molecular weight is 6000 and 10,000 g/mol, two crystallization peaks of PEGFs are observed during cooling, which is because the molecular weight of PEG of 2000 g/mol is equivalent to the molecular weight of PEF, so the two segments are well compatible. When the PEG molecular weight is 6000 and 10,000 g/mol, the PEG segment is long, and it is difficult for some PEG to begin the polycondensation reaction with PEF esterification during the polycondensation stage, resulting in some PEG coexisting with the copolyester in the form of a blend. During the cooling process, PEG present in a blended form is likely to undergo phase separation, so two crystallization peaks appear.
Figure 10b shows the melting curve of PEGFs with PEG of different molecular weights (mass content: 40%). When the molecular weight of PEG increases gradually, the melting point of PEGFs increases first and then decreases, but always exceeds the melting point of PEF, indicating that the introduction of PEG segments destroys the regularity of PEF segments and decreases the melting point of PEGFs. When the molecular weight of PEG reaches 10,000 g/mol, substantial phase separation occurs in the PEGFs, which leads to a decrease in the melting point of the PEGFs.
3.3. Hydrophilicity of PEGF Copolyester
When PEF is applied to the fiber field, there are still problems such as poor hydrophilicity of the polyester. To improve the hydrophilicity of PEF, this research introduced a flexible PEG segment into the PEF segment to increase the amorphous area in the PEGF so that water molecules can easily penetrate into the structure of the PEGFs. The molecular weight and content of PEG all affect the hydrophilicity of PEGFs. In this research, the hydrophilicity of PEGFs with different PEG molecular weights (600, 2000, 6000, 10,000 g/mol) and different contents (10, 20, 40, 60 wt %) was characterized. The results are shown in
Figure 11 and
Figure 12.
Figure 11 shows the change in contact angle on the surface of PEGFs with different PEG contents (PEG
Mn = 2000 g/mol). As seen from
Figure 11, when the molecular weight of PEG is the same, the surface contact angle of the PEGFs decreases from 80.1 to 63.3° with increasing the PEG content, but both are lower than the surface contact angle of the PEF polyester. This is mainly because after the introduction of PEG, the PEGFs form a block structure in which the hydrophilic and hydrophobic groups coexist, and the partial molecular chain of the PEG loosely arranges, so that surface water molecules can easily enter the surface of the PEGFs and increase the hydrophilicity of the PEGFs.
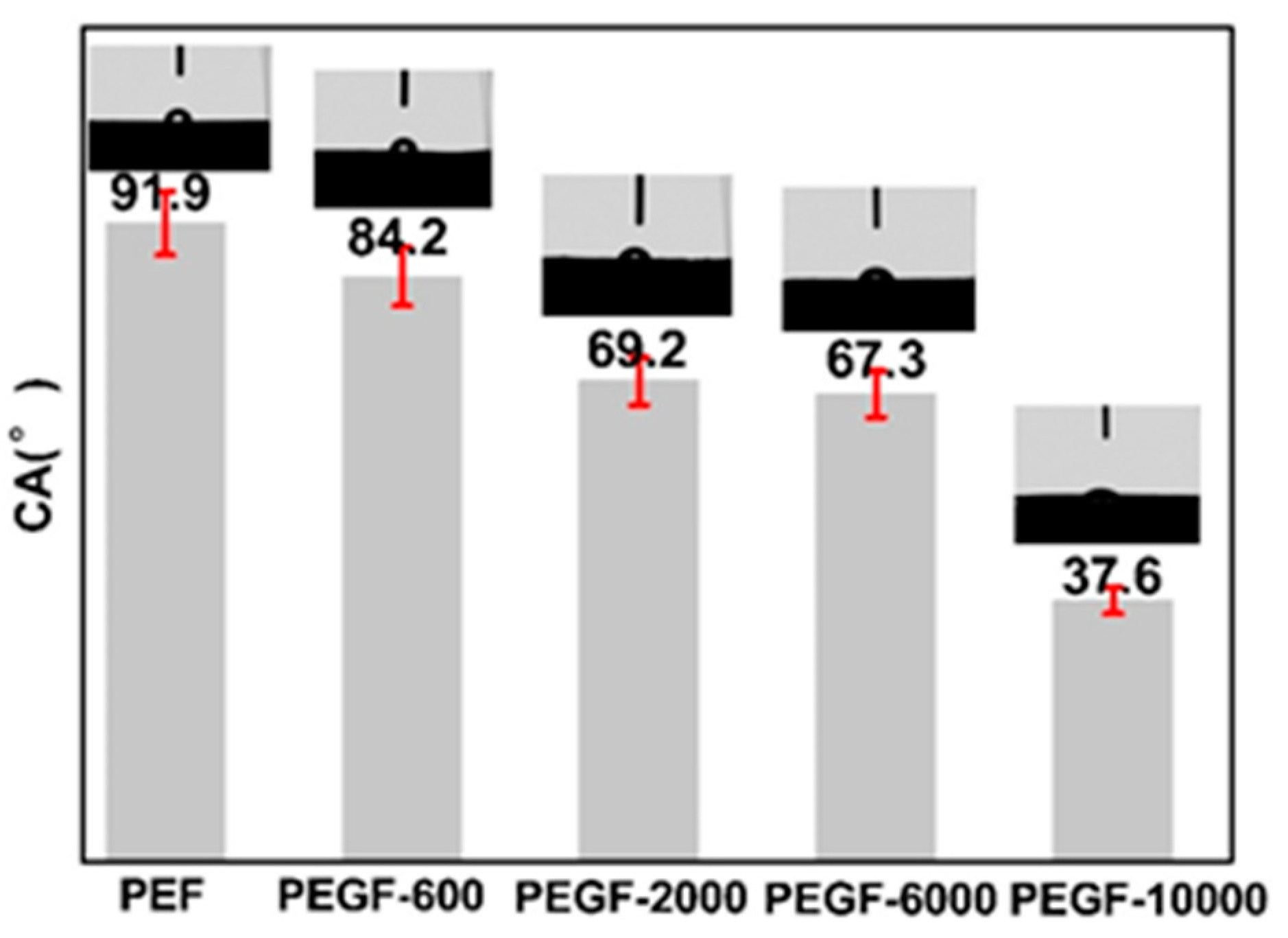
Figure 12 shows the change of contact angle on the surface of PEGFs with different PEG molecular weights (mass content: 40%). When the molecular weight of PEG increases, the surface contact angle of PEGFs decreases from 84.2 to 37.6°. Compared with the PEG content, the molecular weight of PEG has a greater influence on the hydrophilicity of PEGFs. The reason for this phenomenon is that when the molecular weight of PEG is 2000 g/mol, the length of the PEG segment is equivalent to the length of the segment of the PEF. In the polycondensation stage, the PEF is easily polycondensed with PEG to form a PEF-PEG copolyester. The hydrophilicity of PEGFs increases only because the PEG segment is loosely arranged, and there is a weak microphase separation between the soft and hard segments, which has less influence on the hydrophilicity of the copolyester. When the molecular weight of PEG reaches 10,000 g/mol, the molecular weight of PEF differs greatly from the molecular weight of PEG. During the polycondensation process, part of the PEF can react with PEG to form PEGFs, but some PEG still exists in the form of polymer blends. When the PEG segment increases, the molecular chain of the copolyester is loosely arranged, and there is a relatively substantial phase separation in the form of a blend of PEG and PEPFs, which greatly improves the hydrophilicity of the PEGFs.
3.4. Degradability of PEGF Copolyester
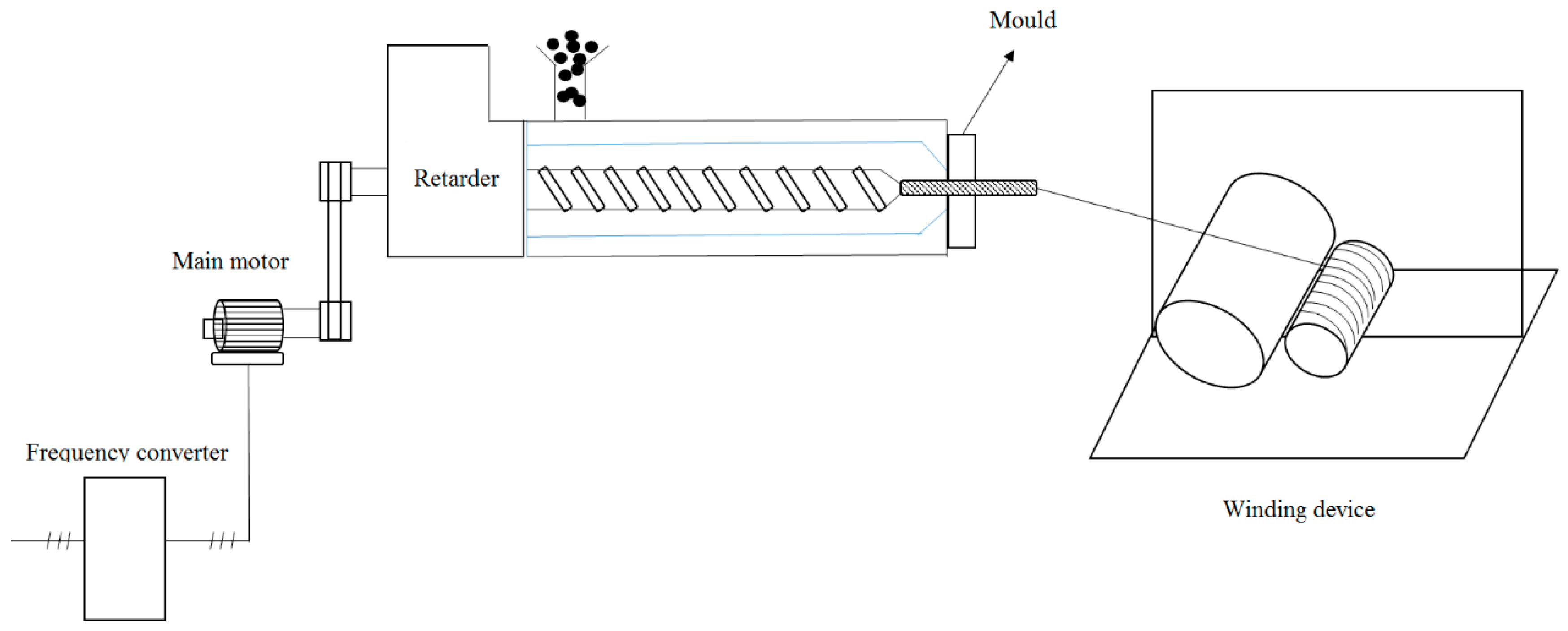
Hydrolytic degradation of PEGFs was performed in a phosphate buffer solution (pH = 7.2) at 37 °C. Samples were taken every 10 days and dried in a vacuum oven at 50 °C for 48 h, and the mass of the sample was measured.
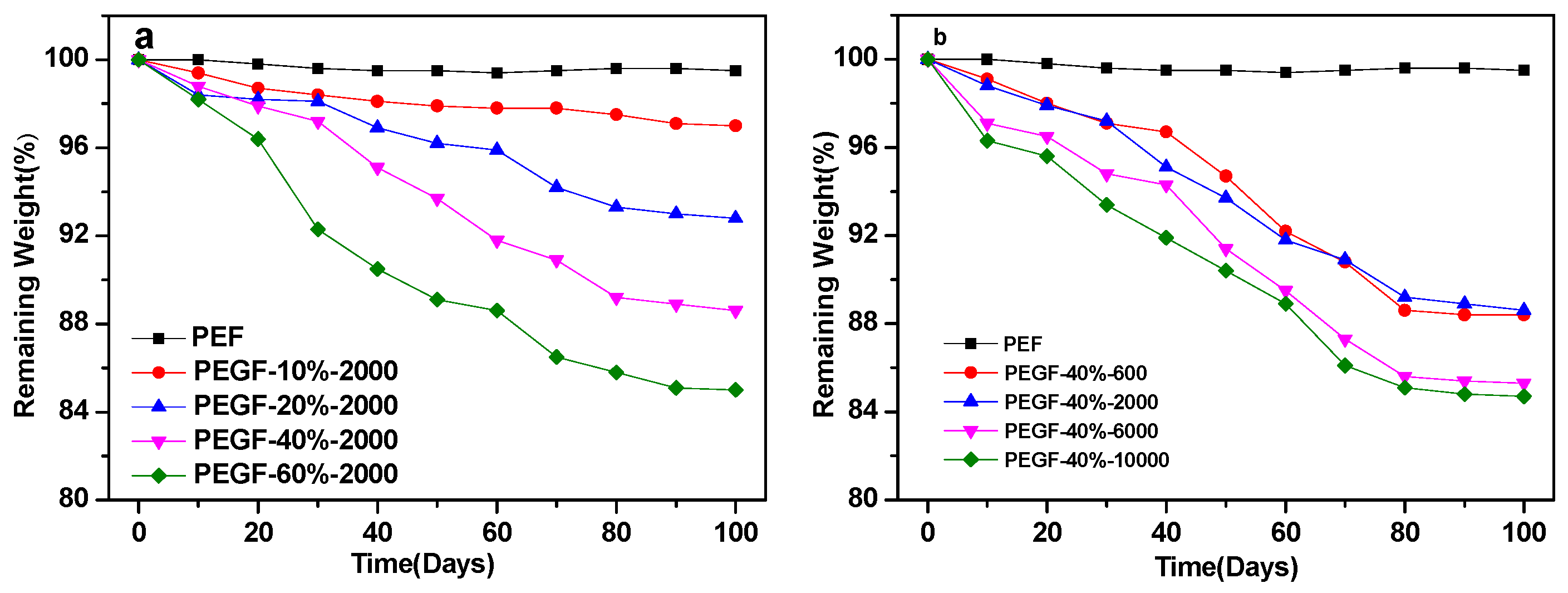
Figure 13 shows the variation of the quality of PEF and its copolyester film with the degradation time. As seen from
Figure 13, the quality change of PEF is not obvious during treatment in the phosphate buffer solution at pH = 7.2, and the mass loss is only approximately 0.5%, which fully indicates that under neutral conditions, PEF is not a degradable polymer. When PEG was introduced into PEF, the quality of PEGFs decreased with the prolongation of degradation time, which indicates that the introduction of PEG endowed the PEGFs with certain degradability, and the content and molecular weight of PEG had a great influence on the mass loss of PEGFs.
As seen from
Figure 13a, when the molecular weight of PEG is 2000 g/mol, the weight loss rate of the PEGFs increases with increasing PEG content. When the PEG content is 60 wt %, the mass loss of the PEGFs is the highest, reaching about 15%. From the degradation results, the change in the degradability of PEGFs is consistent with the change in hydrophilicity of PEGFs. This is mainly because the introduction of PEG improves the hydrophilicity of PEGFs, and water molecules easily enter the interior of the molecule. The ester bond in the molecular chain is attacked by water molecules, a hydrolysis reaction occurs, and the molecular chain is broken.
As seen from
Figure 13b, when the PEG content reaches 40 wt %, the PEGFs experience an obvious mass loss. With the increase in the molecular weight of PEG, the mass loss of PEGFs is more obvious and the degradation is more substantial. When the molecular weight of PEG is 10,000 g/mol, the mass loss of PEGFs is the highest, at 15.3%. When the molecular weight of PEG is 600 and 2000 g/mol, the change in quality of the two is basically the same as the degradation time increases. When the molecular weight of PEG is 6000 and 10,000 g/mol, the change in quality of both are the same. This is mainly because when the molecular weight of PEG is low, the length of the segment is equivalent to that of PEF. The compatibility between soft and hard segments is better, the attack probability of water molecules on ester bonds is not large, and the mass loss is relatively small. When the molecular weight of PEG reaches 6000 and 10,000 g/mol, the molecular chain lengths between the soft and hard segments are significantly different. These two are prone to serious phase separation, which increases the probability of attack by water molecules on ester bonds, and the quality loss is more substantial.
Table 3 shows the change in intrinsic viscosity after 100 days of degradation of PEGFs. From the table, after 100 days of degradation treatment, the intrinsic viscosity of PEF changes very little, only 0.1 dL/g, while the intrinsic viscosity of PEGF copolyester changes greatly. When the molecular weight of PEG is 2000 g/mol, the intrinsic viscosity of PEGFs increases with increasing PEG content. When the PEG content reaches 60 wt %, the intrinsic viscosity decreases by 0.66 dL/g. The conversion of PEGFs from high-molecular-weight polymers to low-molecular-weight polymers leads to the degradation of PEGFs. When the PEG content is 40 wt %, as the molecular weight of PEG increases, the intrinsic viscosity of PEGFs also decreases. When the molecular weight of PEG is 10,000 g/mol, the intrinsic viscosity of the PEGFs decreases by at most 0.45 dL/g. From the trend of intrinsic viscosity change, the introduction of PEG endows PEGFs with certain degradability, and its change trend is consistent with the trend of the change in quality of PEGFs.
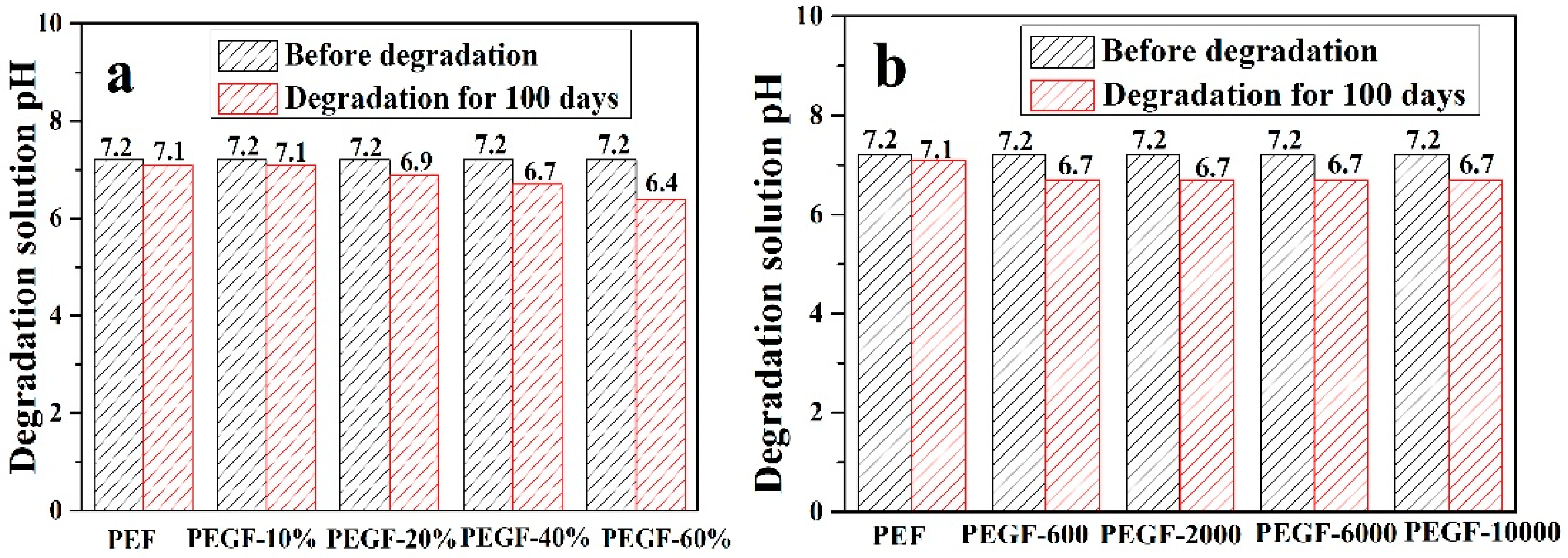
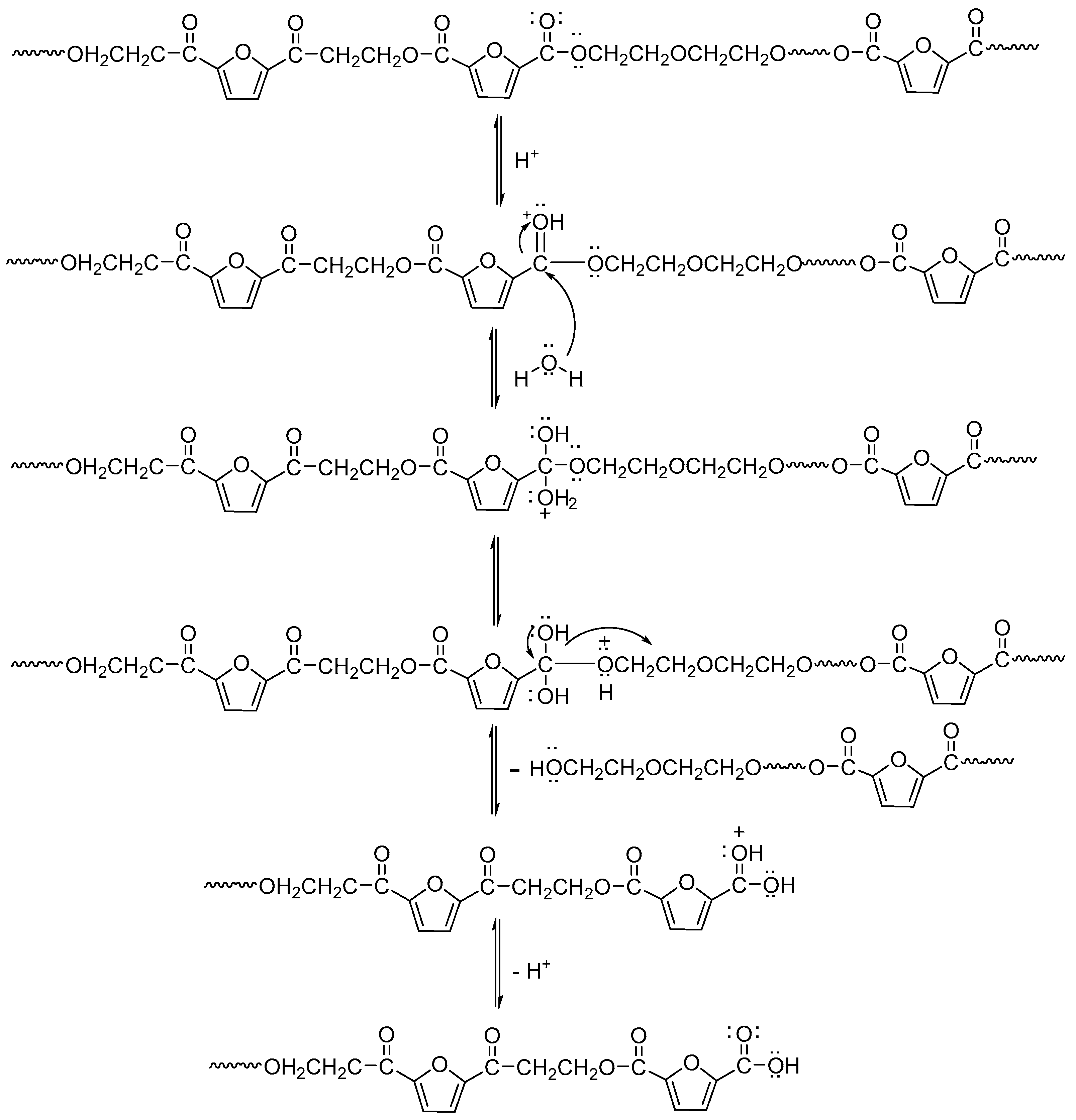
Figure 14 shows the change in the pH value of the degradation solution after 100 days of degradation of PEGFs. With the prolongation of degradation time, the pH value of PEF and its copolyester degradation solution shows a decreasing trend, and the degradation solution gradually changes from neutral to weakly acidic, which indicates that the degradation mechanism of PEEFs is acidic hydrolysis. The mechanism is shown in
Figure 15. As seen from
Figure 14a, when the molecular weight of the PEG is 2000 g/mol and the PEG content is increased, the pH of the PEGF degradation solution is gradually increased. As seen from
Figure 14b, with the increase of the PEG molecular weight, the pH value of the PEGF degradation solution is the same, which indicates that the content of PEG has a major influence on the pH value of the PEGF degradation solution.
To further explain the degradation mechanism of PEGFs and the change in structure of PEGFs during hydrolysis, in this research, the most degraded samples (PEGF-60%-2000 and PEGF-40%-10000) were selected for the analysis of the surface topography of polyester film, nuclear magnetic resonance, and PEGF degradation experiments. Samples were selected after 0, 40, 70, and 100 days of PEGF degradation.
As shown in
Figure 16, the area was integrated to calculate the content of PEG in PEGFs. The results are shown in
Table 4. As seen from
Table 4, when the degradation time increases, the PEG content in PEGFs decreases gradually. Peaks a and b are integrated. The ratio of peak area do not change with the prolongation of degradation time, which indicates that during the degradation of PEGFs, the ester bond linking the PEG moiety is mainly hydrolyzed and broken, and the PEGFs are partially is degraded.
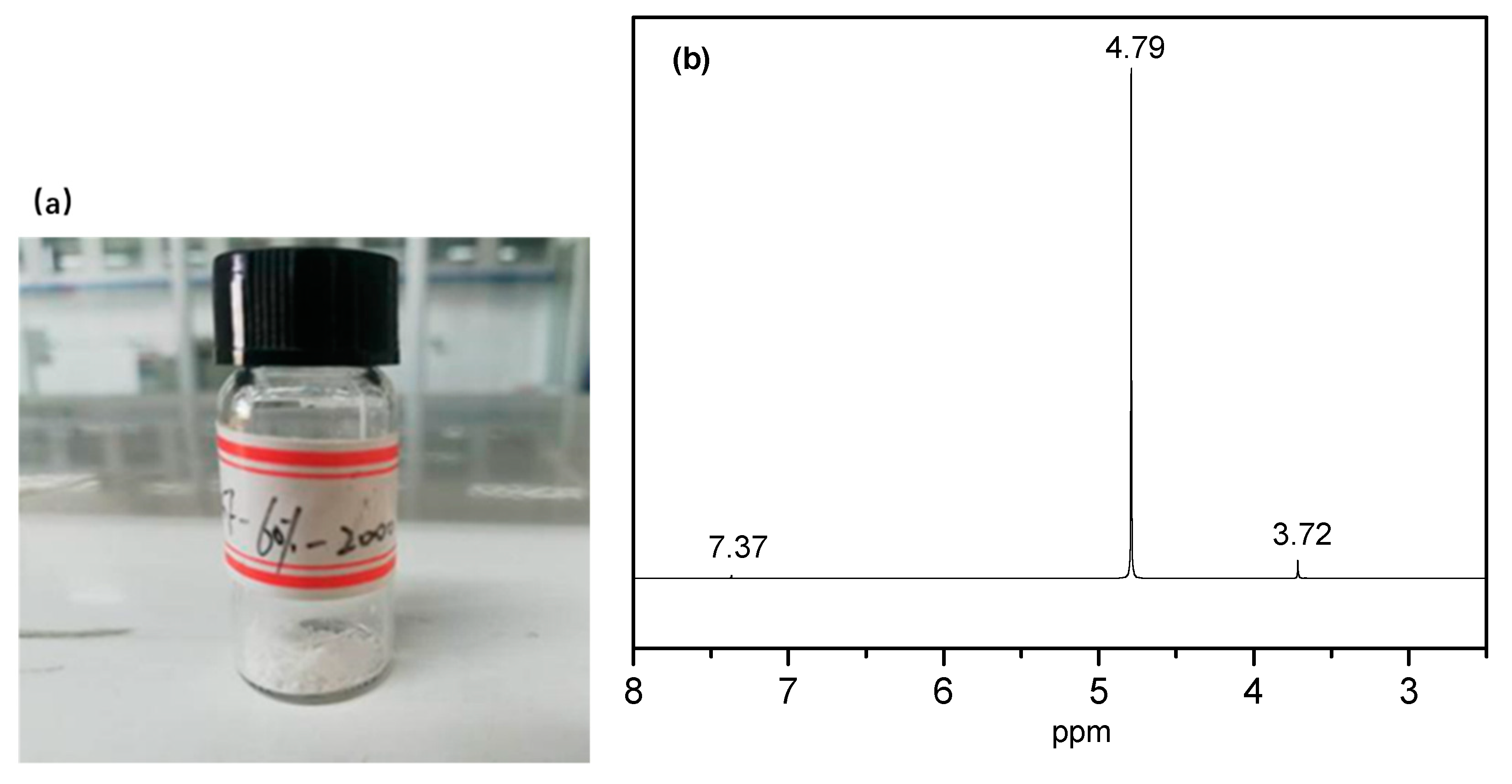
The degradation solution of the PEGF-60%-2000 copolyester was subjected to rotary evaporation at 70 °C to obtain a white flocculent material, which was subjected to nuclear magnetic resonance characterization. As seen from
Figure 17, there are three proton peaks in the nuclear magnetic resonance curve, where
δ = 4.79 ppm is the proton peak of deuterated water, and δ = 7.36 ppm is the proton peak in phosphate. By inspection,
δ = 3.72 ppm is the proton peak of –CH
2– in the PEG segment, which indicates that in the degradation of PEGFs, mainly occurs by PEG partial chain scission.
As seen in
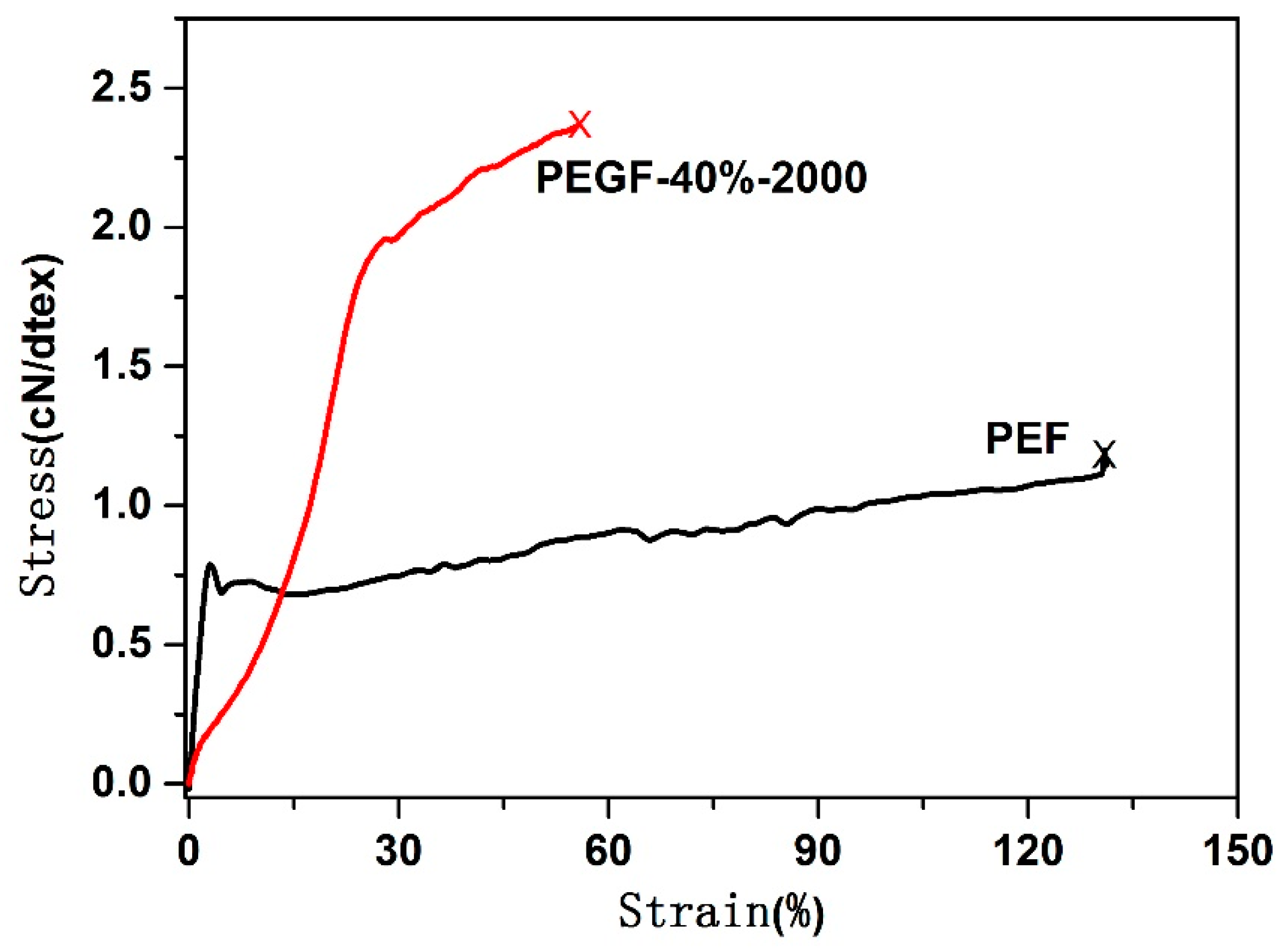
Table 4, at the beginning of degradation, the content of PEG changes slightly, but its mass loss is substantial. In the later stage of degradation, the content of PEG changes greatly, but its mass loss is small. In the beginning of degradation, although the copolyester has a certain hydrophilicity, the internal structure of the PEGFs is still very regular, and it is difficult for water molecules to interact with each ester bond. Some PEG-linked short-chain PEFs enter the degradation solution together, making the weight loss of the PEGFs more obvious. In the late stage of degradation, the molecular weight of PEGFs is low, and there are many pores inside the molecule. The ester bond linked to PEG is easily attacked by water molecules, which causes the ester bond at both ends of the PEG to break, allowing the PEG molecule to enter the degradation liquid, and the remaining short chain PEF is still present in the PEGFs. The mechanical properties of the PEGFs gradually deteriorate, showing the fragile nature of the material. The result is shown in Figure 19.
Figure 18 shows the nuclear magnetic resonance curve of the PEGF-40%-10000 degradation sample after treatment for different numbers of days.
Table 5 shows changes in the PEG content. The PEG content in the PEGFs gradually decreases when the degradation time increases, but when the degradation time increases, the changes in the PEG content in each stage are almost the same. In the late stage of degradation, the change is slightly reduced. This phenomenon is completely different from that observed for the PEGF-60%-2000 copolyester. The reason for this phenomenon is that when the molecular weight of PEG reaches 10,000 g/mol, the segment of PEG is longer, and the PEF esterification chain length is much different from that of PEG. Thus, part of the PEG segment forms an ester with PEF, and part of the PEG segment forms a blend with PEGFs. With the prolongation of degradation time, the PEG segment in the form of blend is gradually degraded and dissolved in the degradation solution, and the PEG in the form of copolyester is gradually hydrolyzed and undergoes chain breakage during degradation. The mechanical properties of PEGFs were analyzed. When the degradation time is prolonged, the PEGF-40%-10000 copolyester is gradually transformed from a PEMF film with good toughness into a fragile film.
The surface morphology of PEGFs was analyzed by SEM.
Figure 19 shows SEM images of the PEF, PEGF-60%-2000, and PEGF-40%-10000 degradation films. With the prolongation of degradation time, there is almost no change in the surface of PEF, indicating that PEF cannot be degraded in a neutral buffer solution. For the PEGF-60%-2000 copolyester, during the pressing film formation, when the surface of the polyester film is naturally cooled from 240 °C to room temperature, the PEG segment rapidly crystallizes. When the degradation time is prolonged, the PEG segment on the surface of the polyester film is gradually reduced, and at the same time the surface has irregularities and a certain depth of pores. For the PEGF-40%-10000 copolyester, when the degradation continues for to 40 days, many pores appear on the surface of the PEGFs. As the degradation time is prolonged, the pores became larger and deeper, which indicates that the degradation of the PEGFs is more substantial.























{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}