Effect of Molecular Structure in the Chain Mobility of Dichalcogenide-Based Polymers with Self-Healing Capacity




Abstract
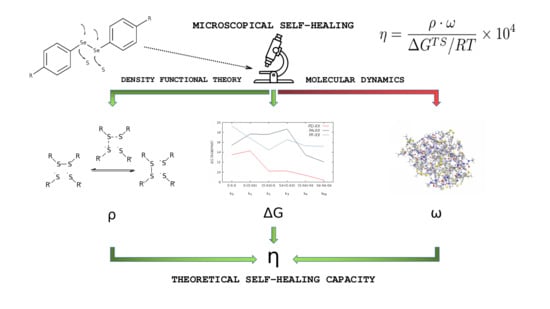
1. Introduction
2. Methods
2.1. Molecular Dynamics Simulations
2.2. Validation of the Computational Protocol
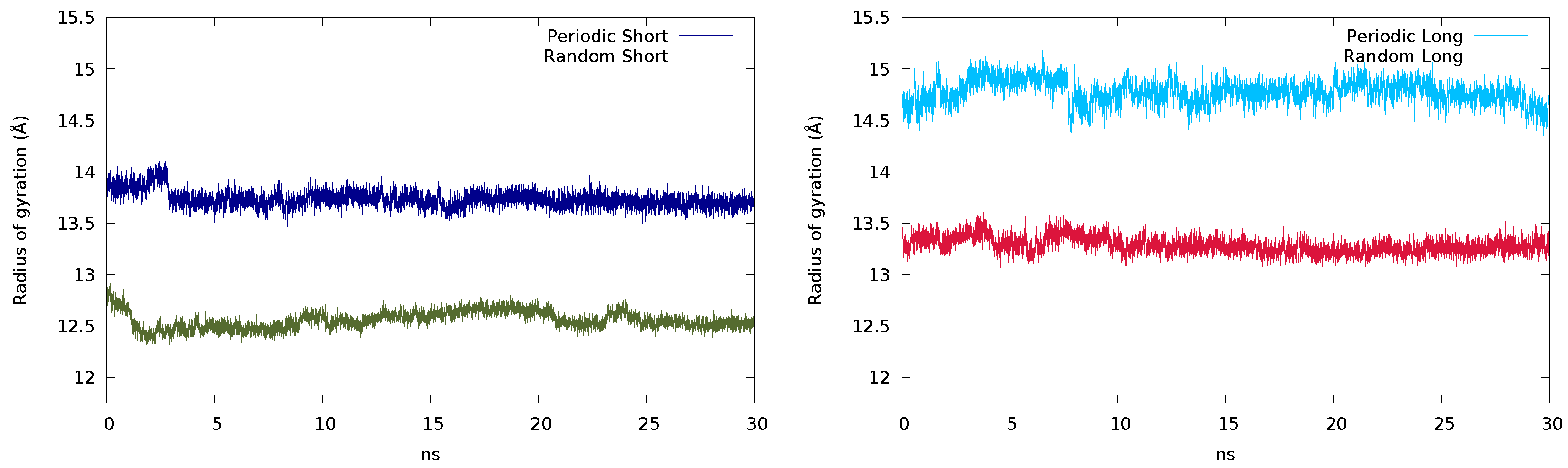
2.2.1. Initial Guess and Chain Length
2.2.2. Size of the Simulation Box
3. Results
3.1. Influence of the Backbone: PD, PA And PF
3.2. Influence of the Substituents (R-R) and Chalcogenide Atoms (X)
3.3. Theoretical Self-Healing Capacity
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| MD | Molecular dynamics |
| RESP | Restrained electrostatic potential |
| HB | Hydrogen bond |
References
- Rowan, S.J.; Cantrill, S.J.; Cousins, G.R.L.; Sanders, J.K.M.; Stoddart, J.F. Dynamic Covalent Chemistry. Angew. Chem. Int. Ed. 2002, 41, 898–952. [Google Scholar] [CrossRef]
- Wojtecki, R.J.; Meador, M.A.; Rowan, S.J. Using the dynamic bond to access macroscopically responsive structurally dynamic polymers. Nat. Mater. 2011, 10, 14–27. [Google Scholar] [CrossRef] [PubMed]
- Jin, Y.; Yu, C.; Denman, R.J.; Zhang, W. Recent advances in dynamic covalent chemistry. Chem. Soc. Rev. 2013, 42, 6634–6654. [Google Scholar] [CrossRef]
- Maeda, T.; Otsuka, H.; Takahara, A. Dynamic covalent polymers: Reorganizable polymers with dynamic covalent bonds. Prog. Polym. Sci. 2009, 34, 581–604. [Google Scholar] [CrossRef]
- Zhang, Z.P.; Rong, M.Z.; Zhang, M.Q. Polymer engineering based on reversible covalent chemistry: A promising innovative pathway towards new materials and new functionalities. Prog. Polym. Sci. 2018, 80, 39–93. [Google Scholar] [CrossRef]
- Wu, D.Y.; Meure, S.; Solomon, D. Self-healing polymeric materials: A review of recent developments. Prog. Polym. Sci. 2008, 33, 479–522. [Google Scholar] [CrossRef]
- Bergman, S.D.; Wudl, F. Mendable polymers. J. Mater. Chem. 2008, 18, 41–62. [Google Scholar] [CrossRef]
- Syrett, J.A.; Becer, C.R.; Haddleton, D.M. Self-healing and self-mendable polymers. Polym. Chem. 2010, 1, 978–987. [Google Scholar] [CrossRef]
- Burattini, S.; Greenland, B.W.; Chappell, D.; Colquhoun, H.M.; Hayes, W. Healable polymeric materials: A tutorial review. Chem. Soc. Rev. 2010, 39, 1973–1985. [Google Scholar] [CrossRef]
- Cordier, P.; Tournilhac, F.; Soulié-Ziakovic, C.; Leibler, L. Self-healing and thermoreversible rubber from supramolecular assembly. Nature 2008, 451, 977–980. [Google Scholar] [CrossRef]
- Burattini, S.; Colquhoun, H.M.; Fox, J.D.; Friedmann, D.; Greenland, B.W.; Harris, P.J.F.; Hayes, W.; Mackay, M.E.; Rowan, S.J. A self-repairing, supramolecular polymer system: Healability as a consequence of donor–acceptor π–π stacking interactions. Chem. Commun. 2009, 44, 6717–6719. [Google Scholar] [CrossRef] [PubMed]
- Burnworth, M.; Tang, L.; Kumpfer, J.R.; Duncan, A.J.; Beyer, F.L.; Fiore, G.L.; Rowan, S.J.; Weder, C. Optically healable supramolecular polymers. Nature 2011, 472, 334–337. [Google Scholar] [CrossRef] [PubMed]
- Hager, M.D.; Greil, P.; Leyens, C.; van der Zwaag, S.; Schubert, U.S. Self-Healing Materials. Adv. Mater. 2010, 22, 5424–5430. [Google Scholar] [CrossRef] [PubMed]
- Billiet, S.; Hillewaere, X.K.D.; Teixeira, R.F.A.; Du Prez, F.E. Chemistry of Crosslinking Processes for Self-Healing Polymers. Macromol. Rapid Commun. 2013, 34, 290–309. [Google Scholar] [CrossRef]
- Hillewaere, X.K.; Du Prez, F.E. Fifteen chemistries for autonomous external self-healing polymers and composites. Prog. Polym. Sci. 2015, 49–50, 121–153. [Google Scholar] [CrossRef]
- Kuhl, N.; Bode, S.; Hager, M.D.; Schubert, U.S. Self-Healing Polymers Based on Reversible Covalent Bonds. Adv. Polym. Sci. 2015, 273, 1–58. [Google Scholar] [CrossRef]
- Chen, X.; Wudl, F.; Mal, A.K.; Shen, H.; Nutt, S.R. New thermally remendable highly cross-linked polymeric materials. Macromolecules 2003, 36, 1802–1807. [Google Scholar] [CrossRef]
- Roling, O.; De Bruycker, K.; Vonhören, B.; Stricker, L.; Körsgen, M.; Arlinghaus, H.F.; Ravoo, B.J.; Du Prez, F.E. Rewritable Polymer Brush Micropatterns Grafted by Triazolinedione Click Chemistry. Angew. Chem. Int. Ed. 2015, 54, 13126–13129. [Google Scholar] [CrossRef]
- Cho, S.; Jeong, S.; Kim, J.M.; Baig, C. Molecular dynamics for linear polymer melts in bulk and confined systems under shear flow. Sci. Rep. 2017, 7, 1–9. [Google Scholar] [CrossRef]
- Schmolke, W.; Perner, N.; Seiffert, S. Dynamically Cross-Linked Polydimethylsiloxane Networks with Ambient-Temperature Self-Healing. Macromolecules 2015, 48, 8781–8788. [Google Scholar] [CrossRef]
- Montarnal, D.; Capelot, M.; Tournilhac, F.; Leibler, L. Silica-like malleable materials from permanent organic networks. Science 2011, 334, 965–968. [Google Scholar] [CrossRef] [PubMed]
- Cromwell, O.R.; Chung, J.; Guan, Z. Malleable and Self-Healing Covalent Polymer Networks through Tunable Dynamic Boronic Ester Bonds. J. Am. Chem. Soc. 2015, 137, 6492–6495. [Google Scholar] [CrossRef] [PubMed]
- Azcune, I.; Odriozola, I. Aromatic disulfide crosslinks in polymer systems: Self-healing, reprocessability, recyclability and more. Eur. Polym. J. 2016, 84, 147–160. [Google Scholar] [CrossRef]
- Takahashi, A.; Goseki, R.; Ito, K.; Otsuka, H. Thermally Healable and Reprocessable Bis(hindered amino)disulfide-Cross-Linked Polymethacrylate Networks. ACS Macro Lett. 2017, 6, 1280–1284. [Google Scholar] [CrossRef]
- Amamoto, Y.; Otsuka, H.; Takahara, A.; Matyjaszewski, K. Self-healing of covalently cross-linked polymers by reshuffling thiuram disulfide moieties in air under visible light. Adv. Mater. 2012, 24, 3975–3980. [Google Scholar] [CrossRef]
- Yoon, J.A.; Kamada, J.; Koynov, K.; Mohin, J.; Nicolay, R.; Zhang, Y.Z.; Balazs, A.C.; Kowalewski, T.; Matyjaszewski, K. Self-Healing Polymer Films Based on Thiol-Disulfide Exchange Reactions and Self-Healing Kinetics Measured Using Atomic Force Microscopy. Macromolecules 2012, 45, 142–149. [Google Scholar] [CrossRef]
- Otsuka, H.; Nagano, S.; Kobashi, Y.; Maeda, T.; Takahara, A. A dynamic covalent polymer driven by disulfide metathesis under photoirradiation. Chem. Commun. 2010, 46, 1150–1152. [Google Scholar] [CrossRef]
- Abdolah Zadeh, M.; van der Zwaag, S.; Garcia, S.J. Adhesion and Long-Term Barrier Restoration of Intrinsic Self-Healing Hybrid Sol–Gel Coatings. ACS Appl. Mater. Interfaces 2016, 8, 4126–4136. [Google Scholar] [CrossRef]
- Martin, R.; Rekondo, A.; de Luzuriaga, A.R.; Casuso, P.; Dupin, D.; Cabañero, G.; Grande, H.J.; Odriozola, I. Dynamic sulfur chemistry as a key tool in the design of self-healing polymers. Smart Mater. Struct. 2016, 25, 084017. [Google Scholar] [CrossRef]
- Xu, W.M.; Rong, M.Z.; Zhang, M.Q. Sunlight driven self-healing, reshaping and recycling of a robust, transparent and yellowing-resistant polymer. J. Mater. Chem. A 2016, 4, 10683–10690. [Google Scholar] [CrossRef]
- Lei, Z.Q.; Xiang, H.P.; Yuan, Y.J.; Rong, M.Z.; Zhang, M.Q. Room-Temperature Self-Healable and Remoldable Cross-linked Polymer Based on the Dynamic Exchange of Disulfide Bonds. Chem. Mater. 2014, 26, 2038–2046. [Google Scholar] [CrossRef]
- Tsarevsky, N.V.; Matyjaszewski, K. Reversible redox cleavage/coupling of polystyrene with disulfide or thiol groups prepared by atom transfer radical polymerization. Macromolecules 2002, 35, 9009–9014. [Google Scholar] [CrossRef]
- Fairbanks, B.D.; Singh, S.P.; Bowman, C.N.; Anseth, K.S. Photodegradable, Photoadaptable Hydrogels via Radical-Mediated Disulfide Fragmentation Reaction. Macromolecules 2011, 44, 2444–2450. [Google Scholar] [CrossRef]
- Lafont, U.; van Zeijl, H.; van der Zwaag, S. Influence of Cross-linkers on the Cohesive and Adhesive Self-Healing Ability of Polysulfide-Based Thermosets. ACS Appl. Mater. Interfaces 2012, 4, 6280–6288. [Google Scholar] [CrossRef] [PubMed]
- Imbernon, L.; Oikonomou, E.K.; Norvez, S.; Leibler, L. Chemically crosslinked yet reprocessable epoxidized natural rubber via thermo-activated disulfide rearrangements. Polym. Chem. 2015, 6, 4271–4278. [Google Scholar] [CrossRef]
- Martin, R.; Rekondo, A.; Ruiz de Luzuriaga, A.; Cabañero, G.; Grande, H.J.; Odriozola, I. The processability of a poly(urea-urethane) elastomer reversibly crosslinked with aromatic disulfide bridges. J. Mater. Chem. A 2014, 2, 5710. [Google Scholar] [CrossRef]
- Rekondo, A.; Martin, R.; Ruiz De Luzuriaga, A.; Cabañero, G.; Grande, H.J.; Odriozola, I. Catalyst-free room-temperature self-healing elastomers based on aromatic disulfide metathesis. Mater. Horiz. 2014, 1, 237–240. [Google Scholar] [CrossRef]
- Martin, R.; Rekondo, A.; Ruiz de Luzuriaga, A.; Santamaria, A.; Odriozola, I. Mixing the immiscible: Blends of dynamic polymer networks. RSC Adv. 2015, 5, 17514–17518. [Google Scholar] [CrossRef]
- Ruiz de Luzuriaga, A.; Martin, R.; Markaide, N.; Rekondo, A.; Cabañero, G.; Rodríguez, J.; Odriozola, I. Epoxy resin with exchangeable disulfide crosslinks to obtain reprocessable, repairable and recyclable fiber-reinforced thermoset composites. Mater. Horiz. 2016, 3, 241–247. [Google Scholar] [CrossRef]
- Johnson, L.M.; Ledet, E.; Huffman, N.D.; Swarner, S.L.; Shepherd, S.D.; Durham, P.G.; Rothrock, G.D. Controlled degradation of disulfide-based epoxy thermosets for extreme environments. Polymer 2015, 64, 84–92. [Google Scholar] [CrossRef]
- Ruiz de Luzuriaga, A.; Matxain, J.M.; Ruipérez, F.; Martin, R.; Asua, J.M.; Cabañero, G.; Odriozola, I. Transient mechanochromism in epoxy vitrimer composites containing aromatic disulfide crosslinks. J. Mater. Chem. C 2016, 4, 6220–6223. [Google Scholar] [CrossRef]
- Aguirresarobe, R.H.; Martin, L.; Fernandez-Berridi, M.J.; Irusta, L. Autonomic healable waterborne organic-inorganic polyurethane hybrids based on aromatic disulfide moieties. Express Polym. Lett. 2017, 11, 266–277. [Google Scholar] [CrossRef]
- Ji, S.; Xia, J.; Xu, H. Dynamic Chemistry of Selenium: Se-N and Se-Se Dynamic Covalent Bonds in Polymeric Systems. ACS Macro Lett. 2016, 5, 78–82. [Google Scholar] [CrossRef]
- Ji, S.; El Mard, H.; Smet, M.; Dehaen, W.; Xu, H. Selenium containing macrocycles: Transformation between Se–N/Se–S/Se–Se bonds. Sci. China Chem. 2017, 60, 1191–1196. [Google Scholar] [CrossRef]
- Ji, S.; Cao, W.; Yu, Y.; Xu, H. Dynamic diselenide bonds: Exchange reaction induced by visible light without catalysis. Angew. Chem. Int. Ed. 2014, 53, 6781–6785. [Google Scholar] [CrossRef]
- Pan, X.; Driessen, F.; Zhu, X.; Du Prez, F.E. Selenolactone as a Building Block toward Dynamic Diselenide-Containing Polymer Architectures with Controllable Topology. ACS Macro Lett. 2017, 6, 89–92. [Google Scholar] [CrossRef]
- Irigoyen, M.; Fernández, A.; Ruiz, A.; Ruipérez, F.; Matxain, J. Diselenide Bonds as an Alternative to Outperform the Efficiency of Disulfides in Self-Healing Materials. J. Org. Chem. 2019, 84, 4200–4210. [Google Scholar] [CrossRef]
- An, X.; Aguirresarobe, R.H.; Irusta, L.; Ruipérez, F.; Matxain, J.M.; Pan, X.; Aramburu, N.; Mecerreyes, D.; Sardon, H.; Zhu, J. Aromatic diselenide crosslinkers to enhance the reprocessability and self-healing of polyurethane thermosets. Polym. Chem. 2017, 8, 3641–3646. [Google Scholar] [CrossRef]
- Suzuki, N.; Takahashi, A.; Ohishi, T.; Goseki, R.; Otsuka, H. Enhancement of the stimuli-responsiveness and photo-stability of dynamic diselenide bonds and diselenide-containing polymers by neighboring aromatic groups. Polymer 2018, 154, 281–290. [Google Scholar] [CrossRef]
- Matxain, J.M.; Asua, J.M.; Ruipérez, F. Design of new disulfide-based organic compounds for the improvement of self-healing materials. Phys. Chem. Chem. Phys. 2016, 18, 1758–1770. [Google Scholar] [CrossRef]
- Nevejans, S.; Ballard, N.; Miranda, J.I.; Reck, B.; Asua, J.M. The underlying mechanisms for self-healing of poly(disulfide)s. Phys. Chem. Chem. Phys. 2016, 18, 27577–27583. [Google Scholar] [CrossRef] [PubMed]
- Formoso, E.; Asua, J.M.; Matxain, J.M.; Ruipérez, F. The role of non-covalent interactions in the self-healing mechanism of disulfide-based polymers. Phys. Chem. Chem. Phys. 2017, 19, 18461–18470. [Google Scholar] [CrossRef] [PubMed]
- Ruipérez, F.; Galdeano, M.; Gimenez, E.; Matxain, J.M. Sulfenamides as Building Blocks for Efficient Disulfide-Based Self-Healing Materials. A Quantum Chemical Study. ChemistryOpen 2018, 7, 248–255. [Google Scholar] [CrossRef] [PubMed]
- Maier, J.A.; Martinez, C.; Kasavajhala, K.; Wickstrom, L.; Hauser, K.E.; Simmerling, C. ff14SB: Improving the Accuracy of Protein Side Chain and Backbone Parameters from ff99SB. J. Chem. Theory Comput. 2015, 11, 3696–3713. [Google Scholar] [CrossRef] [PubMed]
- Case, D.A.; Babin, V.; Berryman, J.T.; Betz, R.M.; Cai, Q.; Cerutti, D.S.; Cheatham, T.E., III; Darden, T.A.; Duke, R.E.; Gohlke, H.; et al. AMBER 14; University of California: San Francisco, CA, USA, 2014. [Google Scholar]
- Torsello, M.; Pimenta, A.C.; Wolters, L.P.; Moreira, I.S.; Orian, L.; Polimeno, A. General AMBER Force Field Parameters for Diphenyl Diselenides and Diphenyl Ditellurides. J. Phys. Chem. A 2016, 120, 4389–4400. [Google Scholar] [CrossRef] [PubMed]
- Bayly, C.I.; Cieplak, P.; Cornell, W.D.; Kollman, P.A. A well-behaved electrostatic potential based method using charge restraints for deriving atomic charges: The RESP model. J. Phys. Chem. 1993, 97, 10269–10280. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian16 Revision C.01; Gaussian Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Martínez, L.; Andrade, R.; Birgin, E.G.; Martínez, J.M. PACKMOL: A package for building initial configurations for molecular dynamics simulations. J. Comput. Chem. 2009, 30, 2157–2164. [Google Scholar] [CrossRef]
- Ruipérez, F. Application of quantum chemical methods in polymer chemistry. Int. Rev. Phys. Chem. 2019, 38, 343–403. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Chain | Guess | HB | HB | HB | HB | HB | HB | HB | HB | ||
|---|---|---|---|---|---|---|---|---|---|---|---|
| 10 ns | 30 ns | ||||||||||
| Short | Periodic | 746,461 | 176 | 149 | 121 | 0.0884 | 2,215,568 | 178 | 148 | 117 | 0.0776 |
| Random | 730,119 | 172 | 146 | 118 | 0.0696 | 2,233,776 | 184 | 149 | 117 | 0.0627 | |
| Long | Periodic | 707,860 | 169 | 142 | 115 | 0.0931 | 2,184,978 | 179 | 146 | 116 | 0.0970 |
| Random | 595,587 | 147 | 119 | 91 | 0.0393 | 1,917,000 | 157 | 128 | 100 | 0.0188 | |
| Box | HB | HB | HB | HB | |
|---|---|---|---|---|---|
| 40 × 30 × 30 | 1,917,000 | 157 | 128 | 100 | 0.0188 |
| 40 × 40 × 40 | 1,900,841 | 158 | 127 | 95 | 0.0490 |
| 50 × 50 × 50 | 1,902,463 | 160 | 127 | 97 | 0.1058 |
| X | R | Backbone | HB | HB | HB | HB | |
|---|---|---|---|---|---|---|---|
| S | R | PD | 1,902,463 | 160 | 127 | 97 | 0.1058 |
| PA | 2,620,182 | 213 | 175 | 142 | 0.0419 | ||
| PF | 1,940,357 | 161 | 129 | 102 | 0.1224 |
| S-S | Se-Se | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Backbone | R | HB | HB | HB | HB | HB | HB | HB | HB | ||
| PD | R | 1,902,463 | 160 | 127 | 97 | 0.1058 | 2,233,772 | 184 | 149 | 115 | 0.0418 |
| R | 1,078,729 | 100 | 72 | 50 | 0.0471 | 921,219 | 87 | 61 | 37 | 0.0538 | |
| R | — | — | — | — | — | 1,359,569 | 119 | 91 | 64 | 0.0307 | |
| R | 731,072 | 76 | 49 | 26 | 0.0212 | 592,646 | 66 | 40 | 16 | 0.0332 | |
| R | 927,209 | 83 | 62 | 42 | 0.0348 | 790,616 | 74 | 53 | 34 | 0.0443 | |
| S-S | Se-Se | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| Backbone | R | ||||||||
| PD | R | 0.0051 | 11.03 | 4.97·10 | 0.1058 | 0.0122 | 8.97 | 1.62·10 | 0.0418 |
| R | 0.0010 | 12.21 | 6.78·10 | 0.0471 | 0.0040 | 8.03 | 7.95·10 | 0.0538 | |
| R | 0.0310 | 10.46 | 1.32·10 | — | 0.0094 | 5.99 | 2.52·10 | 0.0307 | |
| R | 0.0101 | 10.98 | 5.40·10 | 0.0212 | 0.0041 | 7.89 | 1.01·10 | 0.0332 | |
| R | 0.0045 | 16.39 | 0.58·10 | 0.0348 | 0.0000 | 7.96 | 8.98·10 | 0.0443 | |
| PA | R | 0.2382 | 18.26 | 2.46·10 | 0.0419 | ||||
| PF | R | 0.0682 | 18.49 | 1.66·10 | 0.1224 | ||||
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Irigoyen, M.; Matxain, J.M.; Ruipérez, F. Effect of Molecular Structure in the Chain Mobility of Dichalcogenide-Based Polymers with Self-Healing Capacity. Polymers 2019, 11, 1960. https://doi.org/10.3390/polym11121960
Irigoyen M, Matxain JM, Ruipérez F. Effect of Molecular Structure in the Chain Mobility of Dichalcogenide-Based Polymers with Self-Healing Capacity. Polymers. 2019; 11(12):1960. https://doi.org/10.3390/polym11121960
Chicago/Turabian StyleIrigoyen, Mikel, Jon M. Matxain, and Fernando Ruipérez. 2019. "Effect of Molecular Structure in the Chain Mobility of Dichalcogenide-Based Polymers with Self-Healing Capacity" Polymers 11, no. 12: 1960. https://doi.org/10.3390/polym11121960
APA StyleIrigoyen, M., Matxain, J. M., & Ruipérez, F. (2019). Effect of Molecular Structure in the Chain Mobility of Dichalcogenide-Based Polymers with Self-Healing Capacity. Polymers, 11(12), 1960. https://doi.org/10.3390/polym11121960