Mesoscopic Detection of the Influence of a Third Component on the Self-Assembly Structure of A2B Star Copolymer in Thin Films


Abstract
1. Introduction
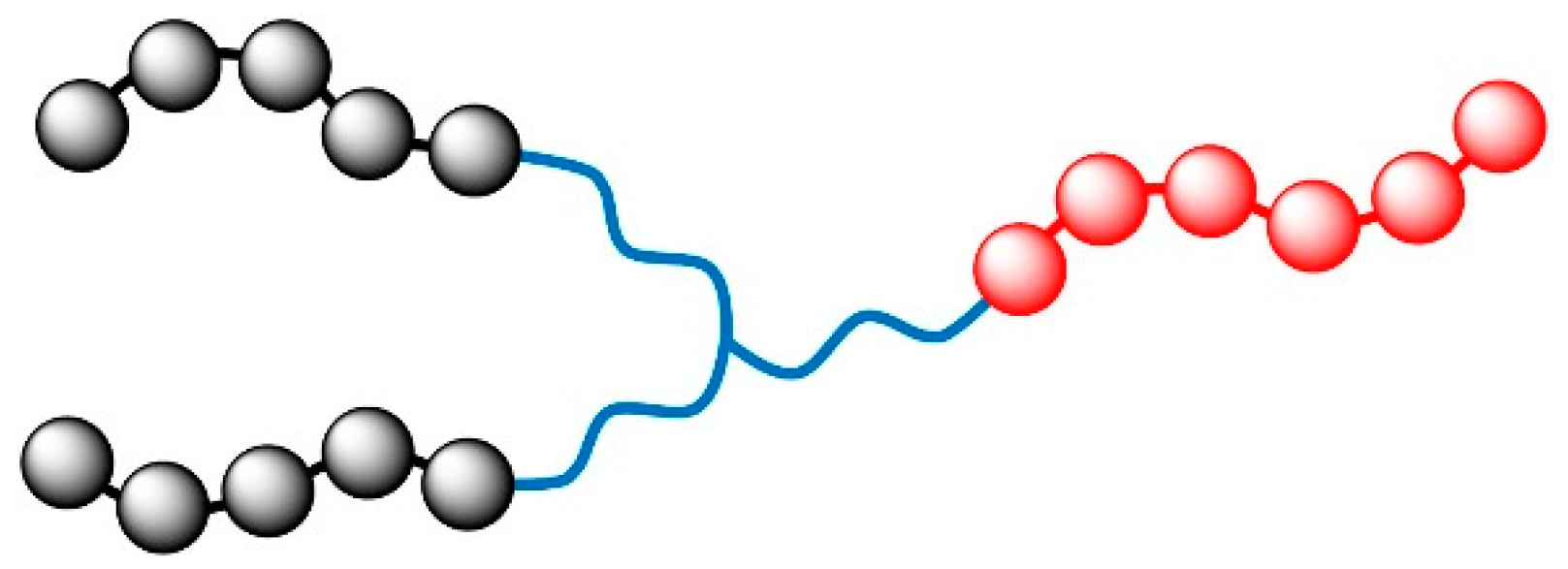
2. Models and Parameter Settings
3. Results and Discussion
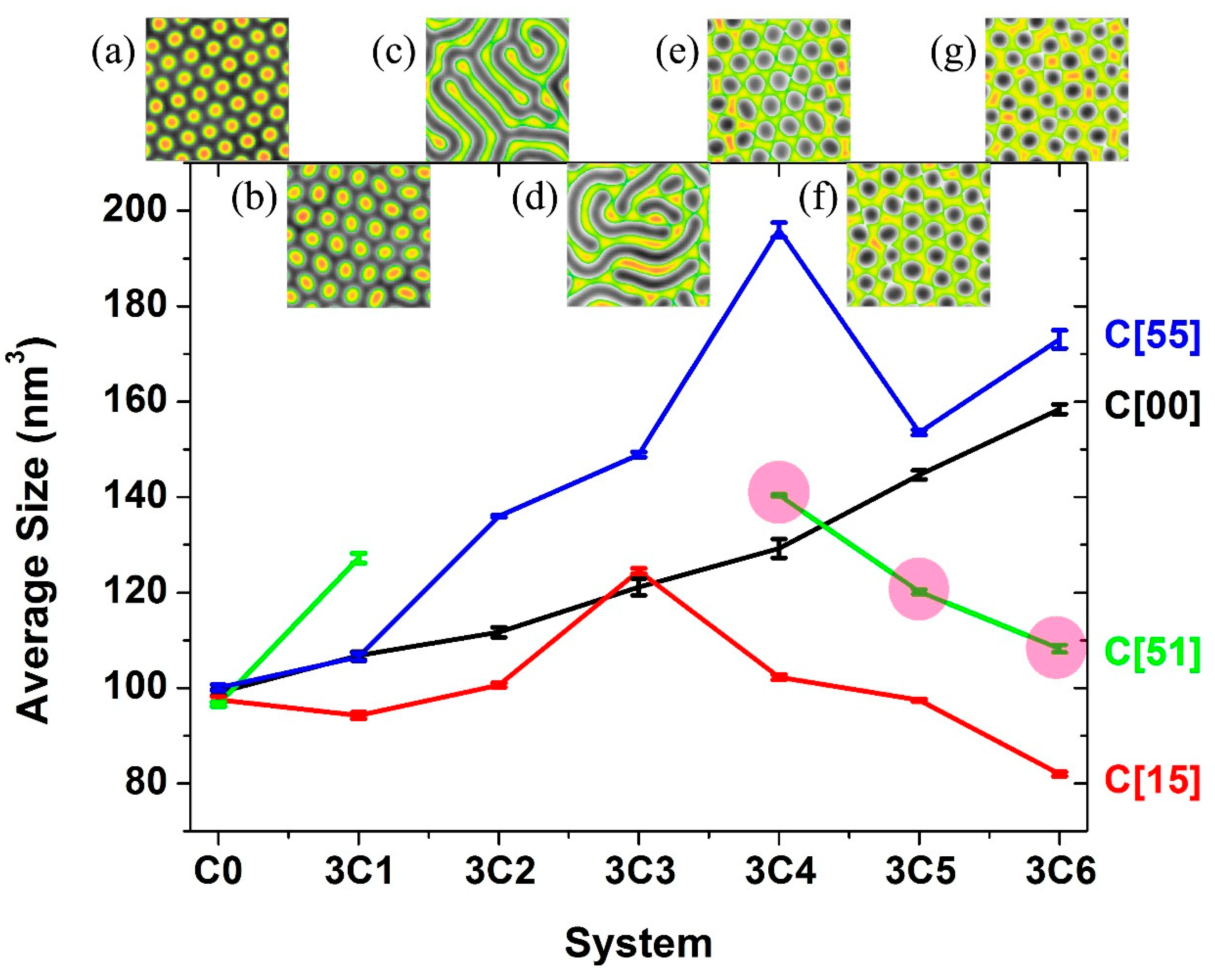
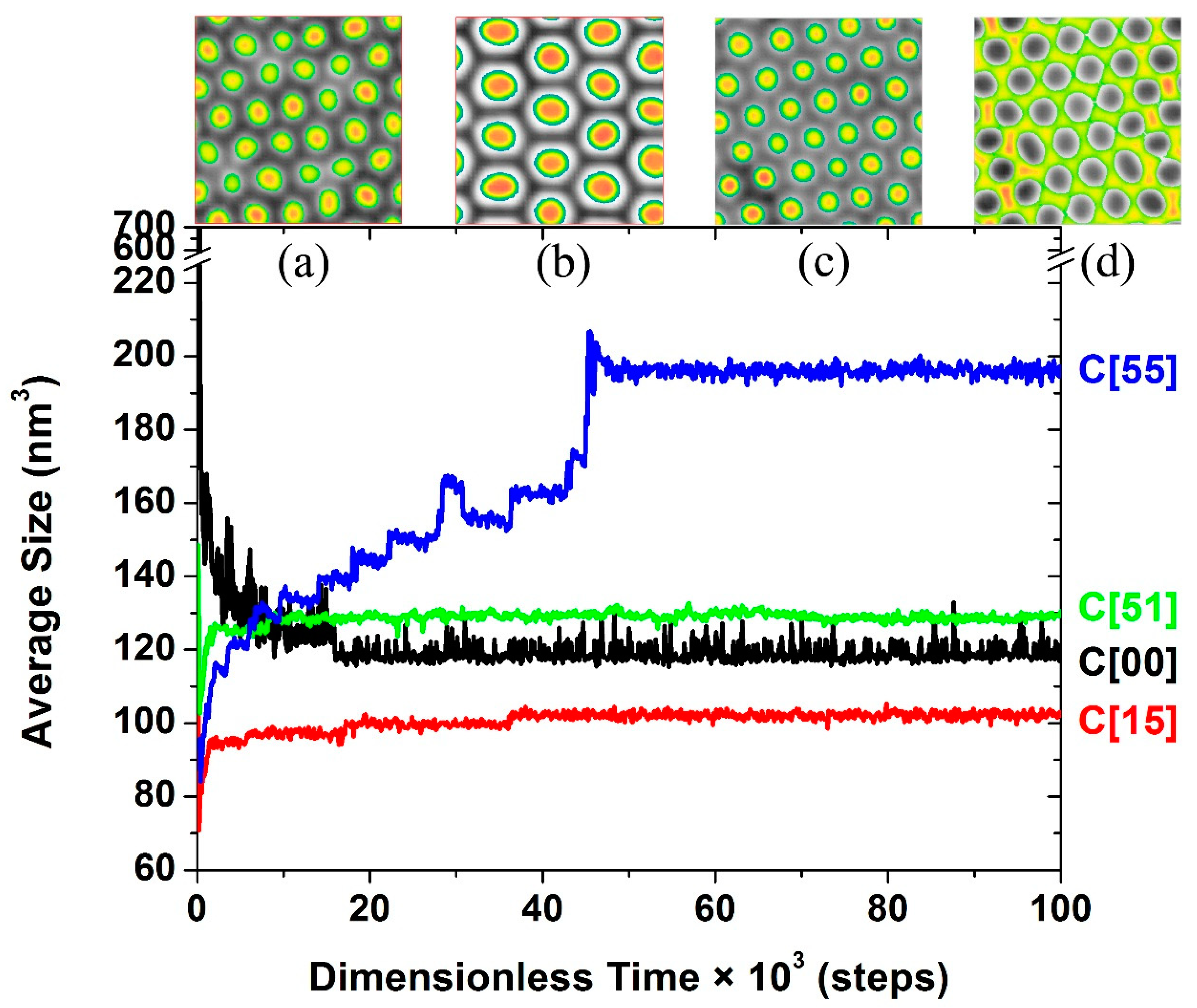
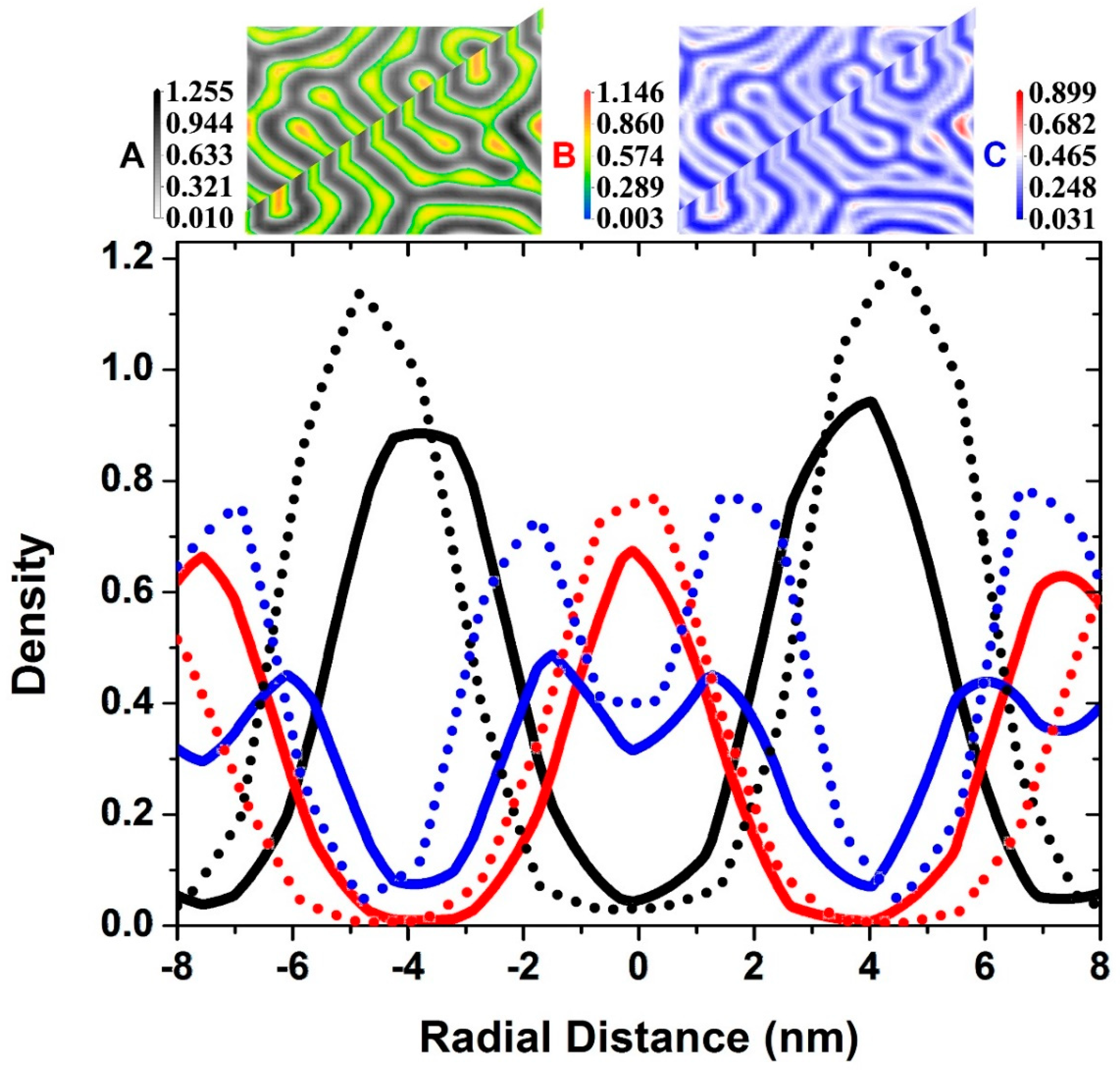
3.1. Self-Assembly under Non-Preferential Interactions
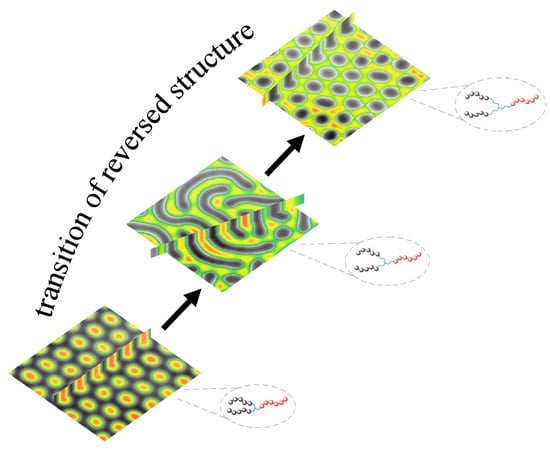
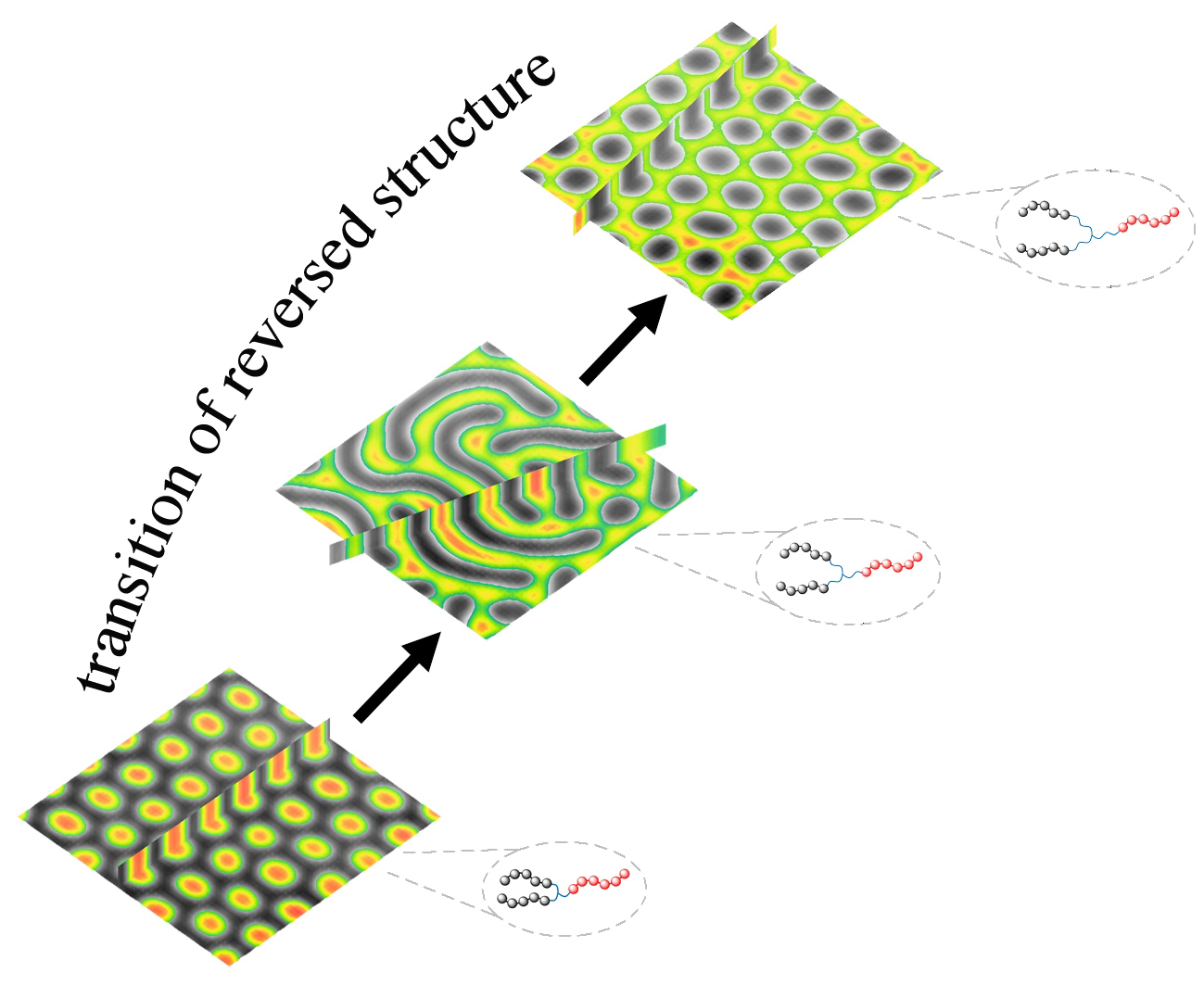
3.2. Two Transitional Systems
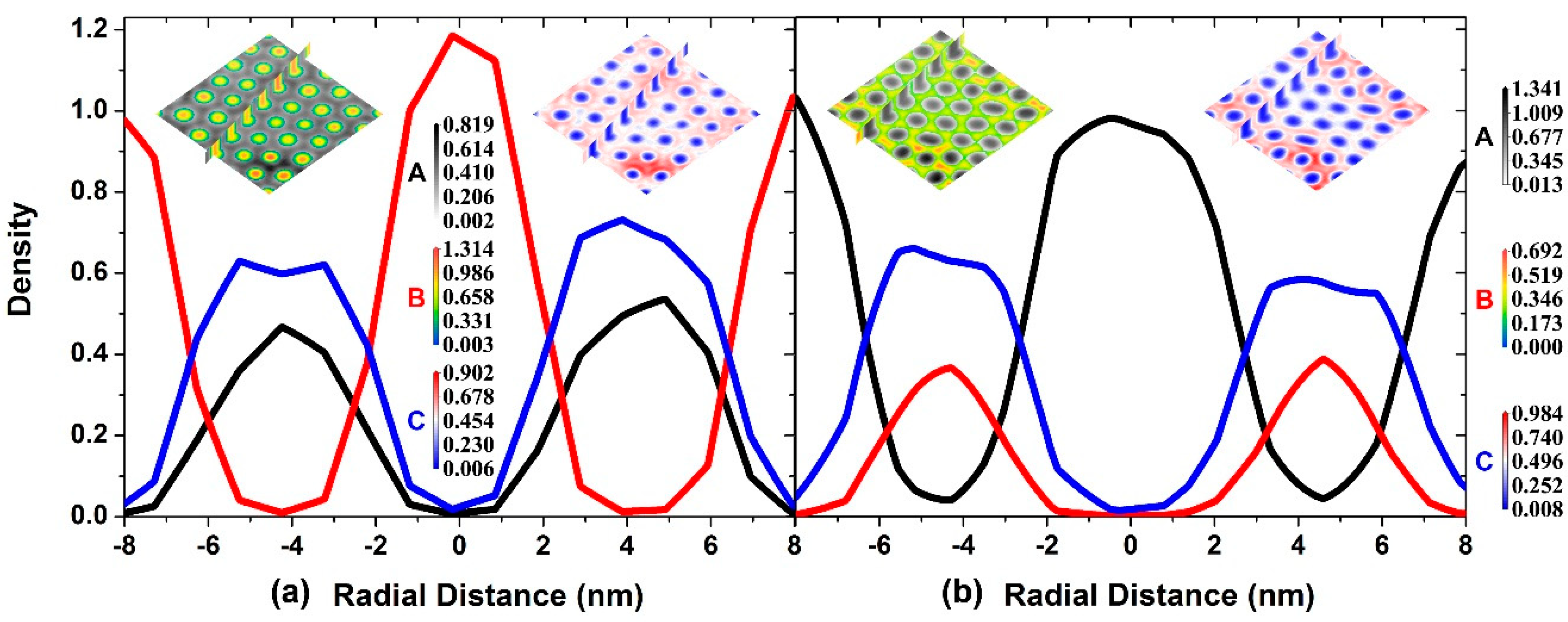
3.3. The Self-Assembly of the Reverse Cylindrical Micellar Structure
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Thurn-Albrecht, T.; Schotter, J.; Kastle, C.A.; Emley, N.; Shibauchi, T.; Krusin-Elbaum, L.; Guarini, K.; Black, C.T.; Tuominen, M.T.; Russell, T.P. Ultrahigh-density nanowire arrays grown in self-assembled diblock copolymer templates. Science 2000, 290, 2126–2129. [Google Scholar] [CrossRef] [PubMed]
- Nunes, S.P. Block copolymer membranes for aqueous solution applications. Macromolecules 2016, 49, 2905–2916. [Google Scholar] [CrossRef]
- Fujikawa, S.; Koizumi, M.; Taino, A.; Okamoto, K. Fabrication and unique optical properties of two-dimensional silver nanorod arrays with nanometer gaps on a silicon substrate from a self-assembled template of diblock copolymer. Langmuir 2016, 32, 12504–12510. [Google Scholar] [CrossRef] [PubMed]
- Higuchi, T.; Sugimori, H.; Jiang, X.; Hong, S.; Matsunaga, K.; Kaneko, T.; Abetz, V.; Takahara, A.; Jinnai, H. Morphological control of helical structures of an ABC-type triblock terpolymer by distribution control of a blending homopolymer in a block copolymer microdomain. Macromolecules 2013, 46, 6991–6997. [Google Scholar] [CrossRef]
- Ludwigs, S.; Boker, A.; Voronov, A.; Rehse, N.; Magerle, R.; Krausch, G. Self-assembly of functional nanostructures from ABC triblock copolymers. Nat. Mater. 2003, 2, 744–747. [Google Scholar] [CrossRef]
- Bates, C.M.; Maher, M.J.; Janes, D.W.; Ellison, C.J.; Willson, C.G. Block copolymer lithography. Macromolecules 2014, 47, 2–12. [Google Scholar] [CrossRef]
- Matsen, M.W.; Schick, M. Stable and unstable phases of a diblock copolymer melt. Phys. Rev. Lett. 1994, 72, 2660–2663. [Google Scholar] [CrossRef]
- Matsen, M.W.; Thompson, R.B. Equilibrium behavior of symmetric ABA triblock copolymer melts. J. Chem. Phys. 1999, 111, 7139–7146. [Google Scholar] [CrossRef]
- Matsen, M.W. Equilibrium behavior of asymmetric ABA triblock copolymer melts. J. Chem. Phys. 2000, 113, 5539–5544. [Google Scholar] [CrossRef]
- Mai, S.-M.; Mingvanish, W.; Turner, S.C.; Chaibundit, C.; Fairclough, J.P.A.; Heatley, F.; Matsen, M.W.; Ryan, A.J.; Booth, C. Microphase-separation behavior of triblock copolymer melts. Comparison with diblock copolymer melts. Macromolecules 2000, 33, 5124–5130. [Google Scholar] [CrossRef]
- Hamersky, M.W.; Smith, S.D.; Gozen, A.O.; Spontak, R.J. Phase behavior of triblock copolymers varying in molecular asymmetry. Phys. Rev. Lett. 2005, 95, 168306. [Google Scholar] [CrossRef] [PubMed]
- Sakurai, S.; Shirouchi, K.; Munakata, S.; Kurimura, H.; Suzuki, S.; Watanabe, J.; Oda, T.; Shimizu, N.; Tanida, K.; Yamamoto, K. Morphology reentry with a change in degree of chain asymmetry in neat asymmetric linear A1BA2 triblock copolymers. Macromolecules 2017, 50, 8647–8657. [Google Scholar] [CrossRef]
- Hadjichristidis, N.; Iatrou, H.; Behal, S.K.; Chludzinski, J.J.; Disko, M.M.; Garner, R.T.; Liang, K.S.; Lohse, D.J.; Milner, S.T. Morphology and miscibility of miktoarm styrene-diene copolymers and terpolymer. Macromolecules 1993, 26, 5812–5815. [Google Scholar] [CrossRef]
- Yang, L.Z.; Hong, S.; Gido, S.P.; Velis, G.; Hadjichristidis, N. I5S miktoarm star block copolymers: Packing constraints on morphology and discontinuous chevron tilt grain boundaries. Macromolecules 2001, 34, 9069–9073. [Google Scholar] [CrossRef]
- Grason, G.M.; Kamien, R.D. Interfaces in diblocks: A study of miktoarm star copolymers. Macromolecules 2004, 37, 7371–7380. [Google Scholar] [CrossRef]
- Xie, N.; Li, W.H.; Qiu, F.; Shi, A.-C. σ phase formed in conformationally asymmetric AB-type block copolymers. ACS Macro Lett. 2014, 3, 906–910. [Google Scholar] [CrossRef]
- Shi, W.C.; Tateishi, Y.C.; Li, W.; Hawker, C.J.; Fredrickson, G.H.; Kramer, E.J. Producing small domain features using miktoarm block copolymers with large interaction parameters. ACS Macro Lett. 2015, 4, 1287–1292. [Google Scholar] [CrossRef]
- Matsen, M.W.; Schick, M. Microphase separation in starblock copolymer melts. Macromolecules 1994, 27, 6761–6767. [Google Scholar] [CrossRef]
- Pang, X.C.; Zhao, L.; Akinc, M.; Kim, J.K.; Lin, Z.Q. Novel amphiphilic multi-arm, star-like block copolymers as unimolecular micelles. Macromolecules 2011, 44, 3746–3752. [Google Scholar] [CrossRef]
- Xu, Y.C.; Li, W.H.; Qiu, F.; Lin, Z.Q. Self-assembly of 21-arm star-like diblock copolymer in bulk and under cylindrical confinement. Nanoscale 2014, 6, 6844–6852. [Google Scholar] [CrossRef] [PubMed]
- Burns, A.B.; Register, R.A. Mechanical properties of star block polymer thermoplastic elastomers with glassy and crystalline end blocks. Macromolecules 2016, 49, 9521–9530. [Google Scholar] [CrossRef]
- Zhang, L.S.; Lin, J.P.; Lin, S.L. Effect of molecular architecture on phase behavior of graft copolymers. J. Phys. Chem. B 2008, 112, 9720–9728. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.Q.; Zhang, L.S.; Lin, J.P. Microphase separation in multigraft copolymer melts studied by random-phase approximation and self-consistent field theory. J. Chem. Phys. 2008, 129, 114905. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.Y.; Li, T.Q.; Mannion, A.M.; Schneiderman, D.K.; Hillmyer, M.A.; Bates, F.S. Tough and sustainable graft block copolymer thermoplastics. ACS Macro Lett. 2016, 5, 407–412. [Google Scholar] [CrossRef]
- Matsen, M.W.; Schick, M. Stable and unstable phases of a linear multiblock copolymer melt. Macromolecules 1994, 27, 7157–7163. [Google Scholar] [CrossRef]
- Spontak, R.J.; Smith, S.D. Perfectly-alternating linear (AB)n multiblock copolymers: Effect of molecular design on morphology and properties. J. Polym. Sci. Part B Polym. Phys. 2001, 39, 947–955. [Google Scholar] [CrossRef]
- Rasmussen, K.Ø.; Kober, E.M.; Lookman, T.; Saxena, A. Morphology and bridging properties of (AB)n multiblock copolymers. J. Polym. Sci. Part B Polym. Phys. 2003, 41, 104–111. [Google Scholar] [CrossRef]
- Nagata, Y.; Masuda, J.; Noro, A.; Cho, D.; Takano, A.; Matsushita, Y. Preparation and characterization of a styrene-isoprene undecablock copolymer and its hierarchical microdomain structure in bulk. Macromolecules 2005, 38, 10220–10225. [Google Scholar] [CrossRef]
- Nap, R.; Sushko, N.; Erukhimovich, I.; ten Brinke, G. Double periodic lamellar-in-lamellar structure in multiblock copolymer melts with competing length scales. Macromolecules 2006, 39, 6765–6770. [Google Scholar] [CrossRef]
- Zhao, B.; Jiang, W.B.; Chen, L.; Li, W.H.; Qiu, F.; Shi, A.-C. Emergence and stability of a hybrid lamella? Sphere structure from linear ABAB tetrablock copolymers. ACS Macro Lett. 2018, 7, 95–99. [Google Scholar] [CrossRef]
- Lynd, N.A.; Oyerokun, F.T.; O’Donoghue, D.L.; Handlin, D.L., Jr.; Fredrickson, G.H. Design of soft and strong thermoplastic elastomers based on nonlinear block copolymer architectures using self-consistent-field theory. Macromolecules 2010, 43, 3479–3486. [Google Scholar] [CrossRef]
- Gao, Y.; Deng, H.L.; Li, W.H.; Qiu, F.; Shi, A.-C. Formation of nonclassical ordered phases of AB-type multi-arm block copolymers. Phys. Rev. Lett. 2016, 116, 068304. [Google Scholar] [CrossRef] [PubMed]
- Pochan, D.J.; Gido, S.P.; Pispas, S.; Mays, J.W.; Ryan, A.J.; Fairclough, J.P.A.; Hamley, I.W.; Terrill, N.J. Morphologies of microphase-separated A2B simple graft copolymers. Macromolecules 1996, 29, 5091–5098. [Google Scholar] [CrossRef]
- Pochan, D.J.; Gido, S.P.; Pispas, S.; Mays, J.W. Morphological transitions in an I2S simple graft block copolymer: from folded sheets to folded lace to randomly oriented worms at equilibrium. Macromolecules 1996, 29, 5099–5105. [Google Scholar] [CrossRef]
- Li, Z.; Kesselman, E.; Talmon, Y.; Hillmyer, M.A.; Lodge, T.P. Multicompartment micelles from ABC miktoarm stars in water. Science 2004, 306, 98–101. [Google Scholar] [CrossRef] [PubMed]
- Saito, N.; Liu, C.; Lodge, T.P.; Hillmyer, M.A. Multicompartment micelles from polyester-containing ABC miktoarm star terpolymers. Macromolecules 2008, 41, 8815–8822. [Google Scholar] [CrossRef]
- Liu, C.; Hillmyer, M.A.; Lodge, T.P. Multicompartment micelles from pH-responsive miktoarm star block terpolymers. Langmuir 2009, 25, 13718–13725. [Google Scholar] [CrossRef]
- Dupont, J.; Liu, G.; Niihara, K.-I.; Kimoto, R.; Jinnai, H. Self assembled ABC triblock copolymer double and triple helices. Angew. Chem. Int. Ed. 2009, 48, 6144–6147. [Google Scholar] [CrossRef] [PubMed]
- Van Horn, R.M.; Zheng, J.X.; Sun, H.-J.; Hsiao, M.-S.; Zhang, W.-B.; Dong, X.-H.; Xu, J.; Thomas, E.L.; Lotz, B.; Cheng, S.Z.D. Solution crystallization behavior of crystalline-crystalline diblock copolymers of poly(ethylene oxide)-block-poly(ε-caprolactone). Macromolecules 2010, 43, 6113–6119. [Google Scholar] [CrossRef]
- Bates, F.S.; Hillmyer, M.A.; Lodge, T.P.; Bates, C.M.; Delaney, K.T.; Fredrickson, G.H. Multiblock polymers: Panacea or Pandora’s Box? Science 2012, 336, 434–440. [Google Scholar] [CrossRef] [PubMed]
- Mu, D.; Li, J.Q.; Feng, S.Y. One-dimensional confinement effect on the self-assembly of symmetric H-shaped copolymers in a thin film. Sci. Rep. 2017, 7, 13610. [Google Scholar] [CrossRef] [PubMed]
- Mu, D.; Li, J.Q.; Feng, S.Y. Mechanistic investigations of confinement effects on the self-assembly of symmetric amphiphilic copolymers in thin films. Phys. Chem. Chem. Phys. 2017, 19, 21938–21945. [Google Scholar] [CrossRef] [PubMed]
- Yu, H.; Qiu, X.; Moreno, N.; Ma, Z.; Calo, V.M.; Nunes, S.P.; Peinemann, K.-V. Self-assembled asymmetric block copolymer membranes: Bridging the gap from ultra- to nanofiltration. Angew. Chem. Int. Ed. 2015, 54, 13937–13941. [Google Scholar] [CrossRef] [PubMed]
- Eslami, H.; Khanjari, N.; Müller-Plathe, F. Self-assembly mechanisms of triblock Janus particles. J. Chem. Theory Comput. 2019, 15, 1345–1354. [Google Scholar] [CrossRef] [PubMed]
- Gadelrab, K.R.; Ding, Y.; Pablo-Pedro, R.; Chen, H.; Gotrik, K.W.; Tempel, D.G.; Ross, C.A.; Alexander-Katz, A. Limits of directed self-assembly in block copolymers. Nano Lett. 2018, 18, 3766–3772. [Google Scholar] [CrossRef] [PubMed]
- Ouaknin, G.; Laachi, N.; Bochkov, D.; Delaney, K.; Fredrickson, G.H.; Gibou, F. Functional level-set derivative for a polymer self consistent field theory Hamiltonian. J. Comput. Phys. 2017, 345, 207–223. [Google Scholar] [CrossRef]
- Xia, Y.; Li, W. Defect-free hexagonal patterns formed by AB diblock copolymers under triangular confinement. Polymer 2019, 166, 21–26. [Google Scholar] [CrossRef]
- Jiang, K.; Zhang, J.; Liang, Q. Self-assembly of asymmetrically interacting ABC star triblock copolymer melts. J. Phys. Chem. B 2015, 119, 14551–14562. [Google Scholar] [CrossRef]
- Gong, H.; Xu, G.; Shi, X. Comparison of aggregation behaviors between branched and linear block polyethers: MesoDyn simulation study. Colloid Polym. Sci. 2010, 288, 1581–1592. [Google Scholar] [CrossRef][Green Version]
- Zhang, Z.-J.; Lu, Z.-Y.; Li, Z.-S. Phase Separation in bimodal molecular weight high density polyethylene with differing branch contents by molecular dynamics and mesoDyn simulation. Chin. J. Polym. Sci. 2009, 27, 493–500. [Google Scholar] [CrossRef]
- Fraaije, J.G.E.M.; van Vlimmeren, B.A.C.; Maurits, N.M.; Postma, M.; Evers, O.A.; Hoffmann, C.; Altevogt, P.; Goldbeck-Wood, G. The Dynamic Mean-Field Density Functional method and its application to the mesoscopic dynamics of quenched block copolymer melts. J. Chem. Phys. 1997, 106, 4260–4269. [Google Scholar] [CrossRef]
- Van Vlimmeren, B.A.C.; Maurits, N.M.; Zvelindovsky, A.V.; Sevink, G.J.A.; Fraaije, J.G.E.M. Simulation of 3D mesoscale structure formation in concentrated aqueous solution of the triblock polymer surfactants (ethylene oxide)13(propylene oxide)30(ethylene oxide)13 and (propylene oxide)19(ethylene oxide)33(propylene oxide)19. Application of dynamic Mean-Field Density Functional theory. Macromolecules 1999, 32, 646–656. [Google Scholar]
- Crank, J.; Nicolson, P. A Practical method for numerical evaluation of solutions of partial differential equations of the heat conduction type. In Mathematical Proceedings of the Cambridge Philosophical Society; Cambridge University Press: Cambridge, UK, 1947; Volume 43, pp. 50–67. [Google Scholar]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Eij (kJ·mol−1) | A | B | C[00] | C[55] | C[15] | C[51] |
|---|---|---|---|---|---|---|
| A | 0 | 4.68 | 0 | 5 | 1 | 5 |
| B | 4.68 | 0 | 0 | 5 | 5 | 1 |
| C[00] | 0 | 0 | 0 | - | - | - |
| C[55] | 5 | 5 | - | 0 | - | - |
| C[15] | 1 | 5 | - | - | 0 | - |
| C[51] | 5 | 1 | - | - | - | 0 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mu, D.; Li, J.-Q.; Cong, X.-S.; Zhang, H. Mesoscopic Detection of the Influence of a Third Component on the Self-Assembly Structure of A2B Star Copolymer in Thin Films. Polymers 2019, 11, 1636. https://doi.org/10.3390/polym11101636
Mu D, Li J-Q, Cong X-S, Zhang H. Mesoscopic Detection of the Influence of a Third Component on the Self-Assembly Structure of A2B Star Copolymer in Thin Films. Polymers. 2019; 11(10):1636. https://doi.org/10.3390/polym11101636
Chicago/Turabian StyleMu, Dan, Jian-Quan Li, Xing-Shun Cong, and Han Zhang. 2019. "Mesoscopic Detection of the Influence of a Third Component on the Self-Assembly Structure of A2B Star Copolymer in Thin Films" Polymers 11, no. 10: 1636. https://doi.org/10.3390/polym11101636
APA StyleMu, D., Li, J.-Q., Cong, X.-S., & Zhang, H. (2019). Mesoscopic Detection of the Influence of a Third Component on the Self-Assembly Structure of A2B Star Copolymer in Thin Films. Polymers, 11(10), 1636. https://doi.org/10.3390/polym11101636