Improved Flame-Retardant and Ceramifiable Properties of EVA Composites by Combination of Ammonium Polyphosphate and Aluminum Hydroxide


Abstract
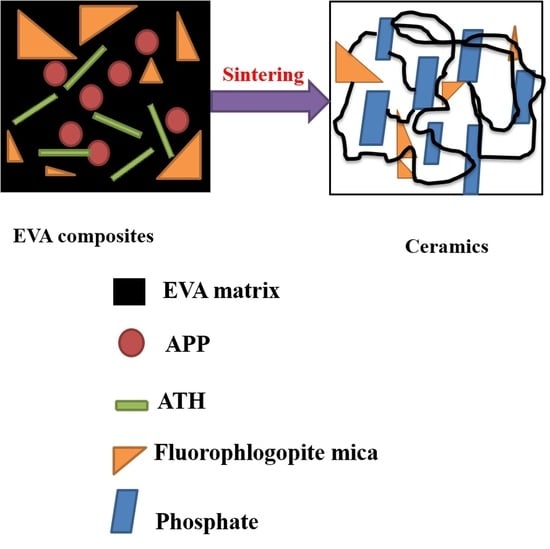
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Sample Preparation
2.3. Sintering Test
2.4. Characterization
3. Results and Discussion
3.1. Flexural Strength and Linear Shrinkage of the Sintered Specimens
3.2. Mechanical Properties and Flammability of EVA Composites
3.3. Combustion Performance of Ceramifiable Flame-Retardant EVA Composites
3.4. Thermal Analysis
3.5. Surface Morphology of the Sintered Specimen
3.6. Linear Shrinkage
3.7. Apparent Porosity and Mechanical Strength of Sintered Sample
3.8. XRD Analysis
3.9. FTIR Analysis
3.10. SEM
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Yuan, B.H.; Bao, C.L.; Qian, X.D.; Jiang, S.H.; Wen, P.Y.; Xing, W.Y. Synergetic dispersion effect of graphene nanohybrid on the thermal stability and mechanical properties of ethylene vinyl acetate copolymer nanocomposite. Ind. Eng. Chem. Res. 2014, 53, 1143–1149. [Google Scholar] [CrossRef]
- Aghjeh, M.R.; Nazari, M.; Khonakdar, H.A.; Jafari, S.H.; Wagenknecht, U.; Heinrich, G. In depth analysis of micro-mechanism of mechanical property alternations in PLA/EVA/clay nanocomposites: A combined theoretical and experimental approach. Mater. Des. 2015, 88, 1277–1289. [Google Scholar] [CrossRef]
- Chen, Y.; Zou, H.W.; Liang, M.; Cao, Y. Melting and crystallization behavior of partially miscible high density polyethylene/ethylene vinyl acetate copolymer (HDPE/EVA) blends. Thermochim. Acta 2014, 586, 1–8. [Google Scholar] [CrossRef]
- Bakar, N.A.; Chee, C.Y.; Abdullah, L.C.; Ratnam, C.T.; Ibrahim, N.A. Thermal and dynamic mechanical properties of grafted kenaf filled poly (vinyl chloride)/ethylene vinyl acetate composites. Mater. Des. 2015, 65, 204–211. [Google Scholar] [CrossRef]
- Castrovinci, A.; Camino, G.; Drevelle, C.; Duquesne, S.; Magniez, C.; Vouters, M. Ammonium polyphosphate–aluminum trihydroxide antagonism in fire retarded butadiene–styrene block copolymer. Eur. Polym. J. 2005, 41, 2023–2033. [Google Scholar] [CrossRef]
- Hu, S.; Chen, F.; Li, J.G.; Shen, Q.; Huang, Z.X.; Zhang, L.M. The ceramifying process and mechanical properties of silicone rubber/ ammonium polyphosphate/ aluminium hydroxide/ mica composites. Polym. Degrad. Stab. 2016, 126, 196–203. [Google Scholar] [CrossRef]
- Guo, J.H.; Chen, X.M.; Zhang, Y. Improving the mechanical and electrical properties of ceramizable silicone rubber/halloysite composites and their ceramic residues by incorporation of different borates. Polymers 2006, 10, 388–399. [Google Scholar] [CrossRef]
- Zhang, X.P.; Guan, Y.Y.; Xie, Y.; Qiu, D. “House-of-cards” structures in silicone rubber composites for superb anti-collapsing performance at medium high temperature. RSC Adv. 2016, 6, 7970–7976. [Google Scholar] [CrossRef]
- Hanu, L.G.; Simon, G.P.; Mansouri, J.; Burford, R.P.; Cheng, Y.B. Development of polymer-ceramic composites for improved fire resistance. J. Mater. Process. Technol. 2004, 153, 401–407. [Google Scholar] [CrossRef]
- Al-Hassany, Z.; Genovese, A.; Shanks, R.A. Fire-retardant and fire-barrier poly(vinyl acetate) composites for sealant application. Express. Polym. Lett. 2010, 4, 79–93. [Google Scholar] [CrossRef]
- Di, H.W.; Deng, C.; Li, R.M.; Dong, L.P.; Wang, Y.Z. A novel EVA composite with simultaneous flame retardation and ceramifiable capacity. RSC Adv. 2015, 5, 51248–51257. [Google Scholar] [CrossRef]
- Lou, F.P.; Cheng, L.H.; Li, Q.Y.; Wei, T.; Guan, X.Y.; Guo, W.H. The combination of glass dust and glass fiber as fluxing agents for ceramifiable silicone rubber composites. RSC Adv. 2017, 7, 38805–38811. [Google Scholar] [CrossRef]
- Hanu, L.G.; Simon, G.P.; Cheng, Y.B. Preferential orientation of muscovite in ceramifiable silicone composites. Mater. Sci. Eng. A 2005, 398, 180–187. [Google Scholar] [CrossRef]
- Mansouri, J.; Burford, R.P.; Cheng, Y.B. Pyrolysis behaviour of silicone-based ceramifying composites. Mater. Sci. Eng. A 2006, 425, 7–14. [Google Scholar] [CrossRef]
- Mansouri, J.; Wood, C.A.; Roberts, K.; Cheng, Y.B.; Burford, R.P. Investigation of the ceramifying process of modified silicone–silicate compositions. J. Mater. Sci. 2007, 42, 6046–6055. [Google Scholar] [CrossRef]
- Wang, J.H.; Ji, C.T.; Yan, Y.T.; Zhao, D.; Shi, L.Y. Mechanical and ceramifiable properties of silicone rubber filled with different inorganic fillers. Polym. Degrad. Stab. 2015, 121, 149–156. [Google Scholar] [CrossRef]
- Li, Y.M.; Deng, C.; Wang, Y.Z. A novel high-temperature-resistant polymeric material for cables and insulated wires via the ceramization of mica-based ceramifiable EVA composites. Compos. Sci. Technol. 2016, 132, 116–122. [Google Scholar] [CrossRef]
- Gong, X.H.; Shen, Y.C.; Wang, T.W. Improved ceramifiable properties of EVA composites with whitened and capsulized red phosphorus (WCRP). RSC Adv. 2016, 6, 96984–96989. [Google Scholar]
- Zhao, D.; Shen, Y.C.; Wang, T.W. Ceramifiable EVA/APP/SGF composites for improved ceramifiable properties. Polym. Degrad. Stab. 2018, 150, 140–147. [Google Scholar] [CrossRef]
- Zhao, H.B.; Liu, B.W.; Wang, X.L.; Chen, L.; Wang, X.L.; Wang, Y.Z. A flame-retardant-free and thermo-cross-linkable copolyester: Flame-retardant and anti-dripping mode of action. Polymer 2014, 55, 2394–2403. [Google Scholar] [CrossRef]
- Wang, W.; Peng, Y.; Zammarano, M.; Zhang, W.; Li, J.Z. Effect of ammonium polyphosphate to aluminum hydroxide mass ratio on the properties of wood-flour/polypropylene composites. Polymers 2017, 9, 615. [Google Scholar] [CrossRef]
- Shao, Z.B.; Deng, C.; Tan, Y.; Yu, L.; Chen, M.J.; Chen, L. Ammonium polyphosphate chemically-modified with ethanolamine as an efficient intumescent flame retardant for polypropylene. J. Mater. Chem. A 2014, 2, 13955–13965. [Google Scholar] [CrossRef]
- Qin, Z.L.; Li, D.H.; Li, Q.; Yang, R.J. Effect of nano-aluminum hydroxide on mechanical properties, flame retardancy and combustion behavior of intumescent flame retarded polypropylene. Mater. Des. 2016, 89, 988–995. [Google Scholar] [CrossRef]
- Zhou, H.M.; Qiao, X.C.; Yu, J.G. Influences of quartz and muscovite on the formation of mullite from kaolinite. Appl. Clay Sci. 2013, 80, 176–181. [Google Scholar] [CrossRef]
- Wang, B.B.; Qian, X.D.; Shi, Y.Q.; Yu, B.; Hong, N.N.; Song, L. Cyclodextrin microencapsulated ammonium polyphosphate: Preparation and its performance on the thermal, flame retardancy and mechanical properties of ethylene vinyl acetate copolymer. Compos. B Eng. 2015, 69, 22–30. [Google Scholar] [CrossRef]
- Réti, C.; Casetta, M.; Duquesne, S.; Bourbigot, S.; Delobel, R. Flammability properties of intumescent PLA including starch and lignin. Polym. Adv. Technol. 2008, 19, 628–635. [Google Scholar] [CrossRef]
- Gao, M.; Wu, W.; Yan, Y. Thermal degradation and flame retardancy of epoxy resins containing intumescent flame retardant. J. Therm. Anal. Calorim. 2009, 95, 605–608. [Google Scholar] [CrossRef]
- Levchik, G.F.; Levchik, S.V.; Lesnikovich, A.I. Thermal behaviour of ammonium polyphosphate-inorganic compound mixtures. Part 1. Talc. Thermochim. Acta 1994, 239, 41–49. [Google Scholar] [CrossRef]
- Aramendía, M.A.; Borau, V.; Jiménez, C.; Marinas, M.J.; Romero, F.J.; Ruiz, J.R. Characterization by XRD, DRIFT, and MAS NMR spectroscopies of a Mg2P2O7Catalyst. J. Colloid Interface Sci. 1998, 202, 456–461. [Google Scholar] [CrossRef]
- Gao, X.; Zhang, A.; Li, S.; Sun, B.; Zhang, L. The resistance to high temperature of magnesia phosphate cement paste containing wollastonite. Mater Struct. 2015, 49, 3423–3434. [Google Scholar] [CrossRef]
- Zhang, Z.; Tang, W. Tunable blue-red emission and energy-transfer properties of Mg3(PO4)2:-Eu2+,Mn2+ phosphors. J. Inorg. Chem. 2015, 2015, 3940–3948. [Google Scholar] [CrossRef]
- Gong, X.H.; Wu, T.Y.; Ma, J.; Zhao, D.; Shen, Y.C.; Wang, T.W. Improved self-supporting property of ceramifying silicone rubber composites by forming crystalline phase at high temperatures. J. Alloys Compd. 2017, 706, 322–329. [Google Scholar] [CrossRef]
- Guo, J.; Gao, W.; Wang, Y.; Li, H.J.; Zhang, X. Effect of glass frit with low softening temperature on the properties, microstructure and formation mechanism of polysiloxane elastomer-based ceramizable composites. Polym. Degrad. Stab. 2017, 136, 71–79. [Google Scholar] [CrossRef]
- Deng, C.L.; Du, S.L.; Zhao, J.; Shen, Z.Q.; Deng, C.; Wang, Y.Z. An intumescent flame retardant polypropylene system with simultaneously improved flame retardancy and water resistance. Polym. Degrad. Stab. 2014, 108, 97–107. [Google Scholar] [CrossRef]
- Yan, Y.W.; Chen, L.; Jian, R.K.; Kong, S.; Wang, Y.Z. Intumescence: An effect way to flame retardance and smoke suppression for polystryene. Polym. Degrad. Stab. 2012, 97, 1423–1431. [Google Scholar] [CrossRef]


















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Oxides | SiO2 | MgO | K2O | Al2O3 | F | CaO | Others |
|---|---|---|---|---|---|---|---|
| Contents/% | 31.5 | 30.2 | 13.2 | 11.3 | 13.3 | 0.28 | 0.22 |
| Compositions/phr | EVA/phr | Mica/phr | APP/phr | ATH/phr |
|---|---|---|---|---|
| EVA0 | 100 | 0 | 0 | 0 |
| EVA1 | 100 | 100 | 100 | 0 |
| EVA2 | 100 | 100 | - | 100 |
| EVA3 | 100 | 100 | 25 | 75 |
| EVA4 | 100 | 100 | 33 | 67 |
| EVA5 | 100 | 100 | 50 | 50 |
| EVA6 | 100 | 100 | 67 | 33 |
| EVA7 | 100 | 100 | 75 | 25 |
| Samples | LOI/% | UL-94 Rating | Tensile Strength/MPa |
|---|---|---|---|
| EVA0 | 20.5 | NC | 13.6 ± 0.6 |
| EVA1 | 28.3 | NC | 6.1 ± 0.7 |
| EVA2 | 27.5 | NC | 5.0 ± 0.8 |
| EVA3 | 28.1 | V-2 | 5.2 ± 0.8 |
| EVA4 | 28.1 | V-1 | 5.4 ± 0.8 |
| EVA5 | 29.7 | V-0 | 6.9 ± 0.9 |
| EVA6 | 29.1 | V-1 | 7.0 ± 0.5 |
| EVA7 | 28.5 | V-2 | 6.9 ± 0.5 |
| Samples | PHRR1 (W g−1) | PHRR2 (W g−1) | THR (kJ g−1) |
|---|---|---|---|
| EVA0 | 51.9 ± 1.7 | 801.8 ± 2.1 | 38.2 ± 0.5 |
| EVA1 | 27.5 ± 2.5 | 270.1 ± 2.3 | 13.1 ± 0.3 |
| EVA2 | 15.6 ± 3 | 281.4 ± 3.2 | 13.6 ± 0.5 |
| EVA5 | 20.1 ± 2 | 258.1 ± 3.4 | 11.5 ± 0.5 |
| Samples | PHRR (kW m−2) | THR (MJ m−2) | TTI (s) | Residue (wt %) | Peak SPR (1 ×10−2 m2 s−1) |
|---|---|---|---|---|---|
| EVA0 | 843.8 | 115.9 | 71 | 8.9 | 7.5 |
| EVA1 | 138.7 | 33.4 | 360 | 67.1 | 2.1 |
| EVA2 | 123.1 | 65.5 | 100 | 59.5 | 0.6 |
| EVA5 | 157.7 | 56.6 | 143 | 67.1 | 3.3 |
| Samples | T5% /°C | Tmax1/°C | Tmax2/°C | Residue at 800 °C/% |
|---|---|---|---|---|
| Nitrogen | ||||
| EVA0 | 335 | 350 | 468 | 0.3 |
| EVA1 | 330 | 345 | 470 | 55.9 |
| EVA2 | 294 | 325 | 460 | 54.1 |
| EVA3 | 298 | 312 | 460 | 56.1 |
| EVA4 | 316 | 315 | 460 | 56.7 |
| EVA5 | 316 | 325 | 460 | 58.1 |
| EVA6 | 318 | 330 | 460 | 55.9 |
| EVA7 | 327 | 335 | 460 | 56.6 |
| Calculate | 320 | 345 | 468 | 48.2 |
| Air | ||||
| EVA0 | 317 | 350 | 460 | 0.1 |
| EVA1 | 336 | 345 | 475 | 55.2 |
| EVA2 | 293 | 325 | 456 | 52.9 |
| EVA3 | 302 | 340 | 470 | 55.6 |
| EVA4 | 304 | 335 | 471 | 54.9 |
| EVA5 | 306 | 345 | 464 | 56.0 |
| EVA6 | 321 | 345 | 471 | 55.6 |
| EVA7 | 329 | 350 | 472 | 55.6 |
| Calculate | 295 | 344 | 460 | 49.2 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lou, F.; Wu, K.; Wang, Q.; Qian, Z.; Li, S.; Guo, W. Improved Flame-Retardant and Ceramifiable Properties of EVA Composites by Combination of Ammonium Polyphosphate and Aluminum Hydroxide. Polymers 2019, 11, 125. https://doi.org/10.3390/polym11010125
Lou F, Wu K, Wang Q, Qian Z, Li S, Guo W. Improved Flame-Retardant and Ceramifiable Properties of EVA Composites by Combination of Ammonium Polyphosphate and Aluminum Hydroxide. Polymers. 2019; 11(1):125. https://doi.org/10.3390/polym11010125
Chicago/Turabian StyleLou, Feipeng, Kai Wu, Quan Wang, Zhongyu Qian, Shijuan Li, and Weihong Guo. 2019. "Improved Flame-Retardant and Ceramifiable Properties of EVA Composites by Combination of Ammonium Polyphosphate and Aluminum Hydroxide" Polymers 11, no. 1: 125. https://doi.org/10.3390/polym11010125
APA StyleLou, F., Wu, K., Wang, Q., Qian, Z., Li, S., & Guo, W. (2019). Improved Flame-Retardant and Ceramifiable Properties of EVA Composites by Combination of Ammonium Polyphosphate and Aluminum Hydroxide. Polymers, 11(1), 125. https://doi.org/10.3390/polym11010125