Revealing the Interface Structure and Bonding Mechanism of Coupling Agent Treated WPC



Abstract
:
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Formulation of Composites
2.3. ATR-FTIR Analysis
2.4. Solid State 13C NMR Analysis
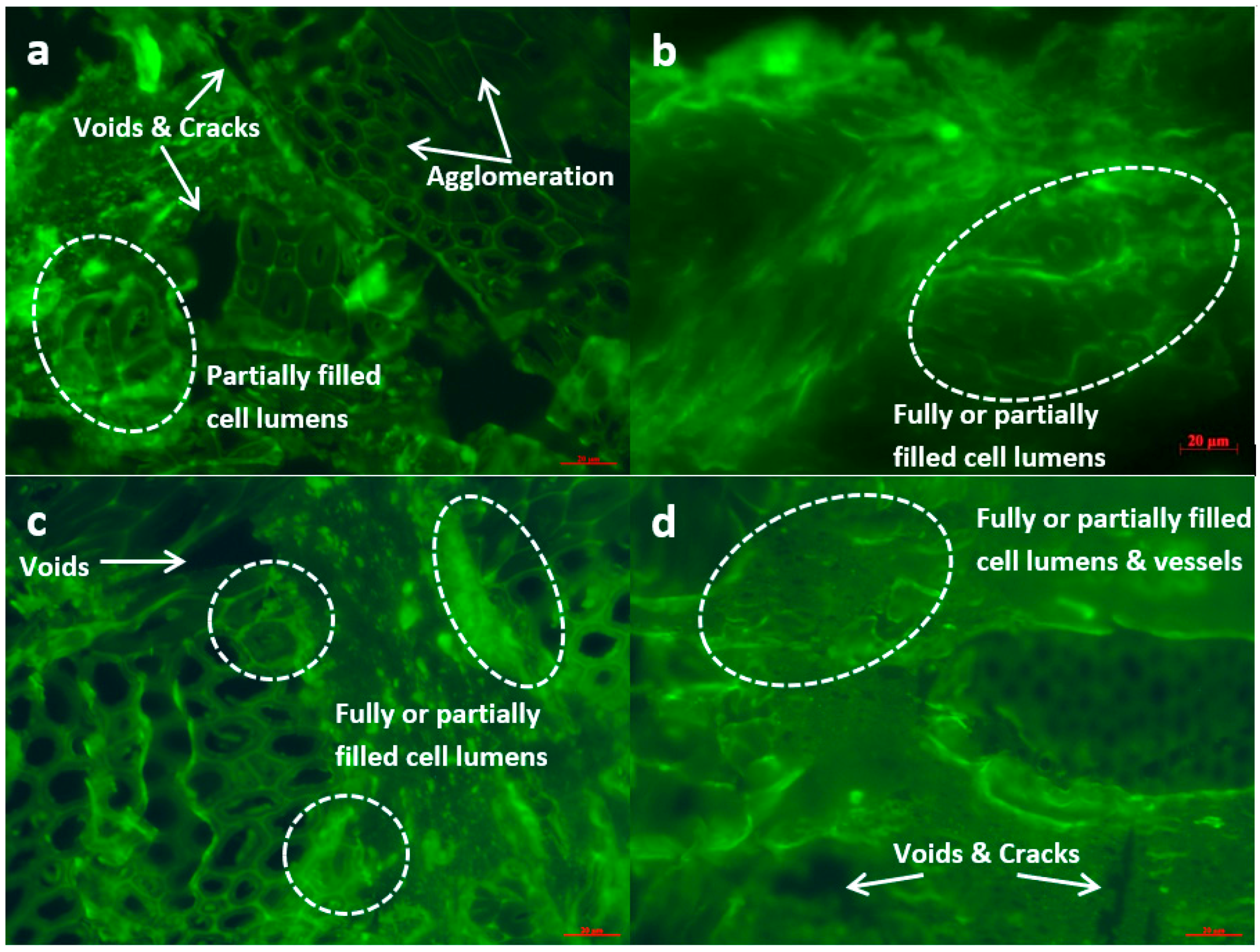
2.5. SEM and FM Analyses
3. Results and Discussion
3.1. Chemical Structure and Bonding
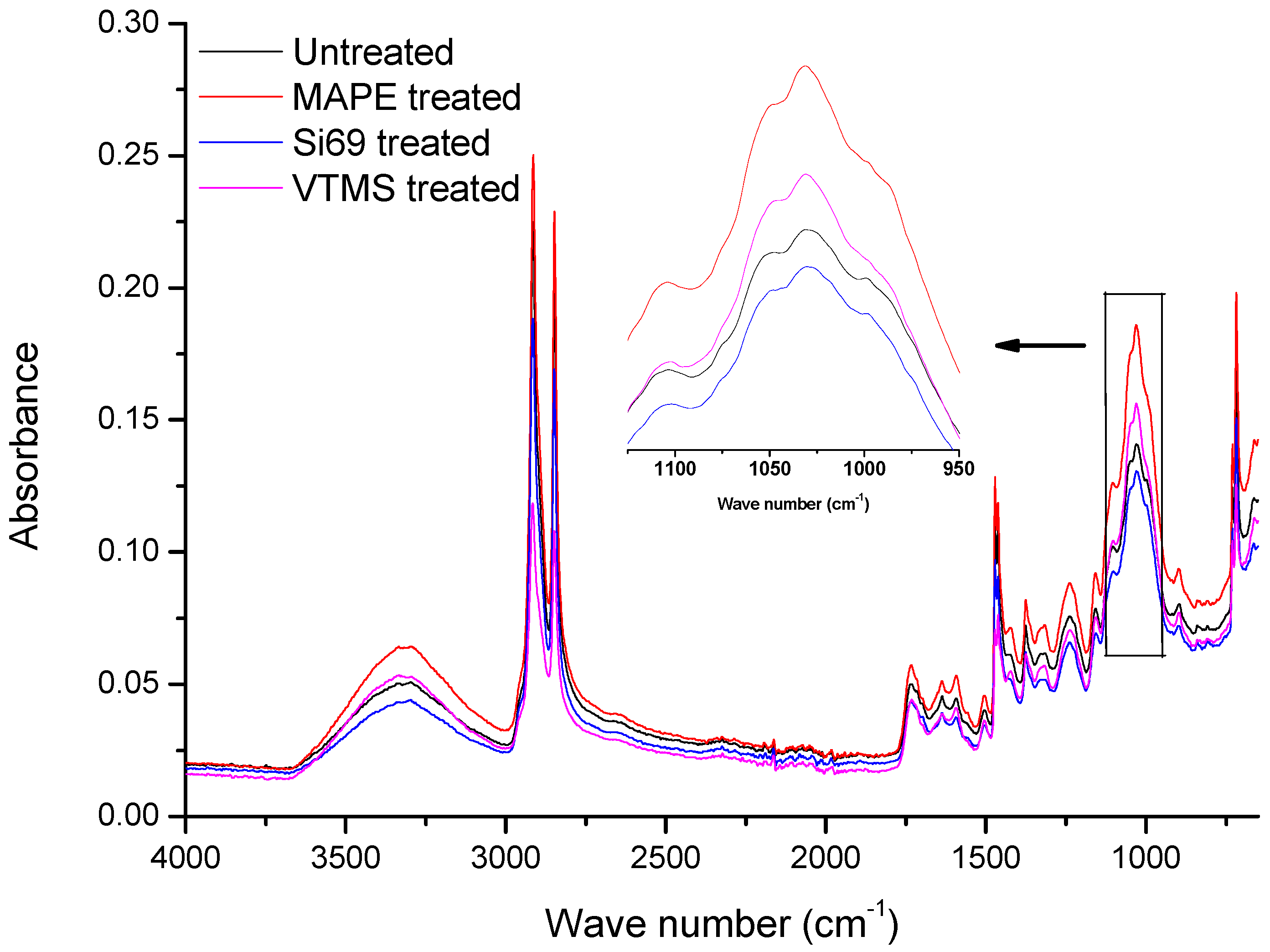
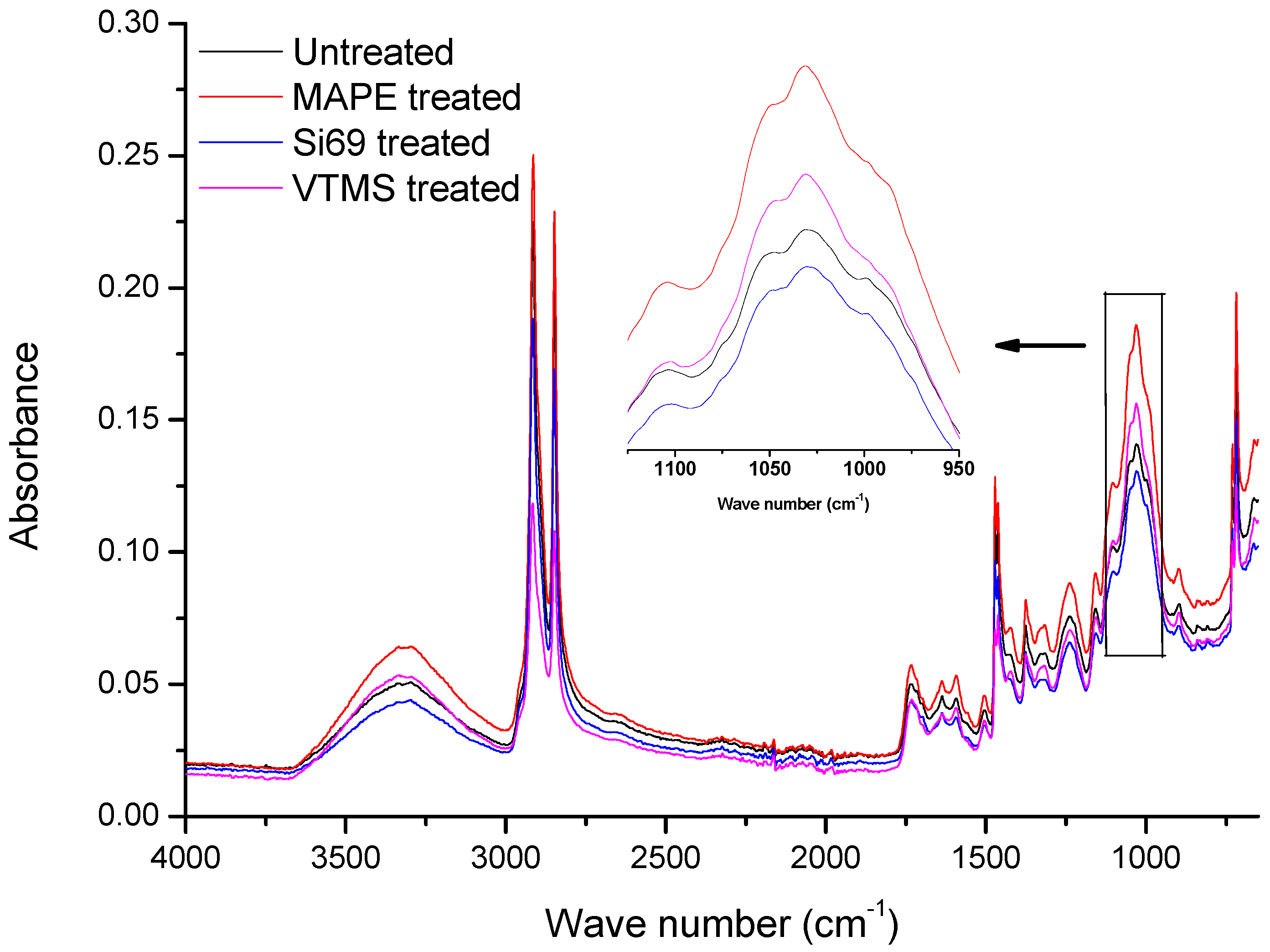
3.1.1. FTIR Analysis
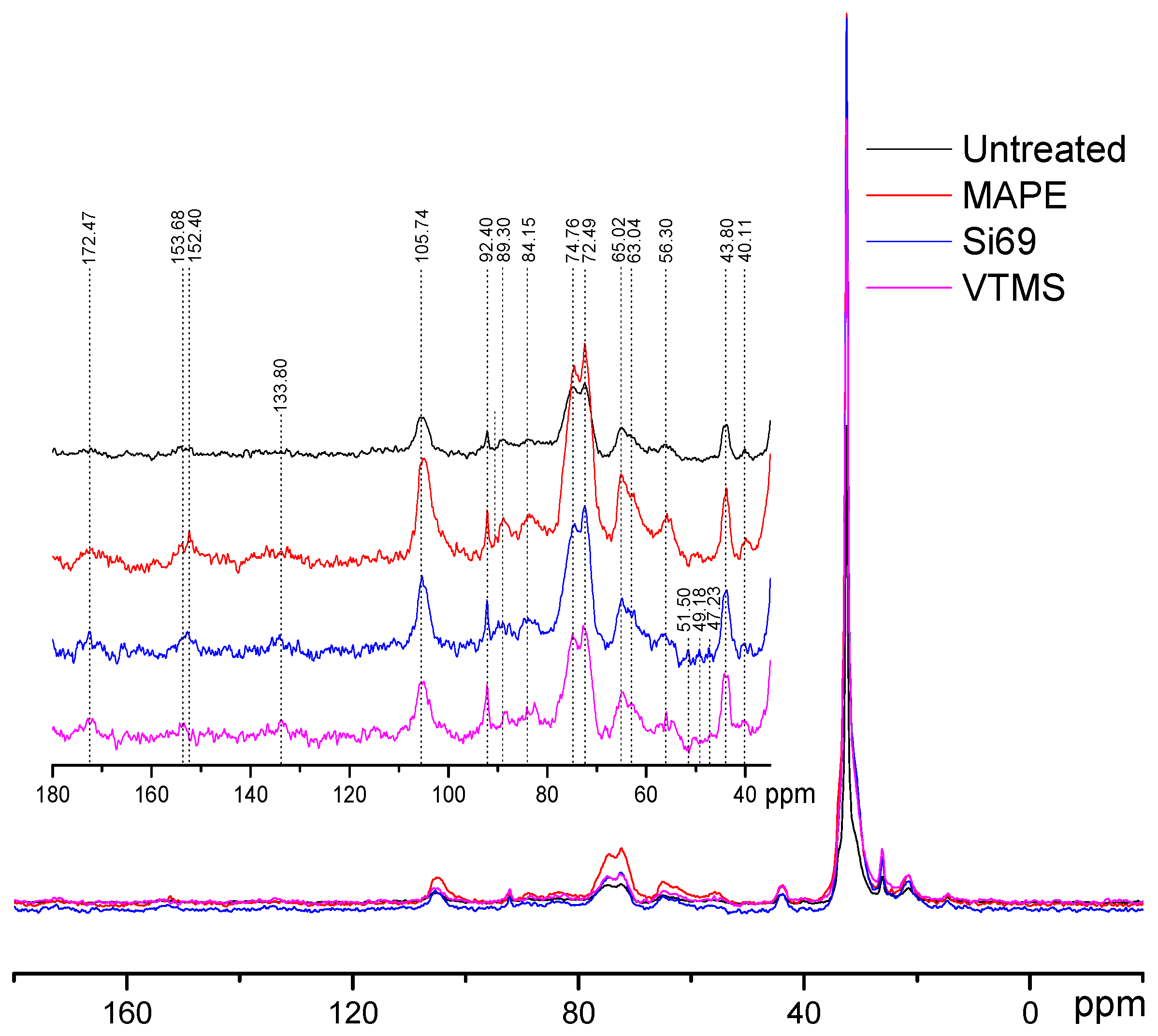
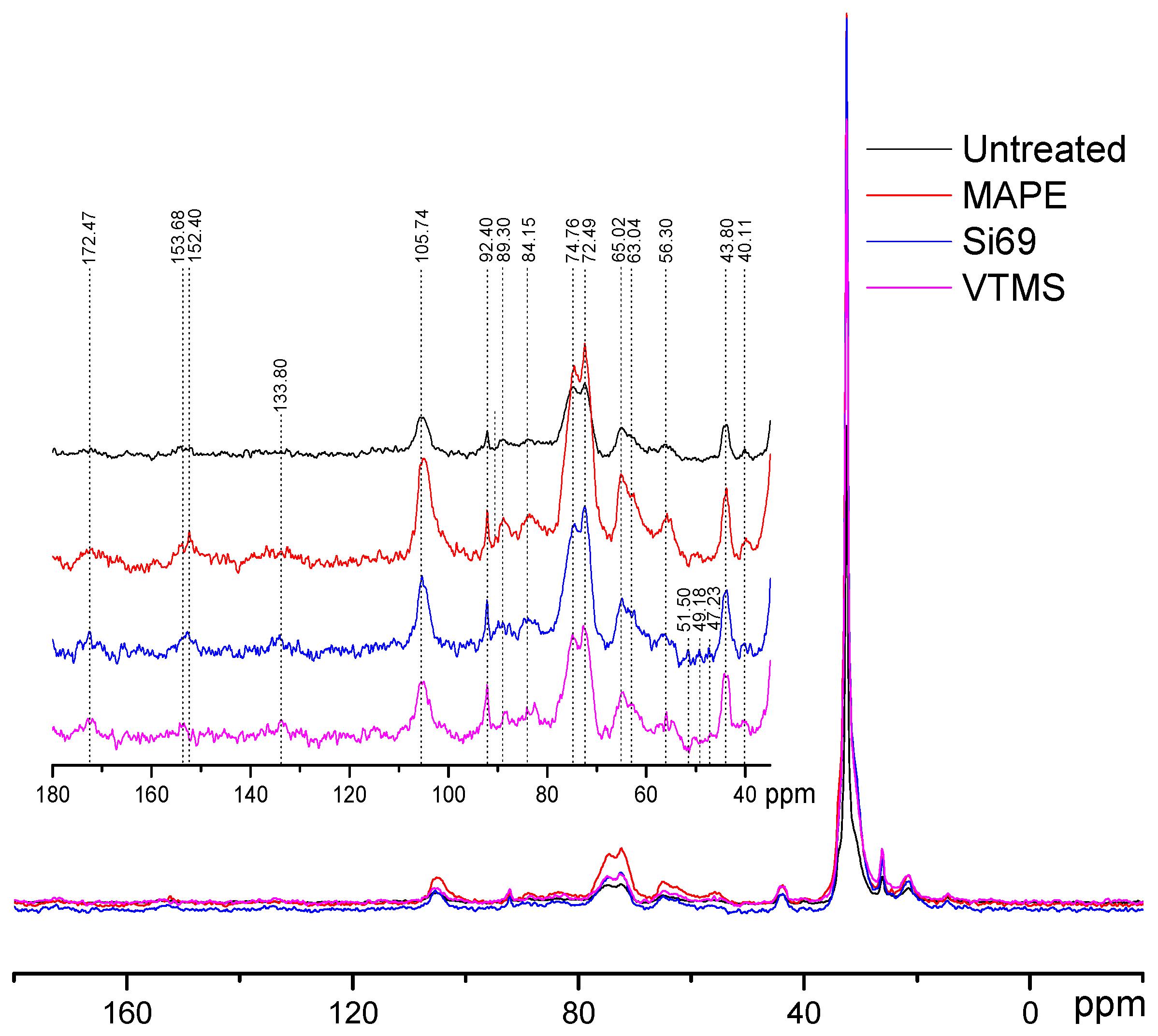
3.1.2. NMR Analysis
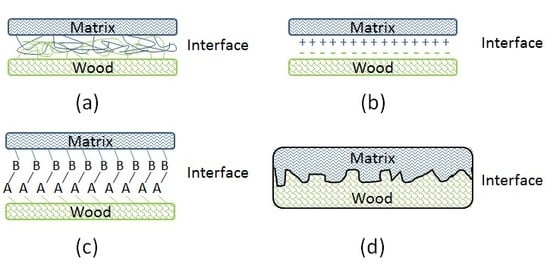
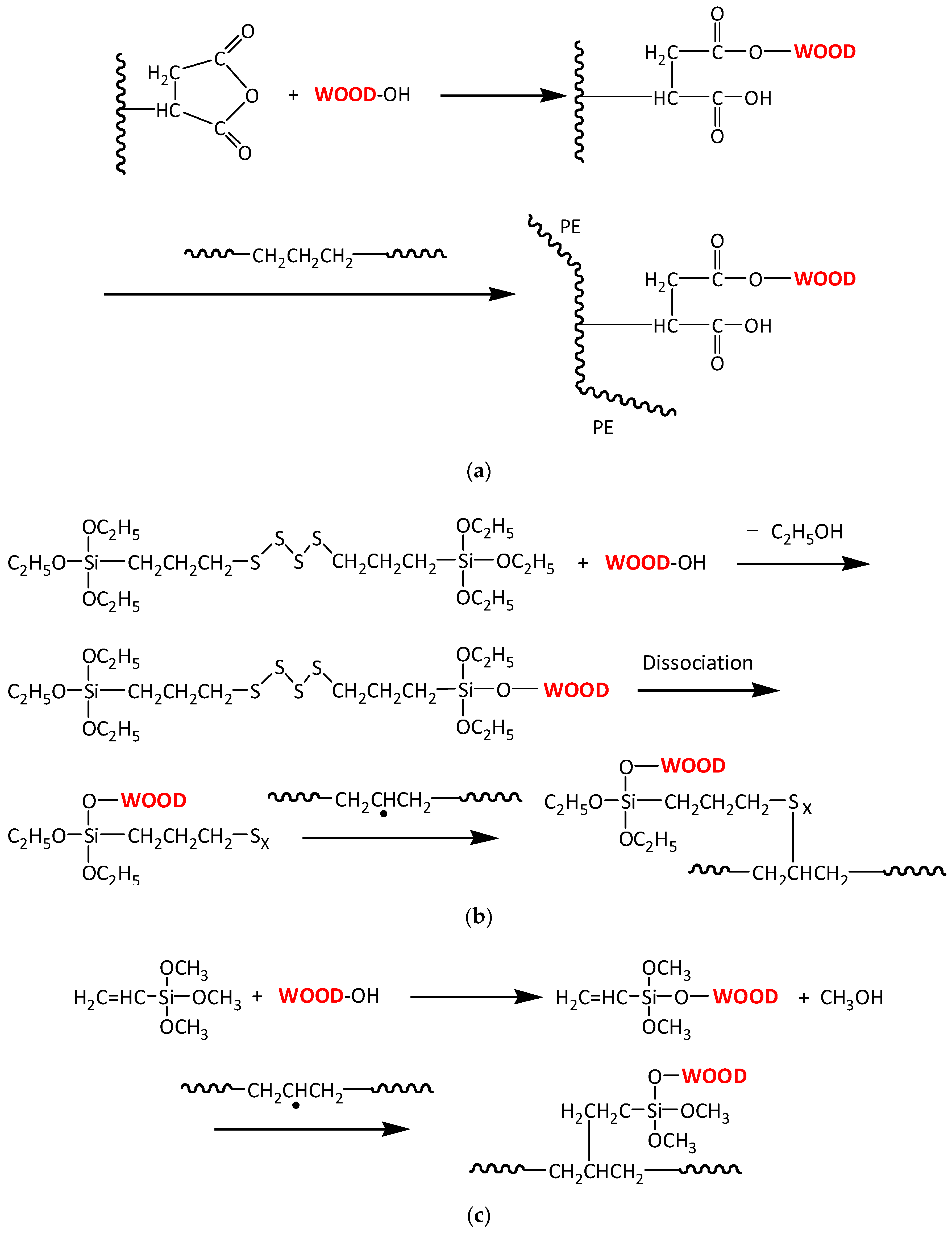
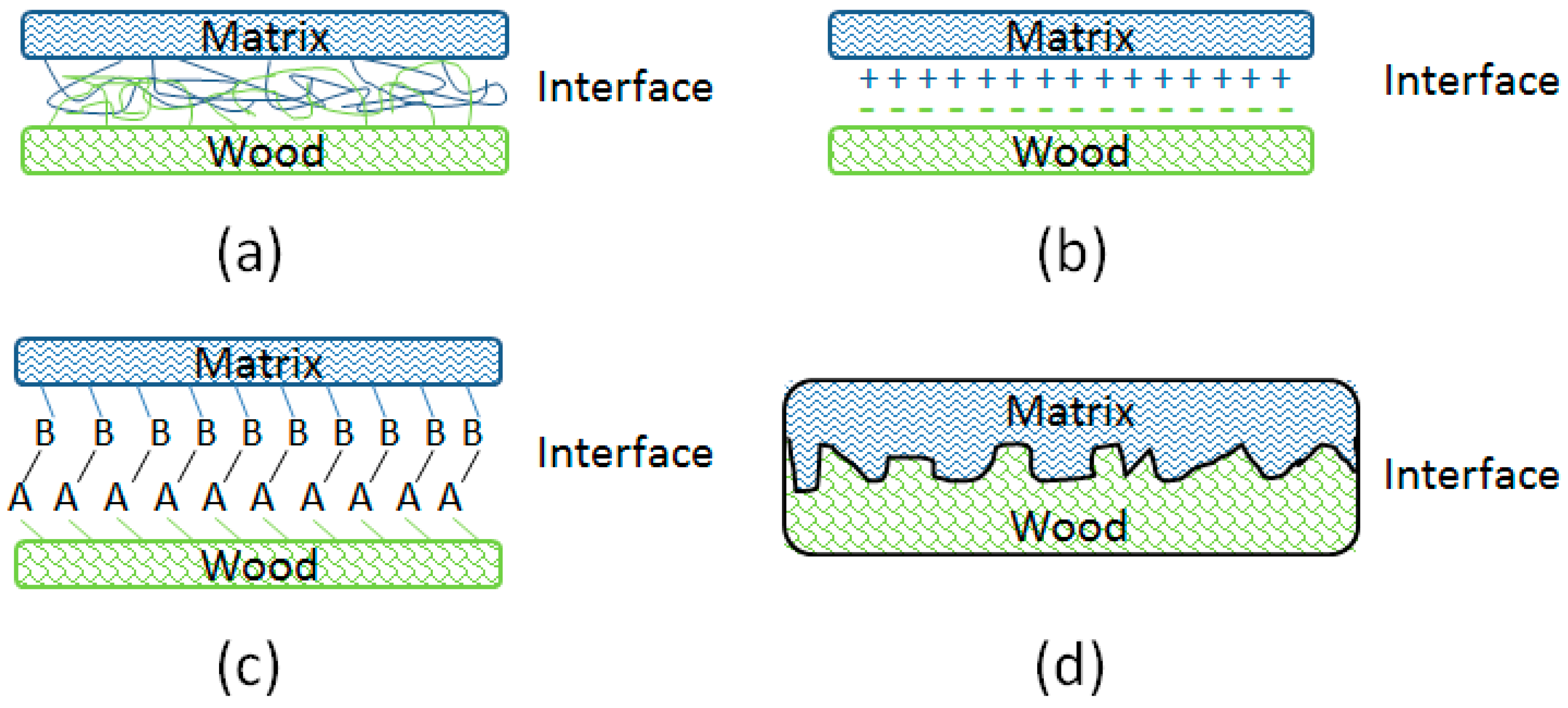
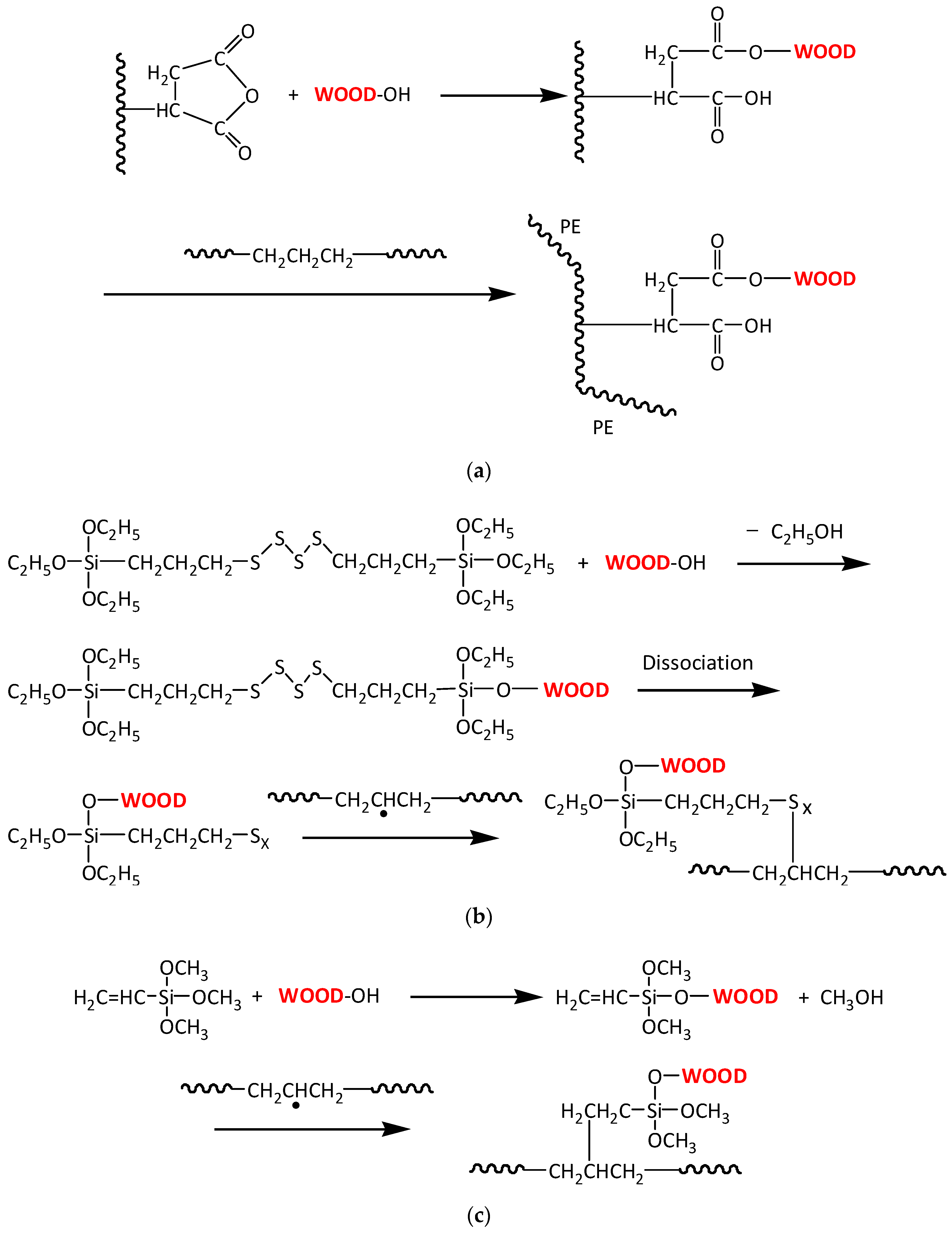
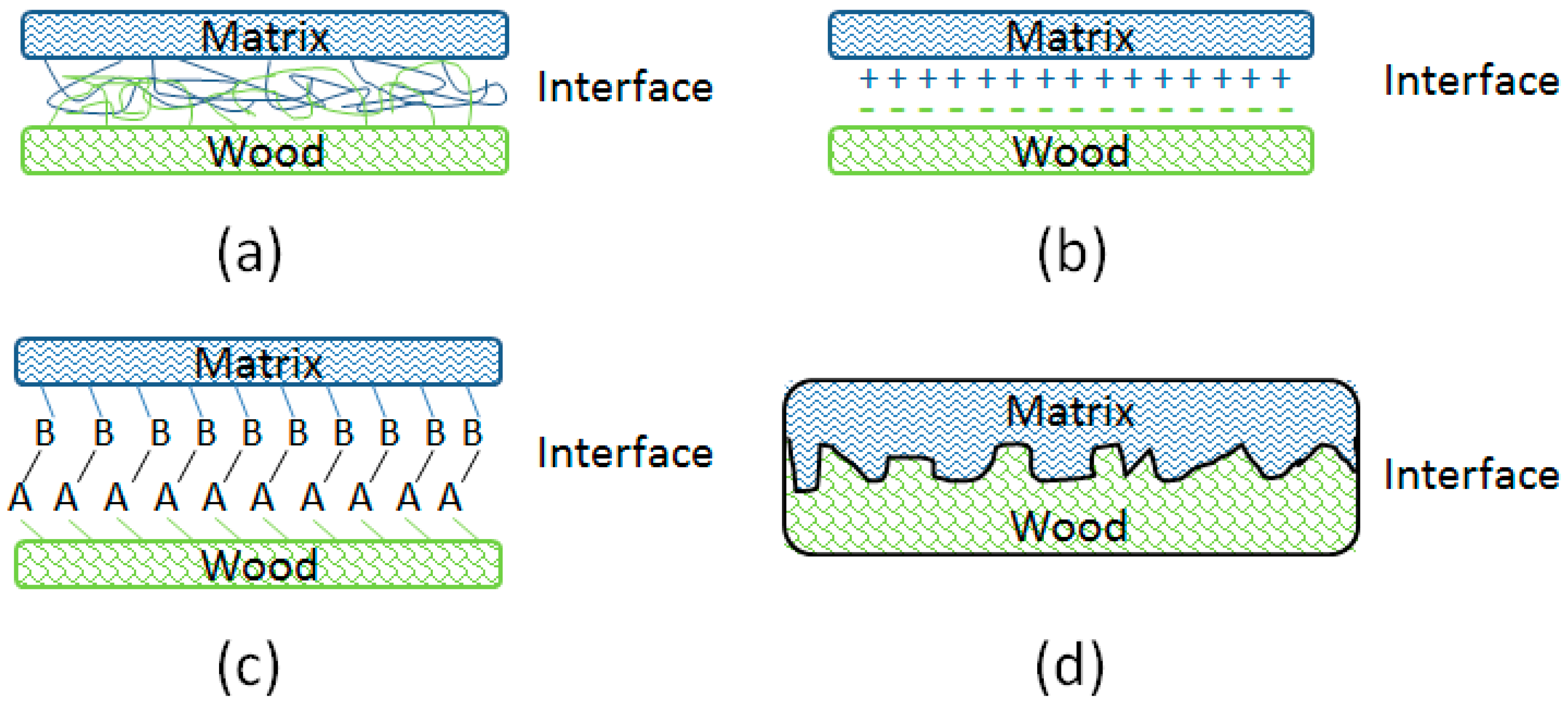
3.2. Interface Bonding Scenarios and Mechanisms
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Kazemi-Najafi, S.; Nikray, S.J.; Ebrahimi, G. A comparison study on creep behavior of wood-plastic composite, solid wood, and polypropylene. J. Compos. Mater. 2012, 46, 801–808. [Google Scholar] [CrossRef]
- Xie, Y.; Hill, C.A.S.; Xiao, Z.; Militz, H.; Mai, C. Silane coupling agents used for natural fiber/polymer composites: A review. Compos. Part A Appl. Sci. Manuf. 2010, 41, 806–819. [Google Scholar] [CrossRef]
- Bledzki, A.K.; Gassan, J.; Theis, S. Wood-filled thermoplastic composites. Mech. Compos. Mater. 1998, 34, 563–568. [Google Scholar] [CrossRef]
- Cantero, G.; Arbelaiz, A.; Llano-Ponte, R.; Mondragon, I. Effects of fibre treatment on wettability and mechanical behaviour of flax/polypropylene composites. Compos. Sci. Technol. 2003, 63, 1247–1254. [Google Scholar] [CrossRef]
- Belgacem, M.N.; Gandini, A. The surface modification of cellulose fibres for use as reinforcing elements in composite materials. Compos. Interfaces 2005, 12, 41–75. [Google Scholar] [CrossRef]
- Nourbakhsh, A.; Ashori, A. Preparation and Properties of Wood Plastic Composites Made of Recycled High-density Polyethylene. J. Compos. Mater. 2009, 43, 877–883. [Google Scholar] [CrossRef]
- Lee, S.; Wang, S.; Pharr, G.M.; Xu, H. Evaluation of interphase properties in a cellulose fiber-reinforced polypropylene composite by nanoindentation and finite element analysis. Compos. Part A Appl. Sci. Manuf. 2007, 38, 1517–1524. [Google Scholar] [CrossRef]
- Lin, J.; Huang, C.; Lin, Z.; Lou, C. Far-infrared emissive polypropylene/wood flour wood plastic composites: Manufacturing technique and property evaluations. J. Compos. Mater. 2016, 50, 2099–2109. [Google Scholar] [CrossRef]
- Pickering, K. Properties and Performance of Natural-Fibre Composites; Woodhead Publishing: Cambridge, UK, 2008. [Google Scholar]
- Bengtsson, M.; Gatenholm, P.; Oksman, K. The effect of crosslinking on the properties of polyethylene/wood flour composites. Compos. Sci. Technol. 2005, 65, 1468–1479. [Google Scholar] [CrossRef]
- Bengtsson, M.; Oksman, K. The use of silane technology in crosslinking polyethylene/wood flour composites. Compos. Part A Appl. Sci. Manuf. 2006, 37, 752–765. [Google Scholar] [CrossRef]
- Bengtsson, M.; Stark, N.M.; Oksman, K. Durability and mechanical properties of silane cross-linked wood thermoplastic composites. Compos. Sci. Technol. 2007, 67, 2728–2738. [Google Scholar] [CrossRef]
- Mohanty, S.; Nayak, S.K. Interfacial, dynamic mechanical, and thermal fiber reinforced behavior of MAPE treated sisal fiber reinforced HDPE composites. J. Appl. Polym. Sci. 2006, 102, 3306–3315. [Google Scholar] [CrossRef]
- Mohanty, S.; Verma, S.K.; Nayak, S.K. Dynamic mechanical and thermal properties of MAPE treated jute/HDPE composites. Compos. Sci. Technol. 2006, 66, 538–547. [Google Scholar] [CrossRef]
- Hassaini, L.; Kaci, M.; Touati, N.; Pillin, I.; Kervoelen, A.; Bruzaud, S. Valorization of olive husk flour as a filler for biocomposites based on poly(3-hydroxybutyrate-co-3-hydroxyvalerate): Effects of silane treatment. Polym. Test. 2017, 59, 430–440. [Google Scholar] [CrossRef]
- Osman, H.; Ismail, H.; Mustapha, M. Effects of Maleic Anhydride Polypropylene on Tensile, Water Absorption, and Morphological Properties of Recycled Newspaper Filled Polypropylene/Natural Rubber Composites. J. Compos. Mater. 2010, 44, 1477–1491. [Google Scholar] [CrossRef]
- Ihemouchen, C.; Djidjelli, H.; Boukerrou, A.; Fenouillot, F.; Barres, C. Effect of compatibilizing agents on the mechanical properties of high-density polyethylene/olive husk flour composites. J. Appl. Polym. Sci. 2013, 128, 2224–2229. [Google Scholar] [CrossRef]
- Carlborn, K.; Matuana, L.M. Functionalization of wood particles through a reactive extrusion process. J. Appl. Polym. Sci. 2006, 101, 3131–3142. [Google Scholar] [CrossRef]
- Kotilainen, R.A.; Toivanen, T.; Alén, R.J. FTIR Monitoring of Chemical Changes in Softwood during Heating. J. Wood Chem. Technol. 2000, 20, 307–320. [Google Scholar] [CrossRef]
- Ihamouchen, C.; Djidjelli, H.; Boukerrou, A.; Krim, S.; Kaci, M.; Martinez, J.J. Effect of surface treatment on the physicomechanical and thermal properties of high-density polyethylene/olive husk flour composites. J. Appl. Polym. Sci. 2012, 123, 1310–1319. [Google Scholar] [CrossRef]
- Clemons, C.M.; Sabo, R.C.; Hirth, K.C. The effects of different silane crosslinking approaches on composites of polyethylene blends and wood Flour. J. Appl. Polym. Sci. 2011, 120, 2292–2303. [Google Scholar] [CrossRef]
- Stael, G.C.; D’Almeida, J.R.M.; Tavares, M.I.B. A solid state NMR carbon-13 high resolution study of natural fiber from sugar cane and their composites with EVA. Polym. Test. 2000, 19, 251–259. [Google Scholar] [CrossRef]
- Martins, M.A.; Forato, L.A.; Mattoso, L.H.C.; Colnago, L.A. A solid state 13C high resolution NMR study of raw and chemically treated sisal fibers. Carbohydr. Polym. 2006, 64, 127–133. [Google Scholar] [CrossRef]
- Grünewald, T.; Grigsby, W.; Tondi, G.; Ostrowski, S.; Petutschnigg, A.; Wieland, S. Chemical Characterization of Wood-Leather Panels by Means of 13C NMR Spectroscopy. BioResources 2013, 8, 2422–2452. [Google Scholar] [CrossRef]
- Renneckar, S.H. Modification of Wood Fiber with Thermoplastics by Reactive Steam-Explosion. Ph.D. Thesis, Virginia Polytechnic Institute and State University, Blacksburg, VA, USA, 2004. [Google Scholar]
- Santoni, I.; Callone, E.; Sandak, A.; Sandak, J.; Dirè, S. Solid state NMR and IR characterization of wood polymer structure in relation to tree provenance. Carbohydr. Polym. 2015, 117, 710–721. [Google Scholar] [CrossRef] [PubMed]
- Wikberg, H.; Maunu, S.L. Characterisation of thermally modified hard- and softwoods by 13C CPMAS NMR. Carbohydr. Polym. 2004, 58, 461–466. [Google Scholar] [CrossRef]
- Sombatsompop, N.; Sungsanit, K.; Thongpin, C. Analysis of low-density polyethylene-g-poly(vinyl chloride) copolymers formed in poly(vinyl chloride)/low-density polyethylene melt blends with gel permeation chromatography and solid-state 13C-NMR. J. Appl. Polym. Sci. 2004, 92, 3167–3172. [Google Scholar] [CrossRef]
- Li, X.; Tabil, L.G.; Panigrahi, S. Chemical Treatments of Natural Fiber for Use in Natural Fiber-Reinforced Composites: A Review. J. Polym. Environ. 2007, 15, 25–33. [Google Scholar] [CrossRef]
- Zaper, A.M.; Koenig, J.L. Solid-state 13C NMR studies of vulcanized elastomers, 4. Sulfur-vulcanized polybutadiene. Macromol. Chem. Phys. 1988, 189, 1239–1251. [Google Scholar] [CrossRef]
- Andreis, M.; Liu, J.; Koenig, J.L. Solid-state carbon-13 NMR Studies of vulcanized elastomers. V. Observation of new structures in sulfur-vulcanized natural rubber. J. Polym. Sci. Part B Polym. Phys. 1989, 27, 1389–1404. [Google Scholar] [CrossRef]
- Silverstein, R.M.; Webster, F.X.; Kiemle, D.J.; Bryce, D.L. Spectrometric Identification of Organic Compounds; Wiley: Hoboken, NJ, USA, 2014. [Google Scholar]
- Brochier Salon, M.; Abdelmouleh, M.; Boufi, S.; Belgacem, M.N.; Gandini, A. Silane adsorption onto cellulose fibers: Hydrolysis and condensation reactions. J. Colloid Interface Sci. 2005, 289, 249–261. [Google Scholar] [CrossRef] [PubMed]
- Fyfe, C.A.; Niu, J. Direct Solid-state 13C NMR Evidence for Covalent Bond Formation between an Immobilized Vinylsilane Linking Agent and Polymer Matrices. Macromolecules 1995, 28, 3894–3897. [Google Scholar] [CrossRef]
- Choi, S. Influence of storage time and temperature and silane coupling agent on bound rubber formation in filled styrene-butadiene rubber compounds. Polym. Test. 2002, 21, 201–208. [Google Scholar] [CrossRef]
- Sitarz, M.; Czosnek, C.; Jeleń, P.; Odziomek, M.; Olejniczak, Z.; Kozanecki, M.; Janik, J.F. SiOC glasses produced from silsesquioxanes by the aerosol-assisted vapor synthesis method. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 2013, 112, 440–445. [Google Scholar] [CrossRef] [PubMed]
- Deguchi, S.; Tsujii, K.; Horikoshi, K. Crystalline-to-amorphous transformation of cellulose in hot and compressed water and its implications for hydrothermal conversion. Green Chem. 2008, 10, 191–196. [Google Scholar] [CrossRef]
- Salon, M.B.; Gerbaud, G.; Abdelmouleh, M.; Bruzzese, C.; Boufi, S.; Belgacem, M.N. Studies of interactions between silane coupling agents and cellulose fibers with liquid and solid-state NMR. Magn. Reson. Chem. 2007, 45, 473–483. [Google Scholar] [CrossRef] [PubMed]
- Grmusa, I.G.; Dunky, M.; Miljkovic, J.; Momcilovic, M.D. Influence of the degree of condensation of urea-formaldehyde adhesives on the tangential penetration into beech and fir and on the shear strength of the adhesive joints. Eur. J. Wood Wood Prod. 2012, 70, 655–665. [Google Scholar] [CrossRef]
- Grmusa, I.G.; Dunky, M.; Miljkovic, J.; Momcilovic, M.D. Influence of the viscosity of UF resins on the radial and tangential penetration into poplar wood and on the shear strength of adhesive joints. Holzforschung 2012, 66, 849–856. [Google Scholar]
- Kalaprasad, G.; Francis, B.; Thomas, S.; Kumar, C.R.; Pavithran, C.; Groeninckx, G.; Thomas, S. Effect of fibre length and chemical modifications on the tensile properties of intimately mixed short sisal/glass hybrid fibre reinforced low density polyethylene composites. Polym. Int. 2004, 53, 1624–1638. [Google Scholar] [CrossRef]
- Mohanty, S.; Nayak, S.K.; Verma, S.K.; Tripathy, S.S. Effect of MAPP as Coupling Agent on the Performance of Sisal-PP Composites. J. Reinf. Plast. Compos. 2004, 23, 2047–2063. [Google Scholar] [CrossRef]
- Zhou, Y.; Fan, M.; Lin, L. Investigation of bulk and in situ mechanical properties of coupling agents treated wood plastic composites. Polym. Test. 2017, 58, 292–299. [Google Scholar] [CrossRef]
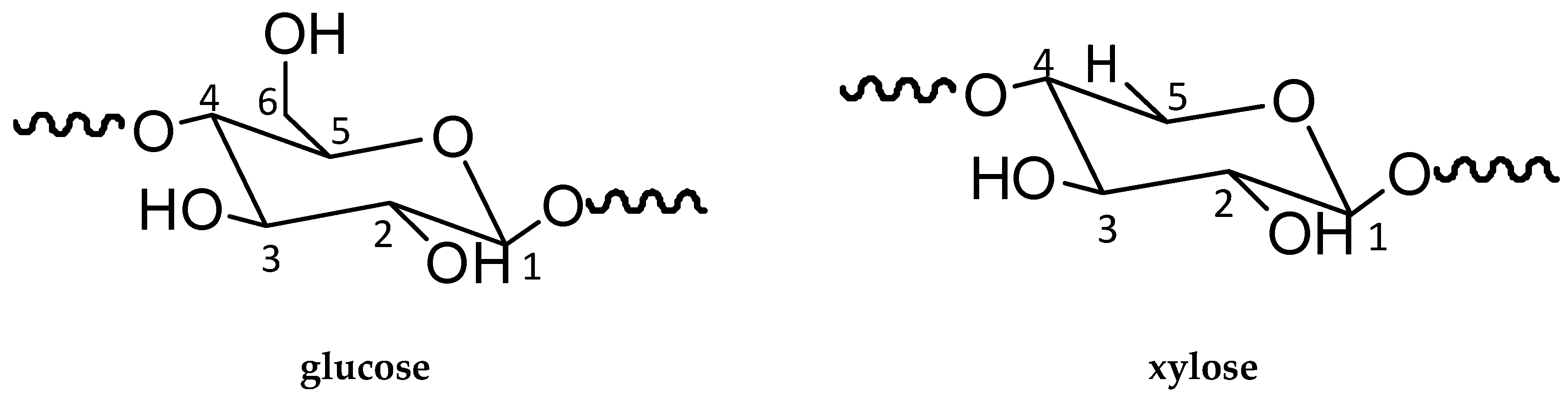



{kind=link}
{kind=link}
{kind=link}
{kind=link}
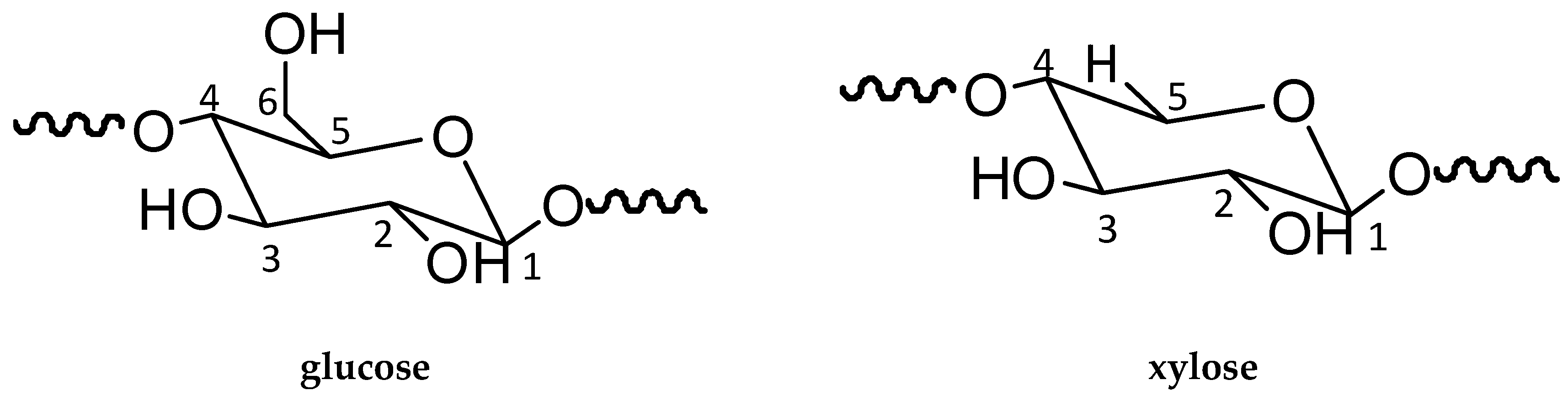{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | Wood (%) | PE (%) | TPW 709 (%) | 12HSA (%) | MAPE (%) | Si69 (%) | VTMS (%) |
|---|---|---|---|---|---|---|---|
| Untreated WPC | 50 | 43 | 3.5 | 3.5 | 0 | 0 | 0 |
| MAPE treated WPC | 50 | 40 | 3.5 | 3.5 | 3 | 0 | 0 |
| Si69 treated WPC | 50 | 40 | 3.5 | 3.5 | 0 | 3 | 0 |
| VTMS treated WPC | 50 | 40 | 3.5 | 3.5 | 0 | 0 | 3 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rao, J.; Zhou, Y.; Fan, M. Revealing the Interface Structure and Bonding Mechanism of Coupling Agent Treated WPC. Polymers 2018, 10, 266. https://doi.org/10.3390/polym10030266
Rao J, Zhou Y, Fan M. Revealing the Interface Structure and Bonding Mechanism of Coupling Agent Treated WPC. Polymers. 2018; 10(3):266. https://doi.org/10.3390/polym10030266
Chicago/Turabian StyleRao, Jiuping, Yonghui Zhou, and Mizi Fan. 2018. "Revealing the Interface Structure and Bonding Mechanism of Coupling Agent Treated WPC" Polymers 10, no. 3: 266. https://doi.org/10.3390/polym10030266
APA StyleRao, J., Zhou, Y., & Fan, M. (2018). Revealing the Interface Structure and Bonding Mechanism of Coupling Agent Treated WPC. Polymers, 10(3), 266. https://doi.org/10.3390/polym10030266