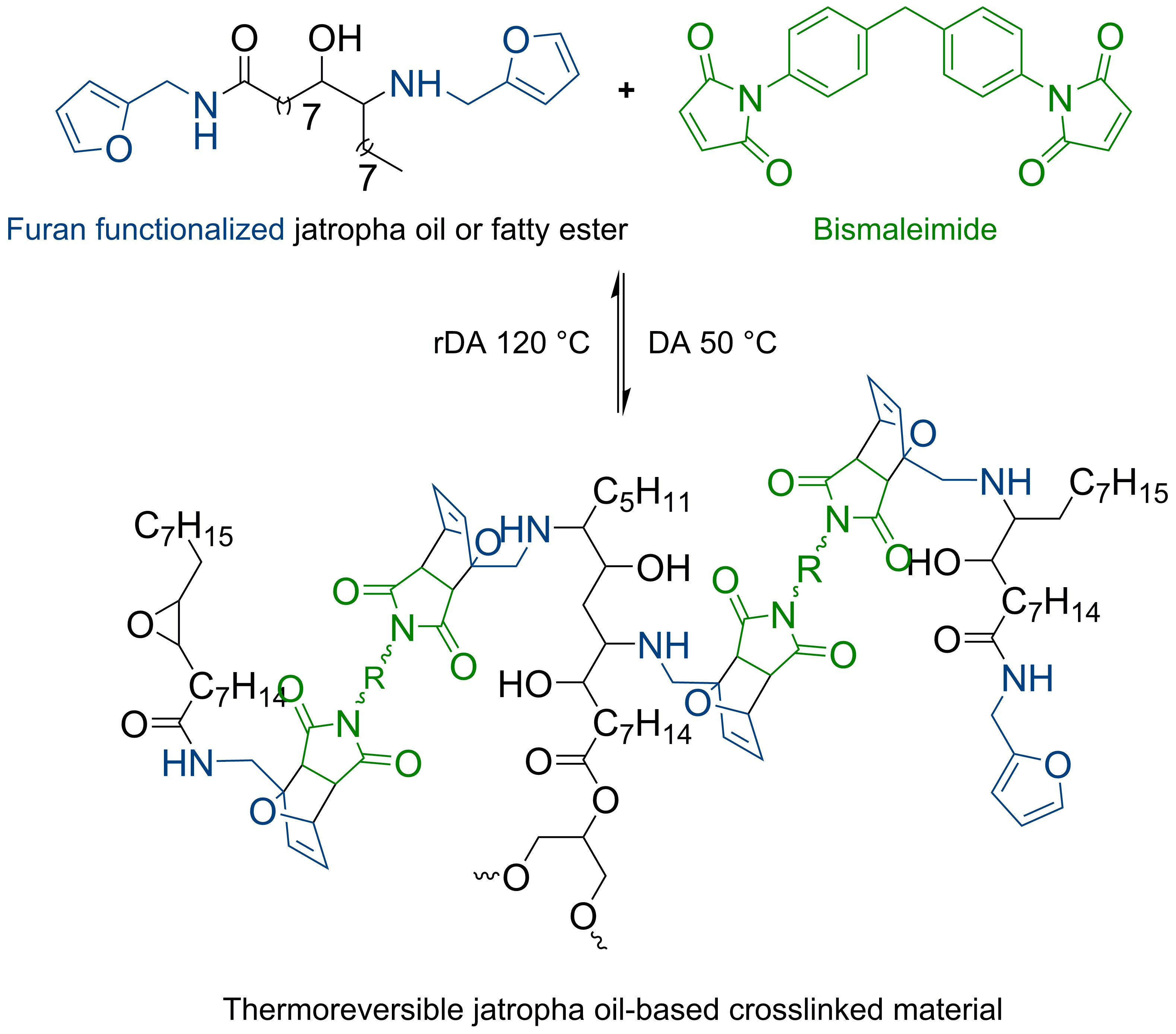
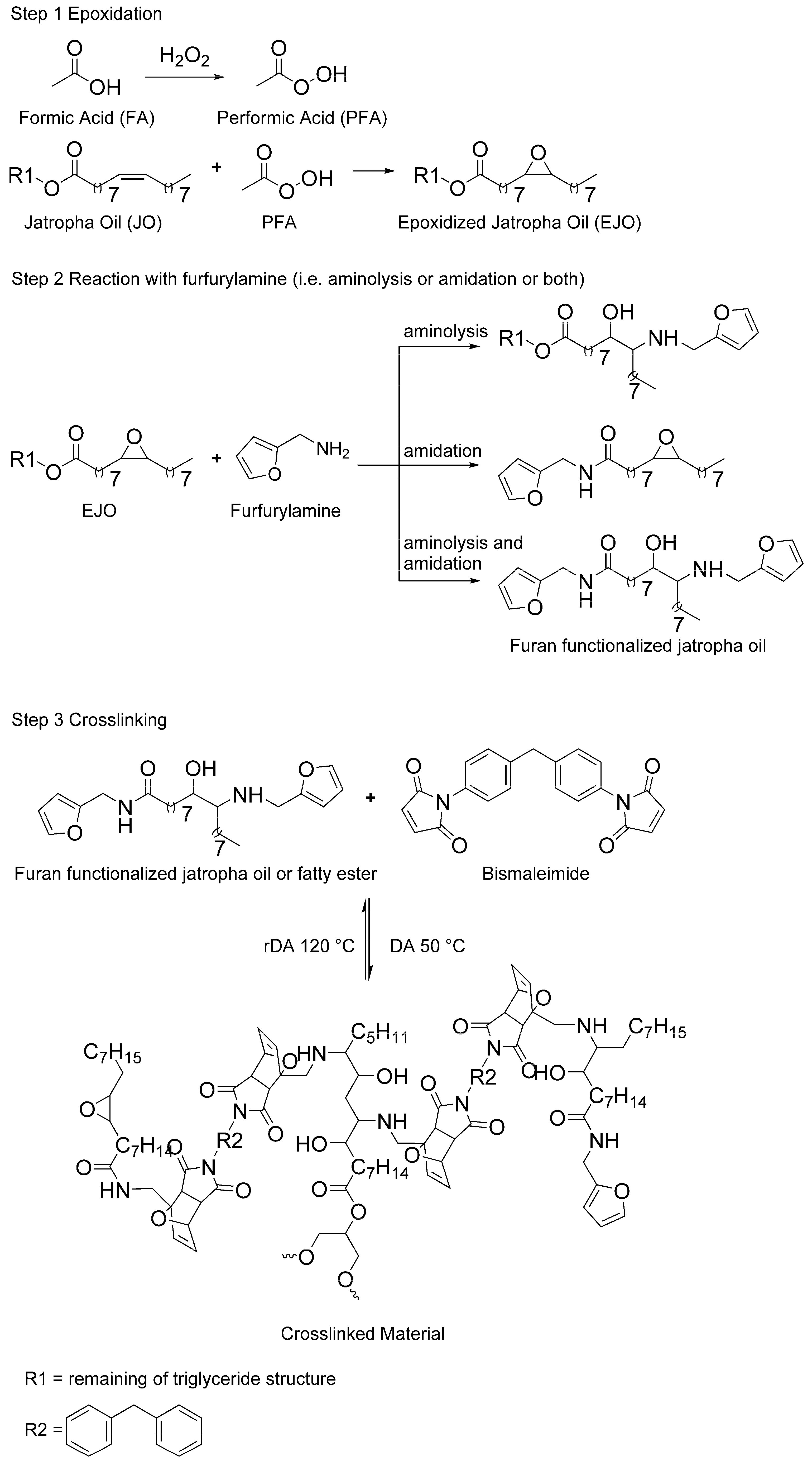
3.1. Furan-Functionalized Methyl Oleate and Methyl Linoleate-Based Polymers
This section discusses the use of methyl oleate (MO) and methyl linoleate (LO) as a preliminary study, with respect to further study of jatropha oil (JO) as a crosslinked polymer precursor. Even though the fatty acid content of JO may vary depending on species, climate, soil quality, etc., MO and LO are chosen based on the fact that both components are the major fatty acid constituents in JO [
9,
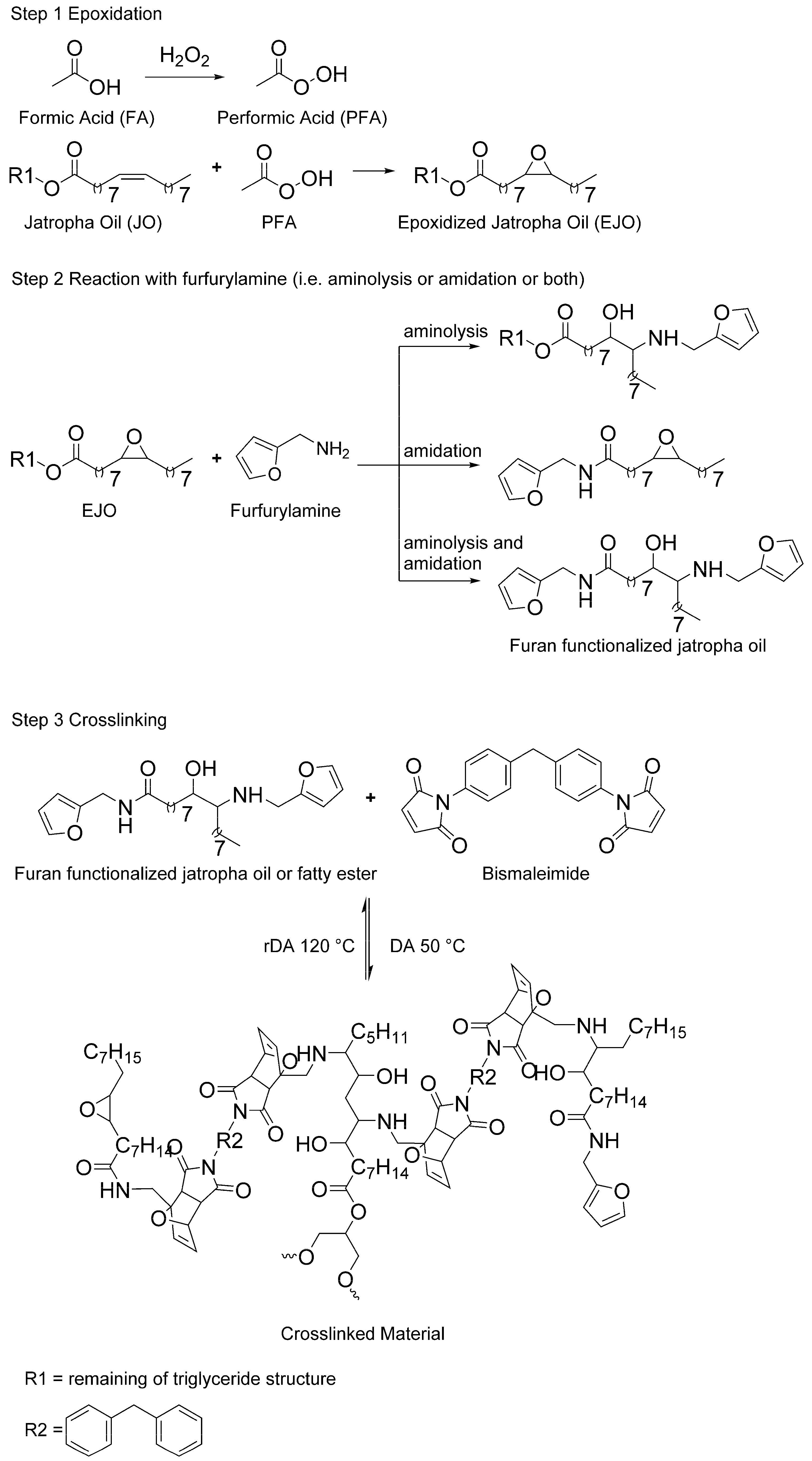
10]. As the first step of the polymer synthesis, MO and LO were epoxidized according to published procedures [
15]. Formic acid was reacted in situ with H
2O
2 to form performic acid, which acts as an oxygen donor for the alkene. The donated oxygen is replenished by H
2O
2, generating water (
Scheme 1). Formic acid can also open the oxirane ring, and hydrolysis can occur in the presence of water. Benzene has been added as diluent of the organic phase (i.e., fatty esters) to minimize oxirane ring opening [
23]. Since the two layers are immiscible, the degree of mixing has a large influence on the reaction. However, at a certain point, a high degree of mixing may promote the unwanted oxirane ring degradation process. In addition, higher reaction temperatures generally favor the degradation reaction [
15,
16].
The opening of the oxirane ring may occur in the presence of water and formic acid and, to a lesser extent, H
2O
2. High chemical activity of formic acid may cause depletion of oxygen in the system if the H
2O
2 is no longer present, resulting in lower conversions [
15,
16]. Therefore, two-step epoxidation reactions were formulated to ensure the presence of H
2O
2. As expected, the epoxidation reaction of MO and LO gives full conversion (NMR spectra not shown for brevity) and, also, good yield (i.e., MO: 76% and LO: 92%). In addition, full conversion gives one oxirane group for epoxidized methyl oleate (ep-MO), and two groups for epoxidized methyl linoleate (ep-LO). This number is rather important for further steps, since the oxirane ring acts as the basis for the furan functionalization reaction.
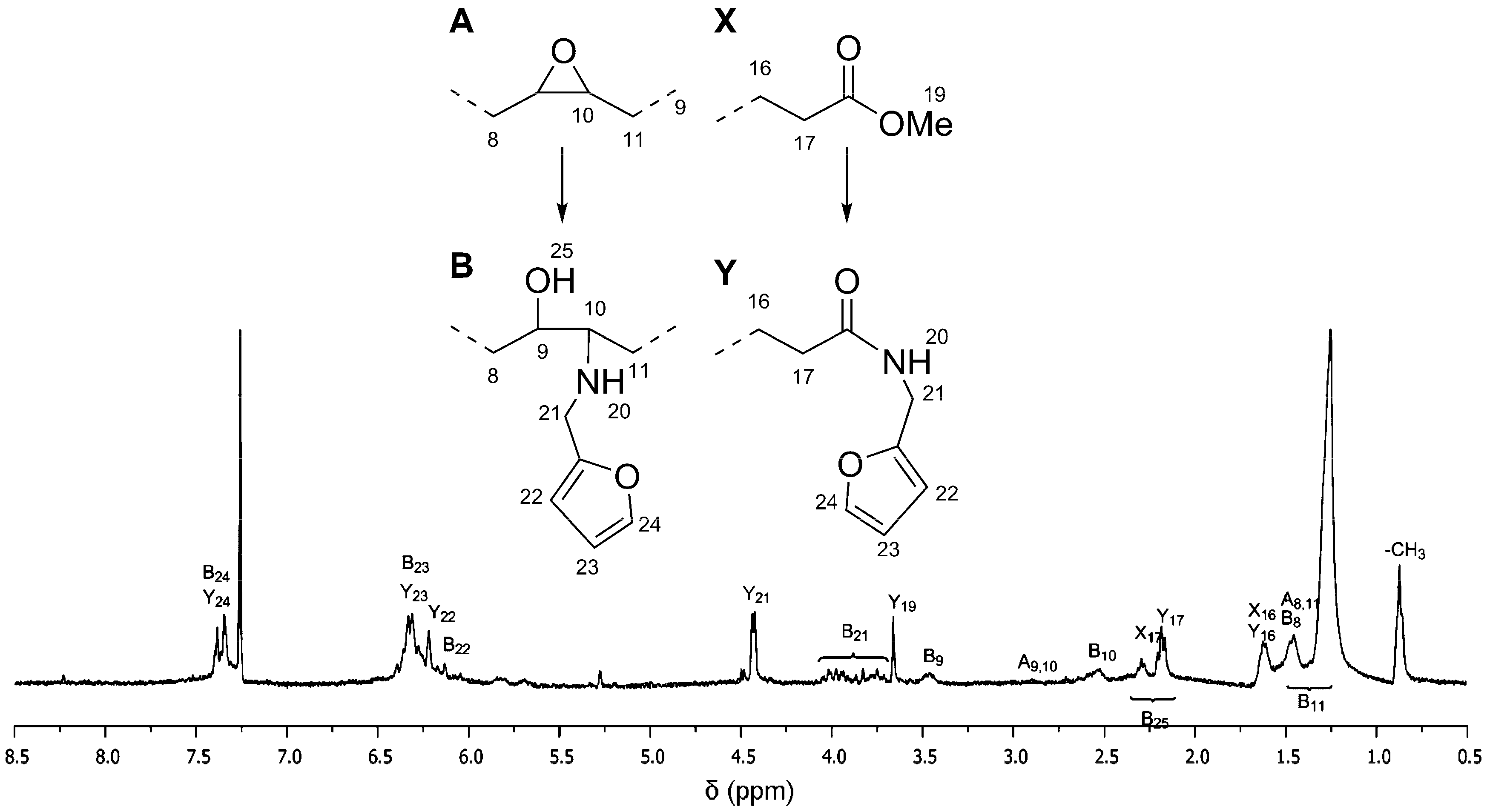
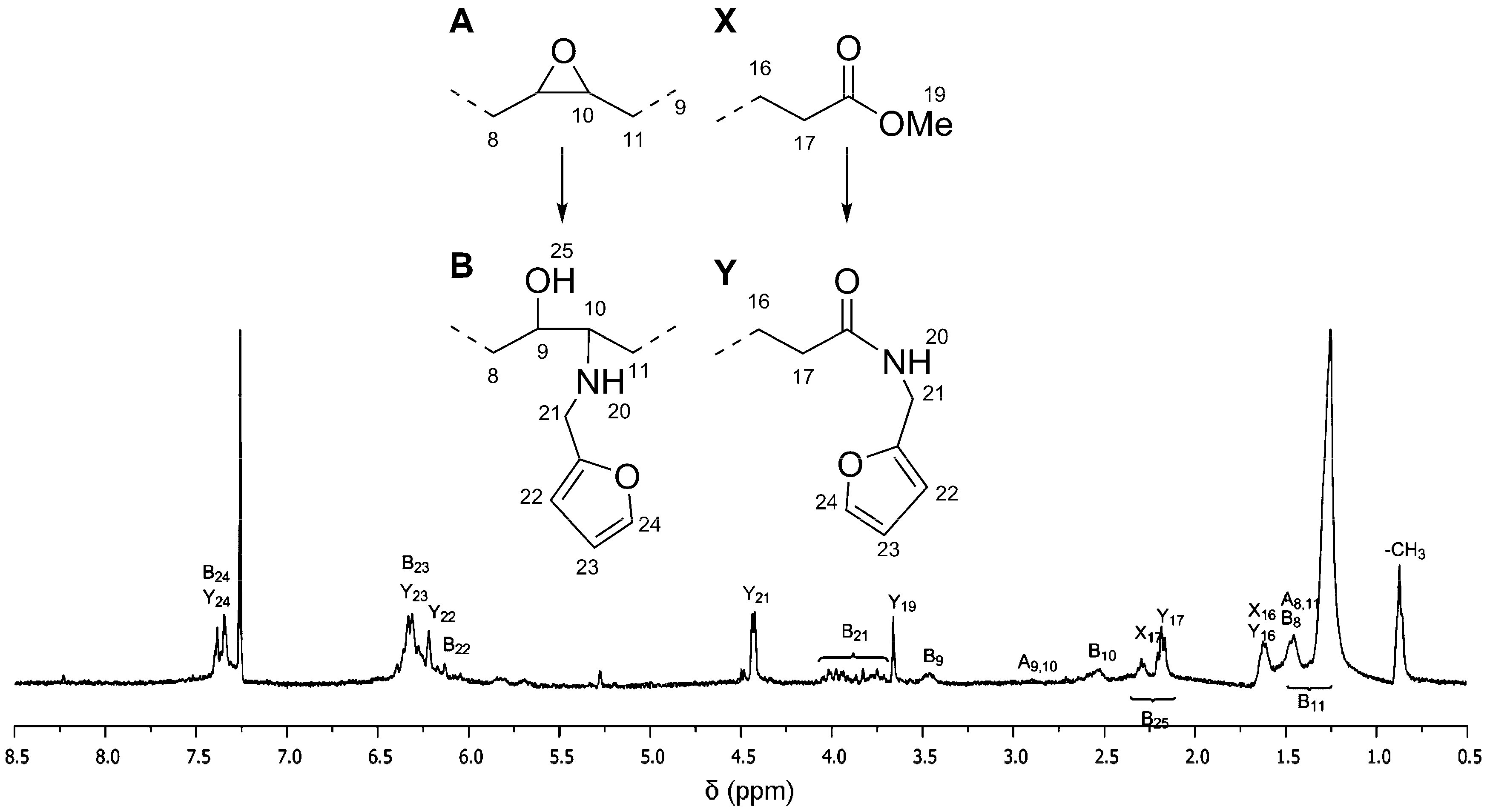
To the best of our knowledge, the reaction between furfurylamine and epoxidized fatty ester has not been reported in literature. The reaction does not spontaneously take place at atmospheric pressure and room temperature. Since the primary amine of furfurylamine molecule is rather weak to undergo ring opening reaction with the oxirane group, this aminolysis reaction requires assistance in the form of a catalyst. In addition, the catalyst is also expected to assist amidation reaction on the ester groups of the epoxidized fatty esters (
Figure 2). Both reactions are rather important, especially for ep-MO, since it requires at least two furan groups in order to assemble a polymeric chain.
Based on a similar aminolysis procedure with different primary amines described in literature [
15,
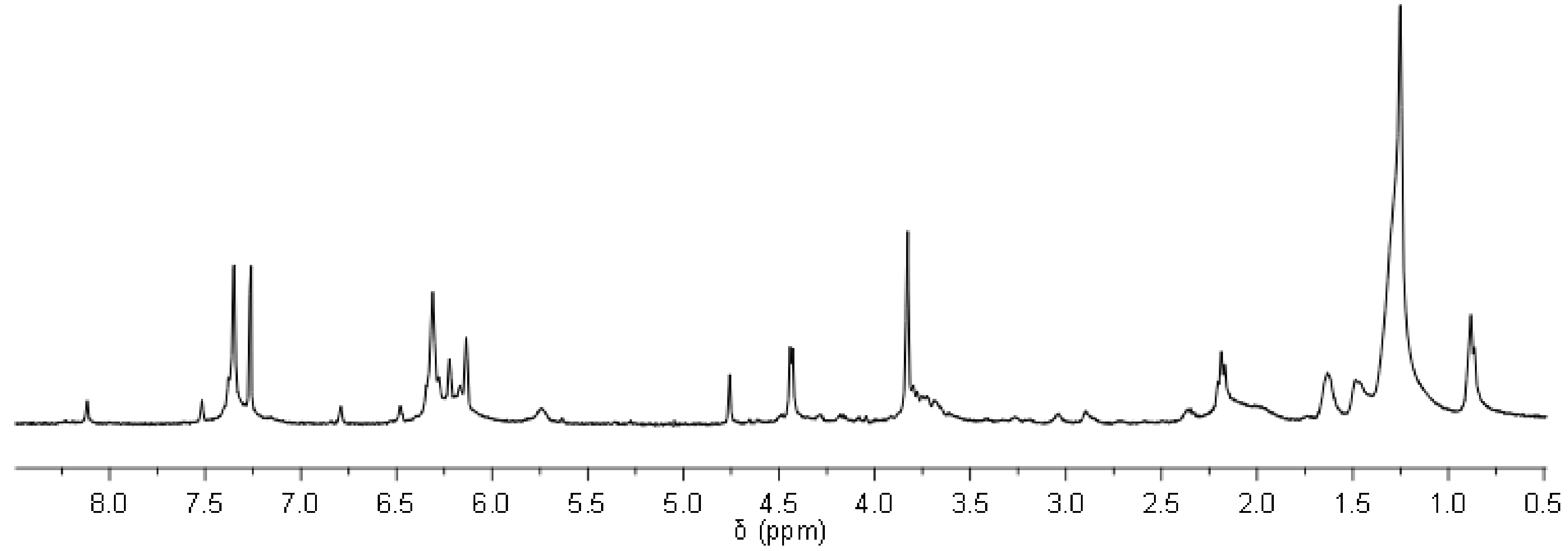
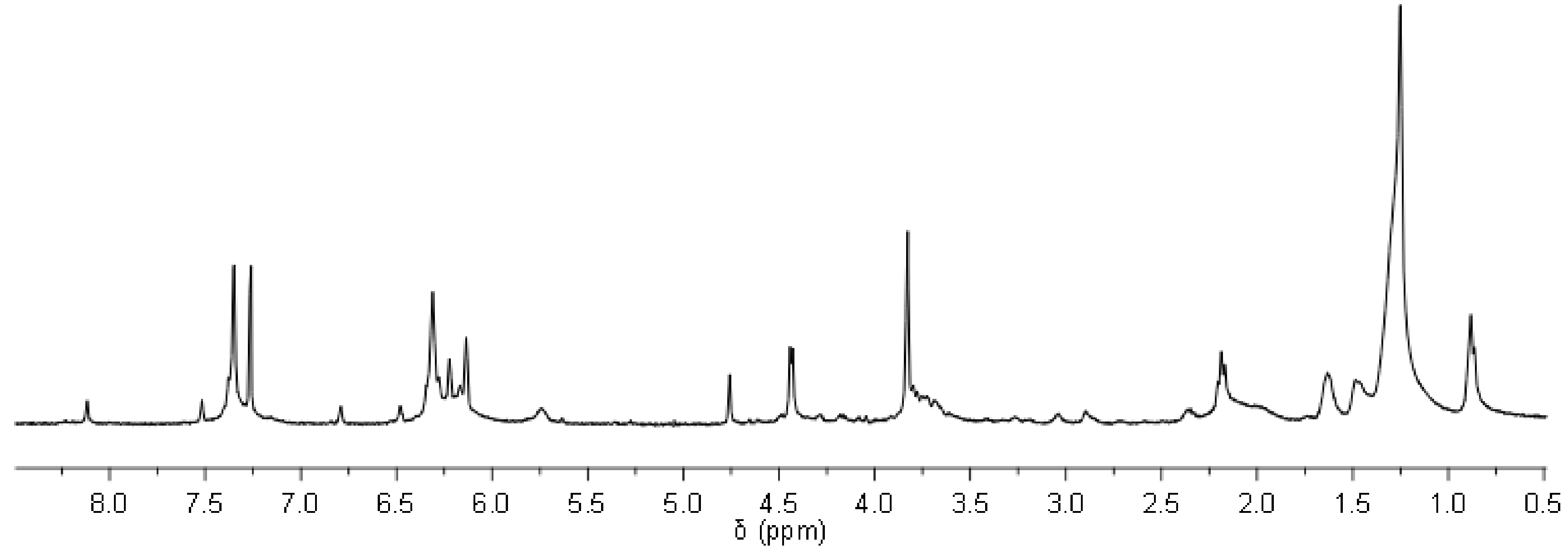
18], oxirane contained ep-MO or ep-LO was reacted with furfurylamine. After 24 h, the reaction gives a conversion of 82% on the ester group and 89% on the epoxide for ep-MO. While ep-LO has a conversion of 83% on the ester group and 75% on the epoxide (confirmed by NMR). The presence of two oxirane ring close to each other on ep-LO is believed to be responsible for lower aminolysis conversion (steric hindrance). These partial conversions are actually desired for later reactions with JO (see below). In addition, the functionalized oleic (FAO) and furan-functionalized linoleic (FALO) chains have approximately 1.7 and 2.4 furan groups/molecule respectively. The mixtures of partially converted FAO (including also stereo-isomers) display a broad range of proton signals in
1H NMR spectra. Therefore, signal assignment is highlighted only to the important peaks (
Figure 2).
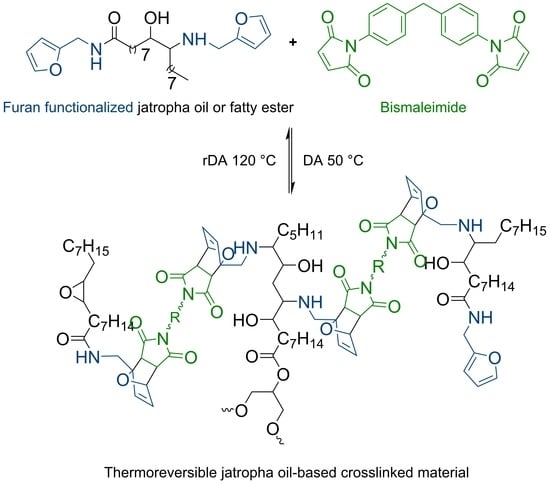
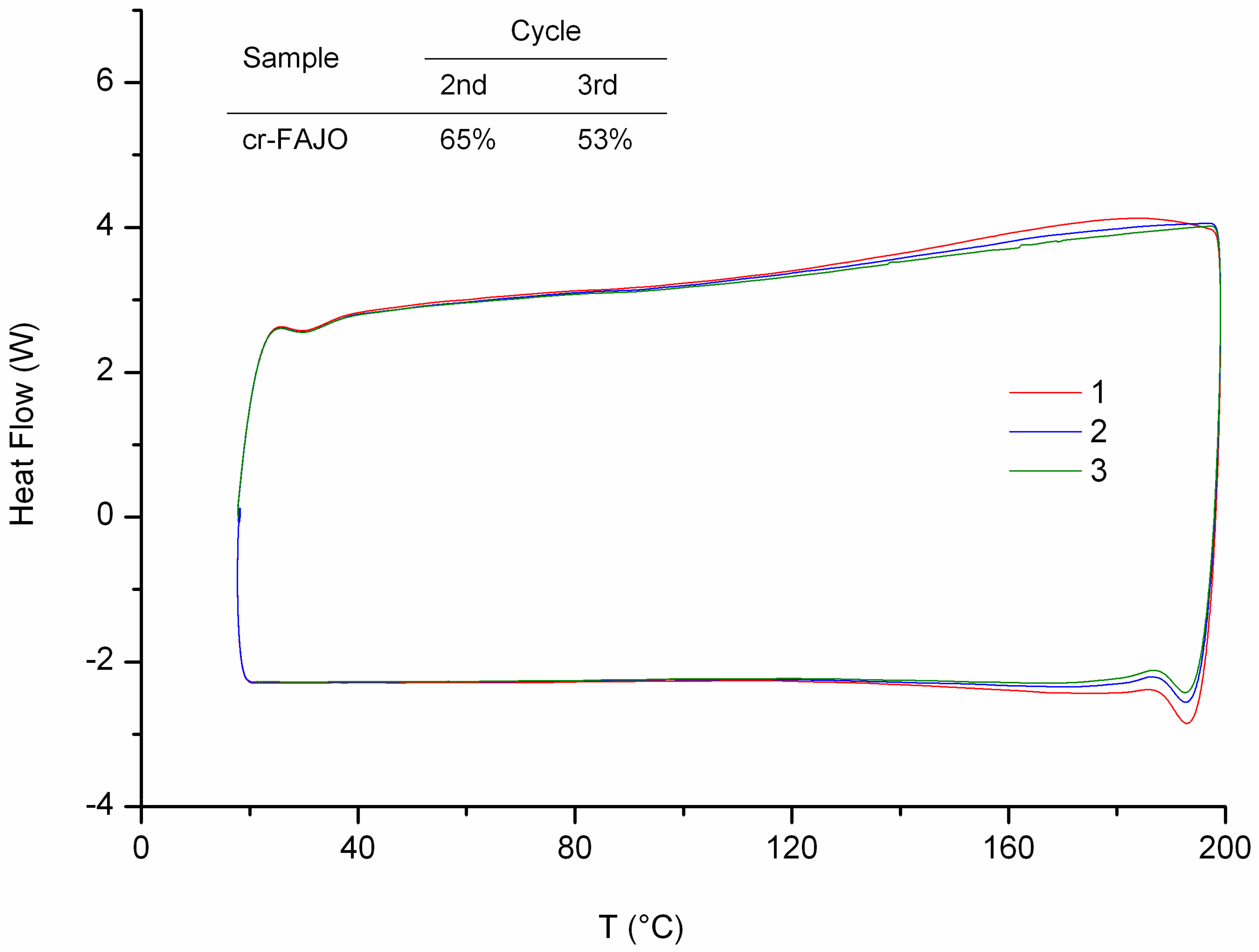
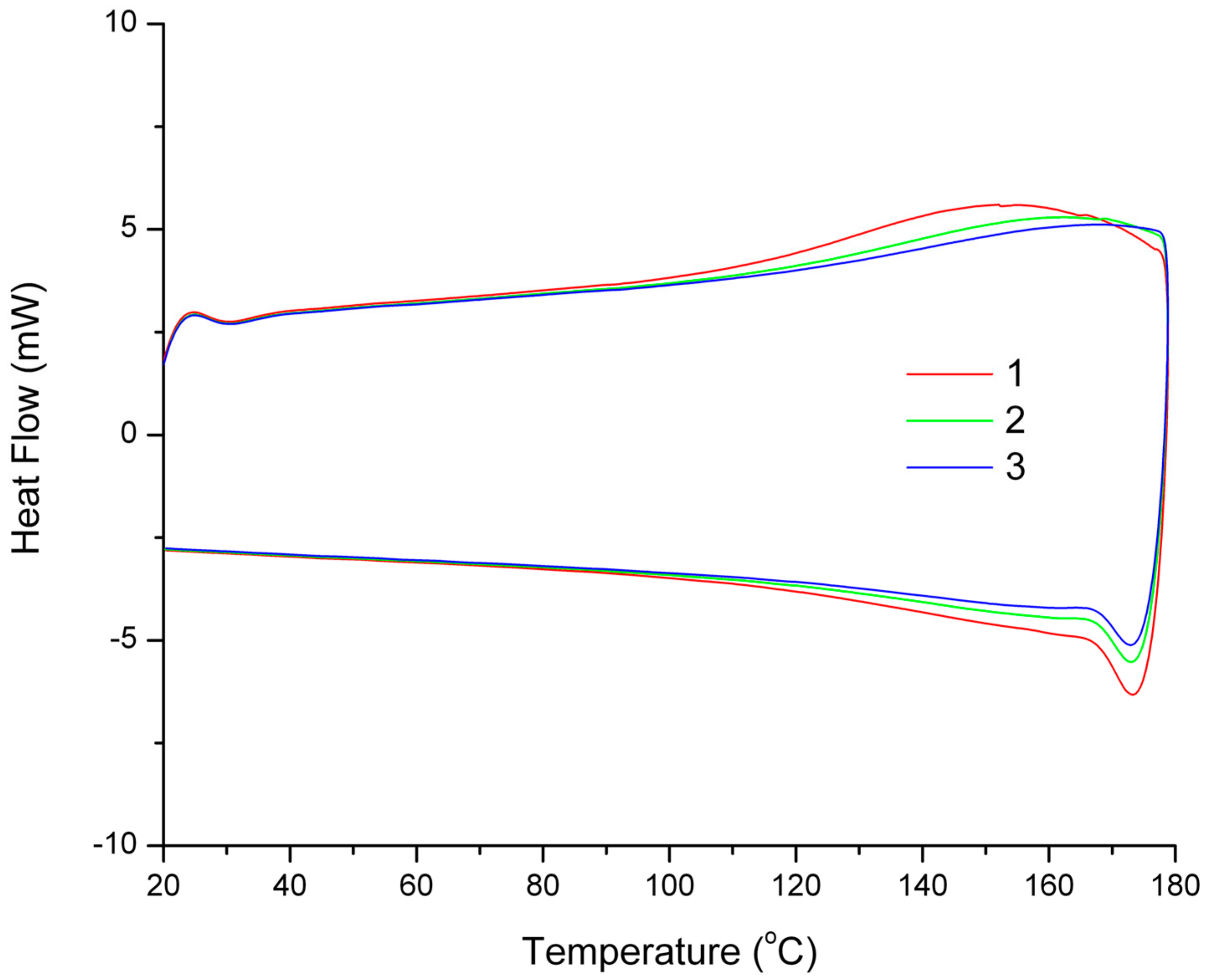
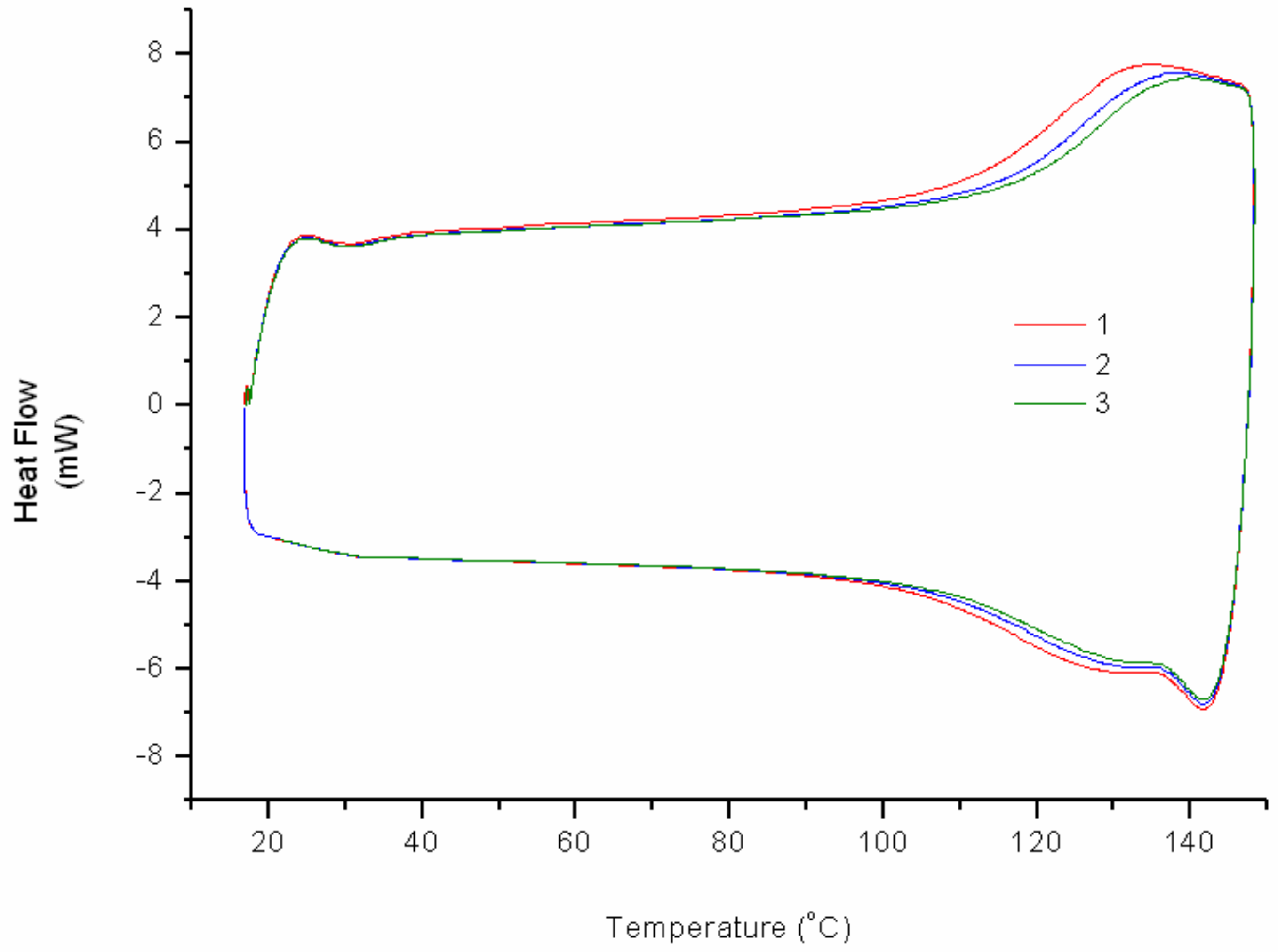
As preliminary study, FAO was reacted with bismaleimide to obtain polymer as a result of the DA adduct formation. With a maximum of two possible adduct site per FAO molecule, the reaction may only give a linear chain. However, poor solubility of the resulted polymer cr-FAO in some solvents (i.e., acetone, THF, chloroform, and DMSO) at room temperature indicates a relatively high molecular weight product was obtained. The DA-rDA thermoreversibility system on cr-FAO was investigated using DSC (
Figure 3). At 110 °C, bonds rupture via rDA reaction route starts taking place indicated by heat uptake. Each consecutive cycle reveals the decrease on the material “thermo-recovering degree” specified by the loss of energy uptake.
The DA-rDA system is an equilibrium system with respect to temperature and also a time-dependent mechanism, especially at lower temperature region. The decrease on thermo-recovering degree most likely caused by the partial failure on molecular reconstitution via DA reaction at given time. The presence of more stable DA adduct isomer (i.e.,
endo and
exo adduct) also contribute to the drop of thermo-recovering degree, as previously reported for similar systems [
7]. In addition, loss of functionality through permanent crosslinking also arises as another possibility. The latter possibility only implies if reduction of weight occurs. The possibility can be eliminated since the thermogram profile at 40 °C to 80 °C shows no apparent decrease of heat uptake. The crosslinking reaction of FALO and bismaleimide (
Im/f = 1) gives much brittle product compared to cr-FAO. The difference caused by the number of possible furan functionality to undergo DA reaction with bisamleimide. A crosslinked product was obtained since the average furan groups per molecules are more than two.
3.2. Furan-Functionalized Jatropha Oil-Based Polymers
Through same method used on epoxidation of the fatty esters (i.e., MO and LO), JO was fully epoxidized resulting high yield of 96% (
Figure 4). Therefore, the full conversion gives an average of 3.3 epoxides group per triglyceride molecules of ep-JO. As mentioned earlier, this sum of oxirane ring per molecules is important since it will undergo aminolysis reaction with furfurylamine as nucleophile. In addition, occurring amidation reaction on the ester groups splits the fatty ester from the original triglyceride molecule.
Reactions of ep-JO with furfurylamine were performed with lower LiBr intake (i.e., 50%). Partial amidation on furan functionalization reaction of ep-JO result in a broad variety of Furfuryl-amine functionalized Jatropha Oil (FAJO) products (i.e., furan-functionalized mono-, di-, triglyceride, and free fatty amide). The multiplet at 3.7 ppm gives strong indication of glycerol or monoglyceride (
Figure 4). However, some signal overlap occurs since aminolysis products give a broad signal between 3.7 to 4.0 ppm. Steric hindrance of the triglyceride structure contributes to partial aminolysis reaction, with remaining unreacted epoxide peaks at 2.9 to 3.2 ppm. A triglyceride contains three ester groups close together making the reaction site bulky for amidation. The stereochemistry of the molecule hinders its accessibility to nucleophile (i.e., furfurylamine) as well as LiBr to assist the reaction. Improving reaction conversion by using an excess of LiBr at certain point might lead to contra-productive results since the metal ion may also interact with two carbonyl groups through a chelation effect [
20]. The amide signal at 4.4 ppm is used to calculate the amidation reaction conversion, while the remaining oxirane group is used for the aminolysis. A total of 4.2 furan groups were found for every triglyceride as combined reaction of amidation (47% conversion) and aminolysis (74% conversion).
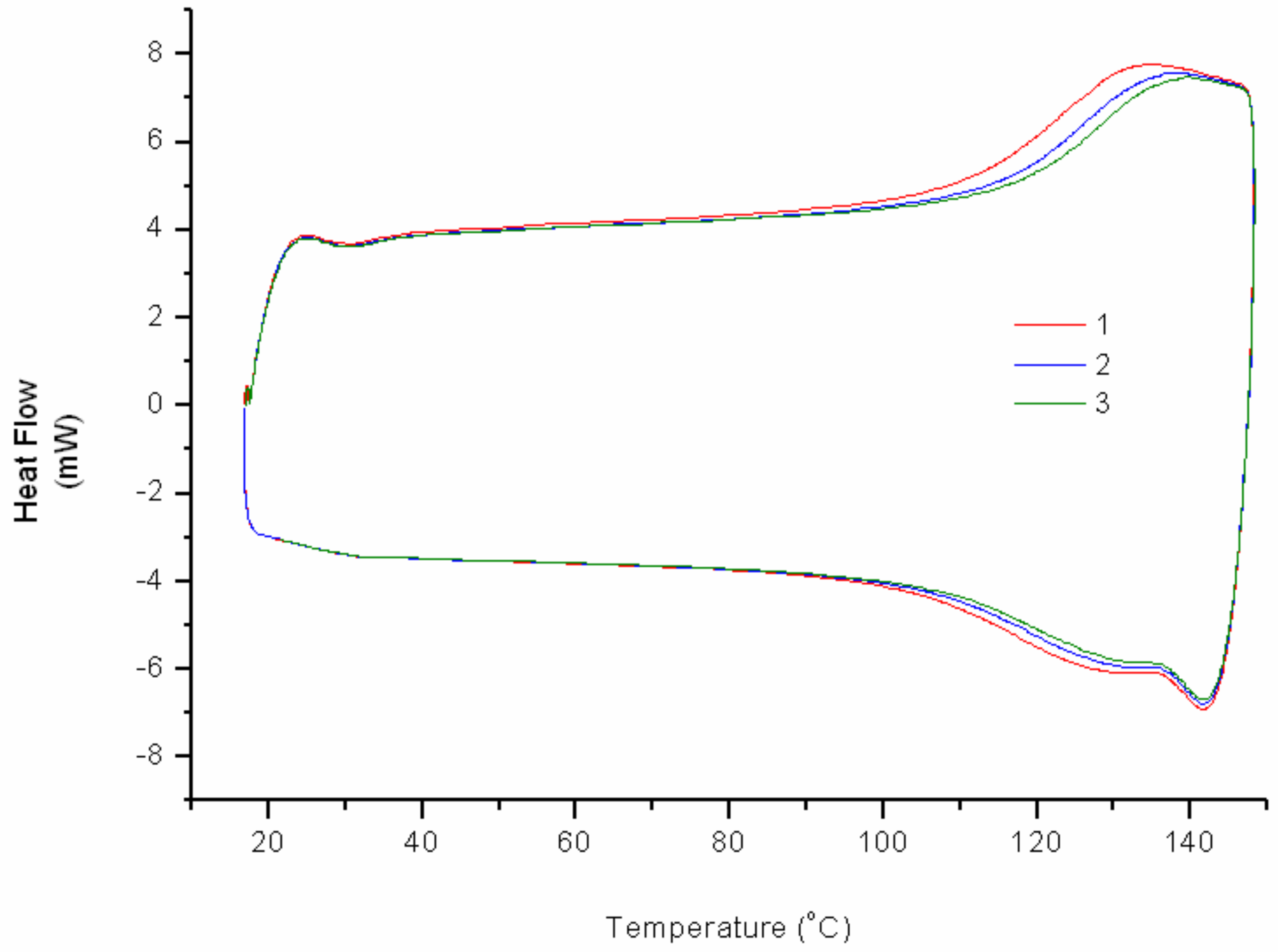
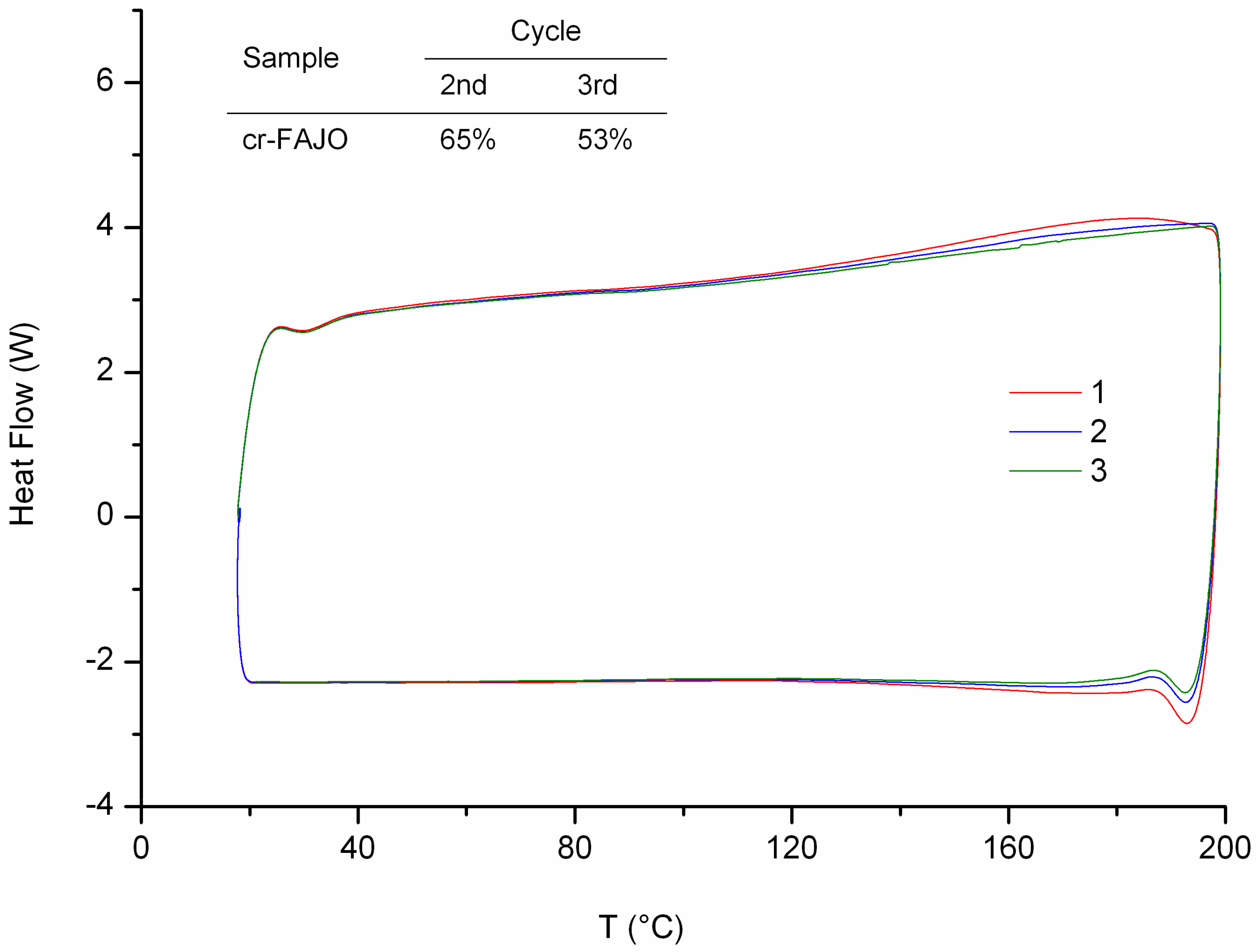
Crosslinking reactions of FAJO using bismaleimide (equimolar ratio) were performed in a reflux setup. The resulted product (cr-FAJO,
Im/f = 1) was analyzed using DSC to investigate its thermoreversibility profile (
Figure 5). An increase of heat flow starts to occur around 125 °C for each cycle which represents the rDA reaction. Similar to cr-FAO and cr-FALO, the thermo-recovering degree of each cycles decrease gradually compared to its previous cycles. The decrease can be explained using same possibilities as for crosslinked fatty esters (i.e., short time for bond reconstitution and formation of most thermodynamically stable adducts isomer) [
6,
7]. In addition, relatively high degree of crosslink on cr-FAJO caused the product to have a brittle nature.
3.3. Crosslinking of Furan-Functionalized Fatty Esters or Jatropha Oil with PK-Furan
The use of furan-functionalized fatty ester or jatropha oil, for partial substitution in well-established PK-Furan thermoreversible polymers, is intended to give improvements to polymer properties and, also, an added value through use of renewable resources. Thermoreversibility of PK-Furan, as reference material, and several poly blends with FAO, FALO, and FAJO, are investigated using DSC (
Table 1). Presented values are the results of normalization with respect to its previous cycle. In addition, for the sake of brevity, only DSC profiles of FAJO-10% in PK-Furan are presented (
Figure 6).
A constant decrease in thermoreversibility can be observed on every sample. The thermos-recovery degree of mixture samples is higher with respect to pure PK-Furan, except for the sample with 10% furan-functionalized linoleate (cr-FALO-10%). This effect can be attributed to the DA adduct bond density of each sample. The samples were mixed based on the percentage of total furan groups reacted. Therefore, the amounts of furan groups per unit weight or volume may differ. For FALO and PK-Furan, the numbers of furan groups per gram are similar (i.e., 4.62 × 10−3 and 4.58 × 10−3 mol/g, while the furan groups content for FAO and FAJO are, respectively, 79% and 68%, compared to PK-Furan. Therefore, the system with higher adduct density will require more energy to decrosslink and, indeed, more time to fully reconstitute. However, it should be highlighted that the DA adducts in PK-Furan samples and furan-functionalized fatty esters have a different fundamental structure. The DA adducts of furan-functionalized fatty esters are similar to crosslinking points in epoxy resin. On the other hand, DA adducts of PK-Furan system eventually form a crosslinked structure of relatively long backbone on alternating polyketone. Therefore, the adduct reconstitution process in PK-Furan will be easier and faster, since the rDA route will leave the polyketone backbone intact. The addition of furan-functionalized compounds will eventually increase the free space on PK-Furan polymer networks. Therefore, heat would penetrate more easily and improve the thermo-recovering efficiency, especially demonstrated by increasing the FAJO content to 20%. On the other hand, the addition of FALO will create a very dense DA adduct network. These dense networks may hinder the reconstitution process from taking place (steric hindrance). Therefore, the cr-FALO-10% sample shows lower efficiency with respect to pure cr-PK-Furan.
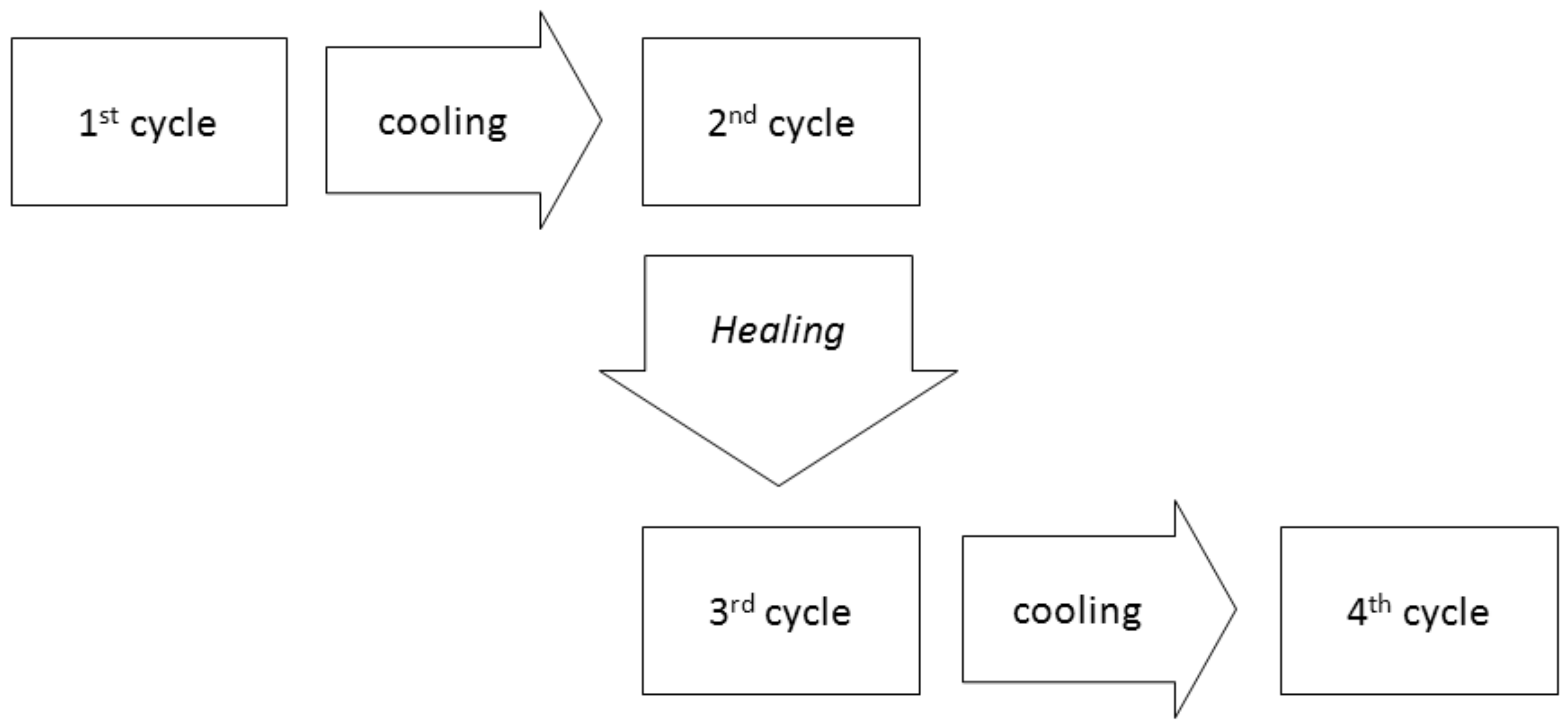
The mechanical properties of the samples were evaluated using DMTA, following the works of Zhang with PK-Furan [
6]. It is found that the second cycle of DMTA measurement of PK-Furan slightly differs from the first one. However, subsequent cycles show no significant differences in the mechanical properties. In addition, after the healing process, the mechanical properties were found to be similar to the first measurement. Therefore, it is possible that the difference may be caused by partial “healing”. To overcome this, we used in our investigation a two-steps measurement strategy. Each step consisted of two cycles of DMTA measurement, and a “healing” process was applied before the second step was conducted.
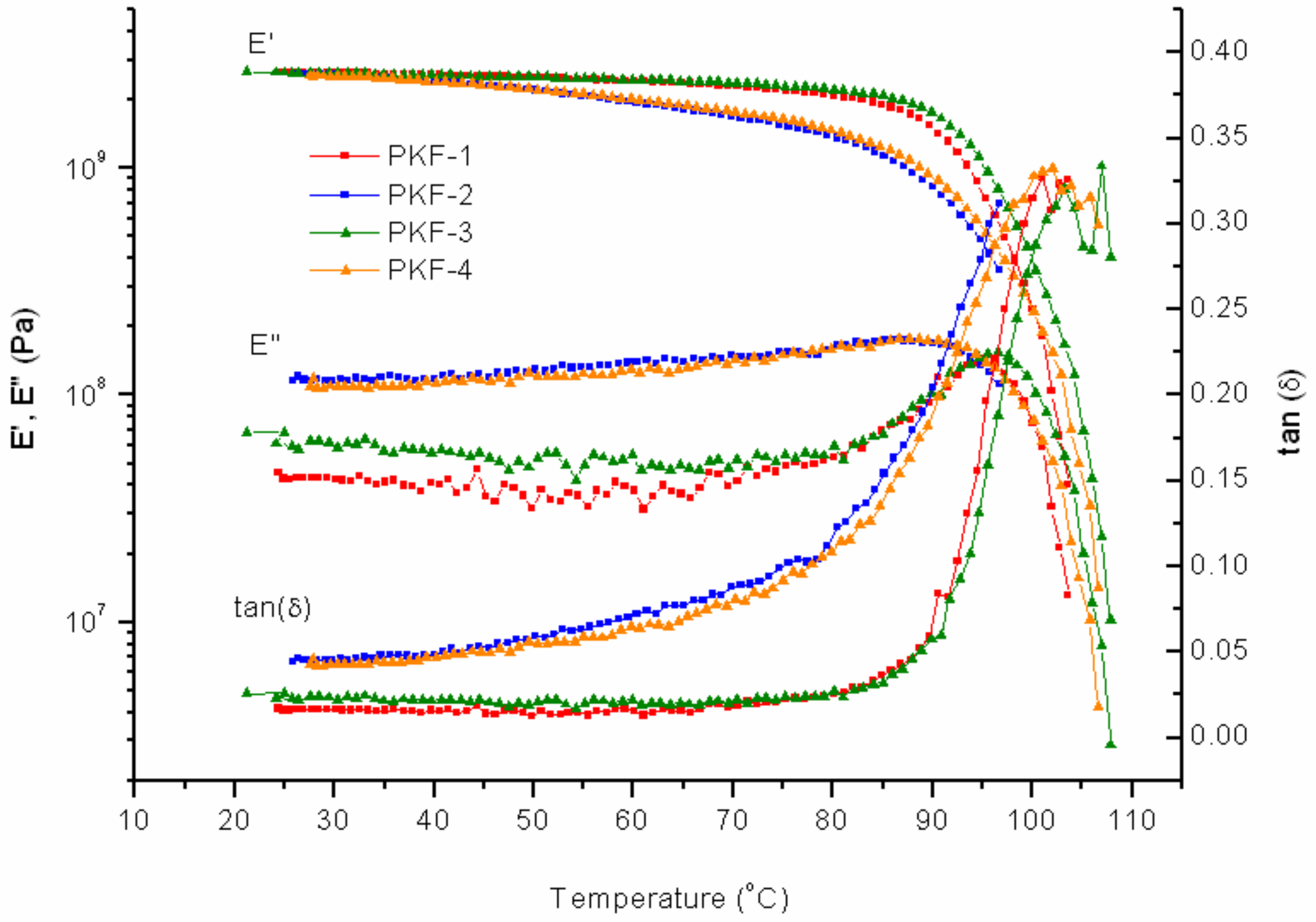
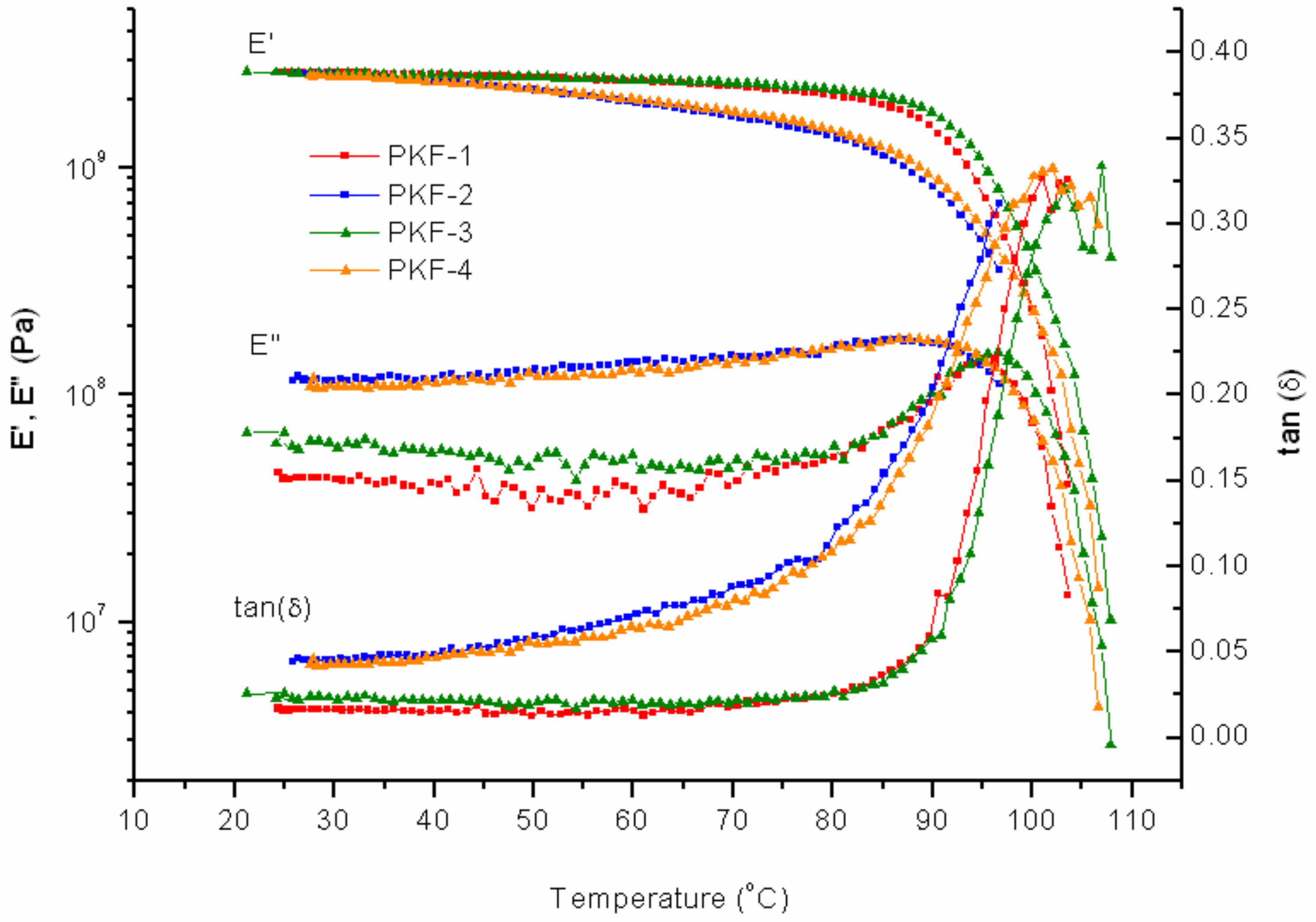
On the first cycle of PK-Furan, the increase in temperature does not affect the elastic storage modulus (
E’’) in a significant way, up to 70 °C (
Figure 7). Due to the high degree of crosslinking (
Im/f = 1), the viscosity effects, which are normally caused by the increase of temperature, are greatly reduced. A temperature increase up to 95 °C, shows an exponential increase of
E’’. Further temperature increase eventually increases the viscous forces, followed by rDA bond ruptures, which reduce the crosslinking and cause deformation. Eventually, the DMTA measures no resistance from the sample with the given strain rate. After the first cycle, the system was cooled down and the measurement was repeated. The initial storage modulus is increased, and its slope is constant. Decrosslinking and deformation effects that occurred in the first cycle become the initial state of the second cycle. The mechanical properties are more dependent on the temperature due to less crosslinking. The same conclusions can be derived from the difference in the curves of tan(δ). The same measurements were repeated after the healing process. Cycles 3 and 4, after the healing process, show similar results compared to cycles 1 and 2, showing a fully healed product (
Figure 7). The results are in agreement with previously reported literature [
5,
6].
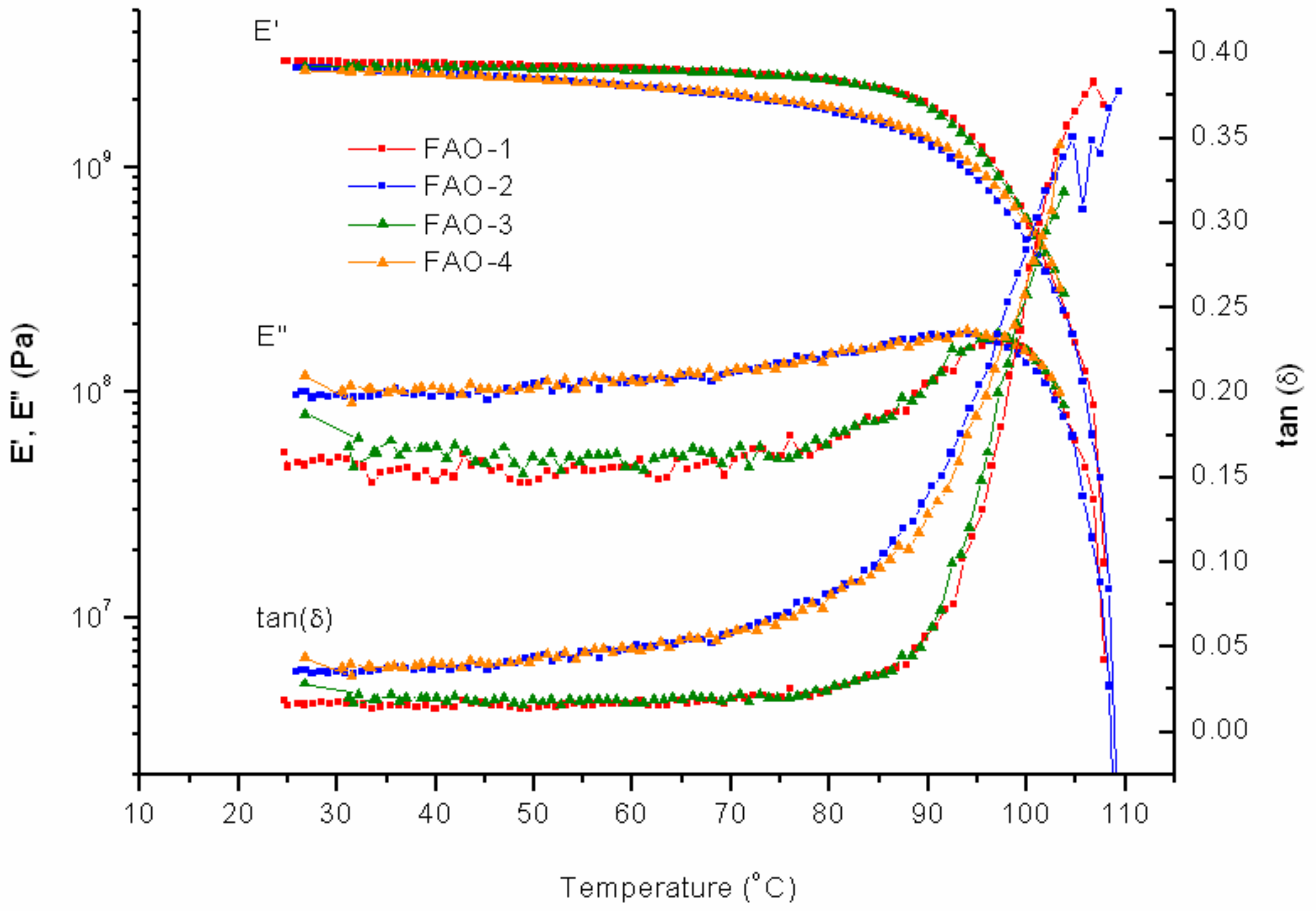
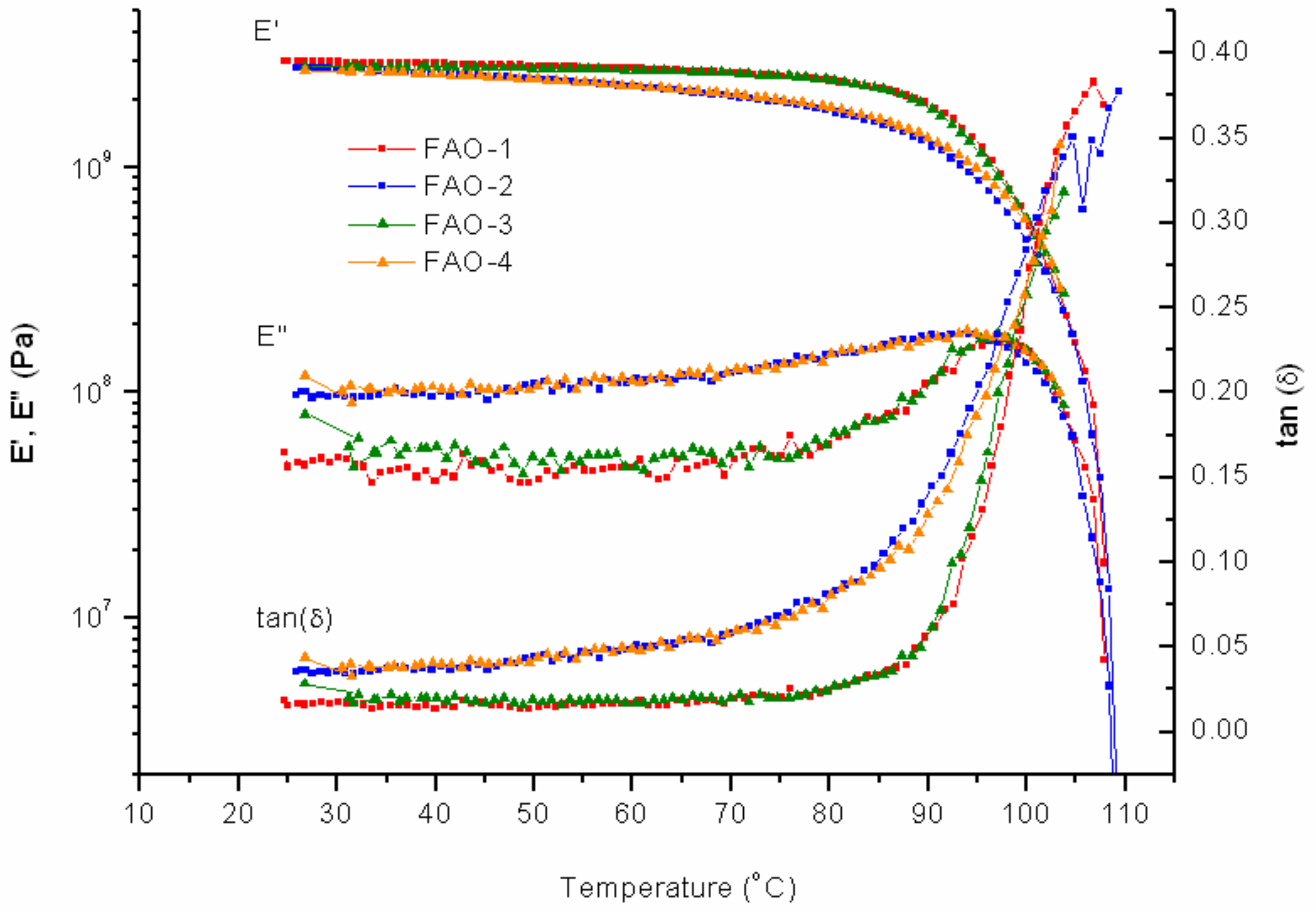
The addition of FAO-10% to PK-Furan does not result in significant changes to the mechanical properties, compared to pure PK-Furan. Measurements after 24 h of healing process also showed full self-healing properties (
Figure 8).
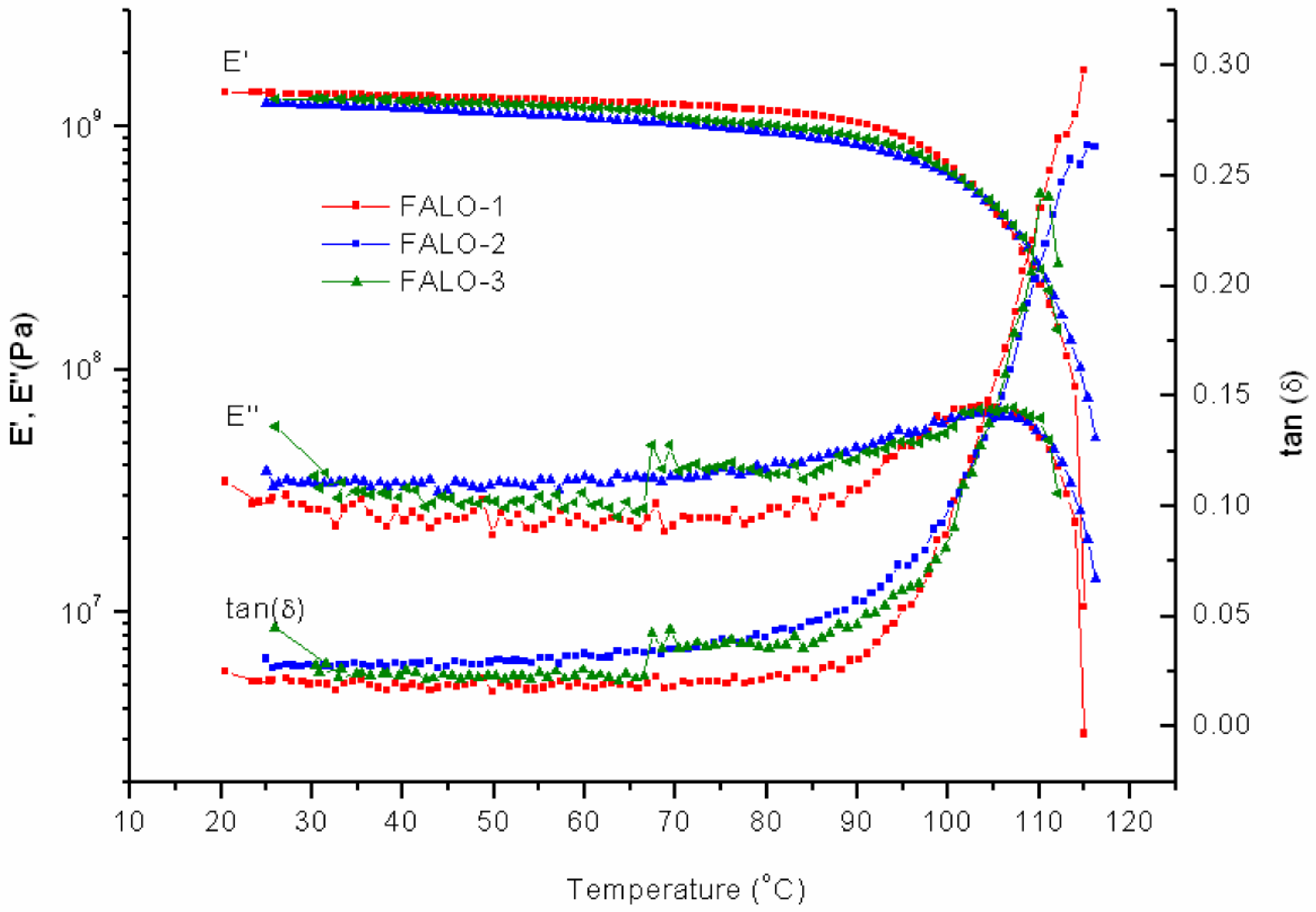
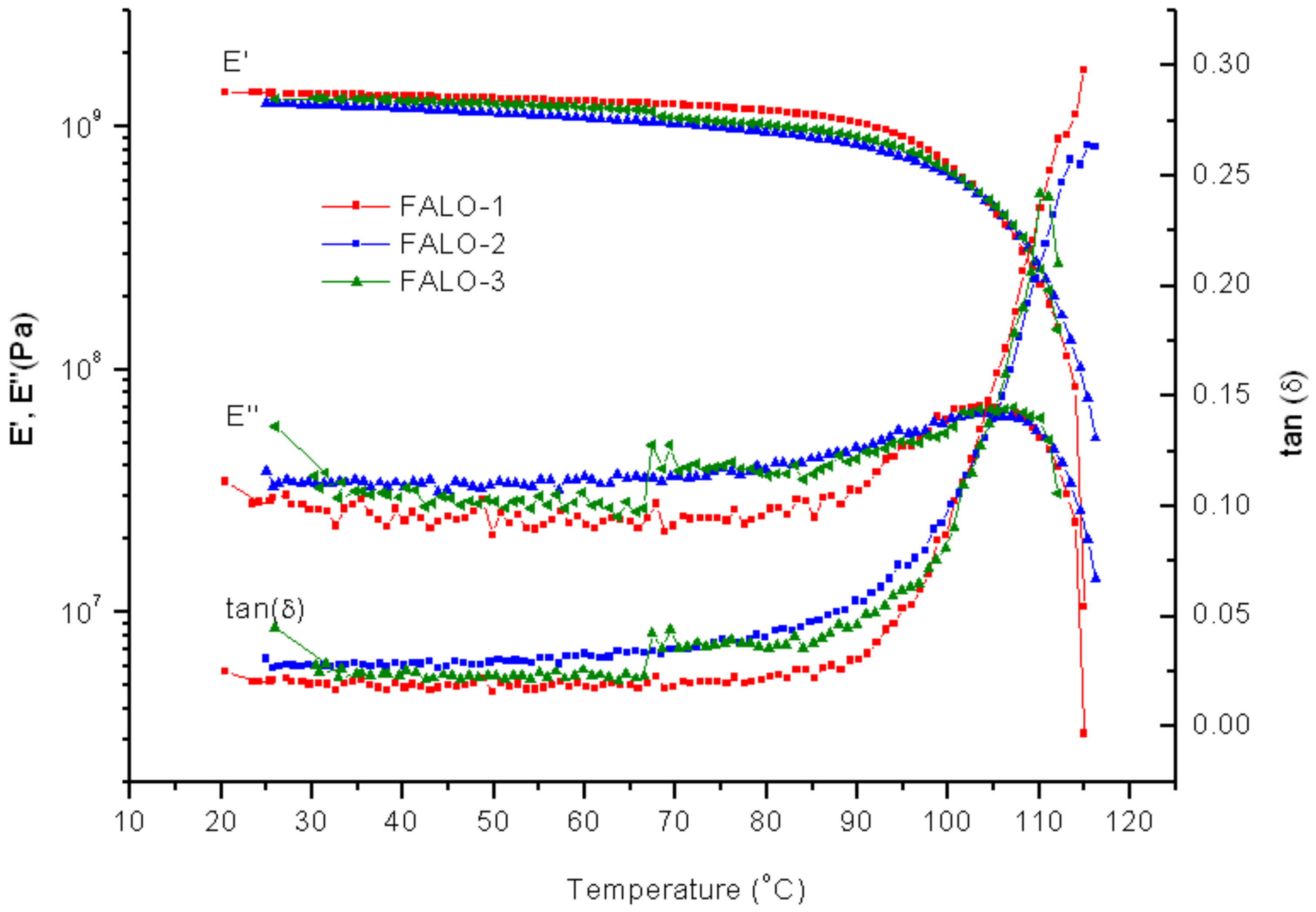
As expected, samples with FALO-10% intake were more fragile compared to any others. The multifunctional furan-functionalized linoleic, resulting in very dense DA adduct, contributes to the brittleness of the networks. From the DMTA measurement, the modulus shows lower values with respect to those of FAO and PK-Furan (
Figure 9). There seems to be a smaller difference between the first and the second cycle. The reduced peak of the storage modulus in the second cycle implies a difference in molecular state. A minor increase of the
E’’ in the second cycle up to 85 °C implies less deformation within the network. Interestingly, a difference with PK-Furan is found when looking at the effect of temperature on the material. The peak of the storage modulus lies higher in temperature than that of PK-Furan, and the decrease in strength for both the loss and the storage modulus shows a less steep slope for FALO, compared to PK-Furan. The result shows that FALO-10% exceeds the PK-Furan in decrosslinking temperature. The third cycles gave an increase to the storage modulus at 67 °C, even if the effect seems small on the loss modulus. The storage modulus peak was improved compared to cycle 2, showing some healing of the product. However, the sample could not regain its initial mechanical properties. Consequently, none of the samples could survive another cycle of measurement.
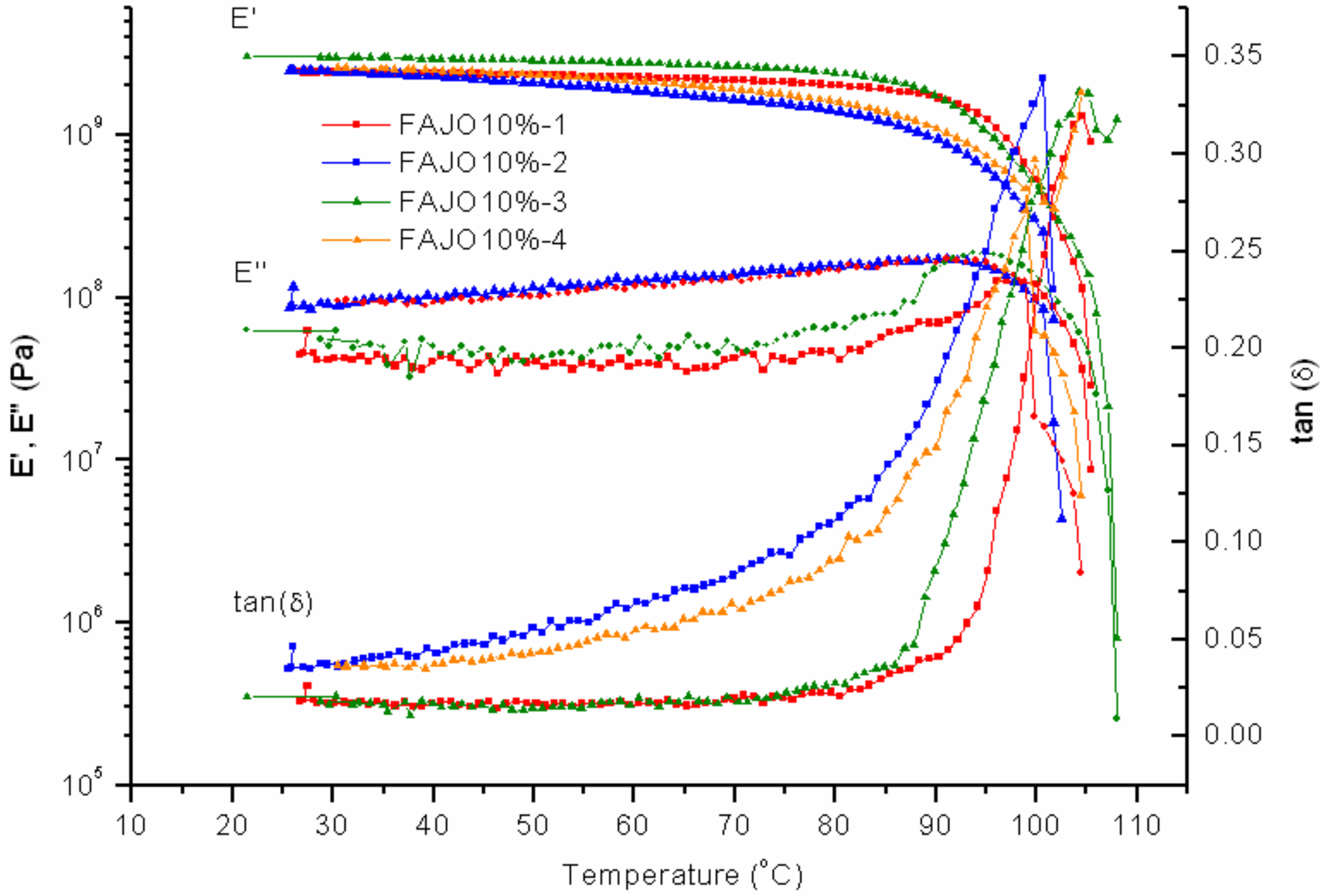
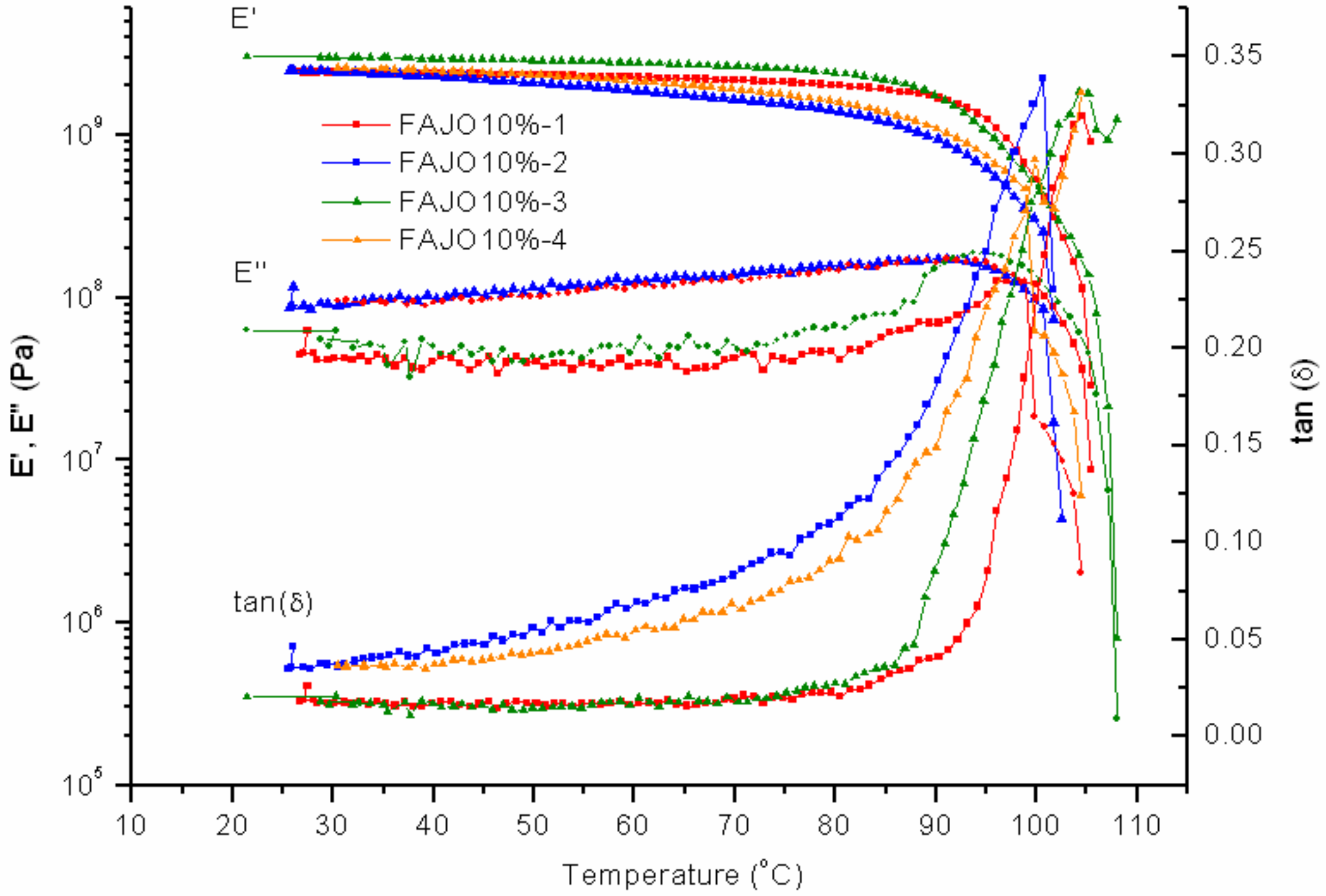
FAJO was mixed with PK-Furan in two different fractions, 10% and 20%. Compared to PK-Furan, the results of FAJO-10% yield no large differences before healing (
Figure 10). A difference can be observed in the storage modulus between the first cycle and third cycle. However, the differences were not further observed after the fourth cycles.
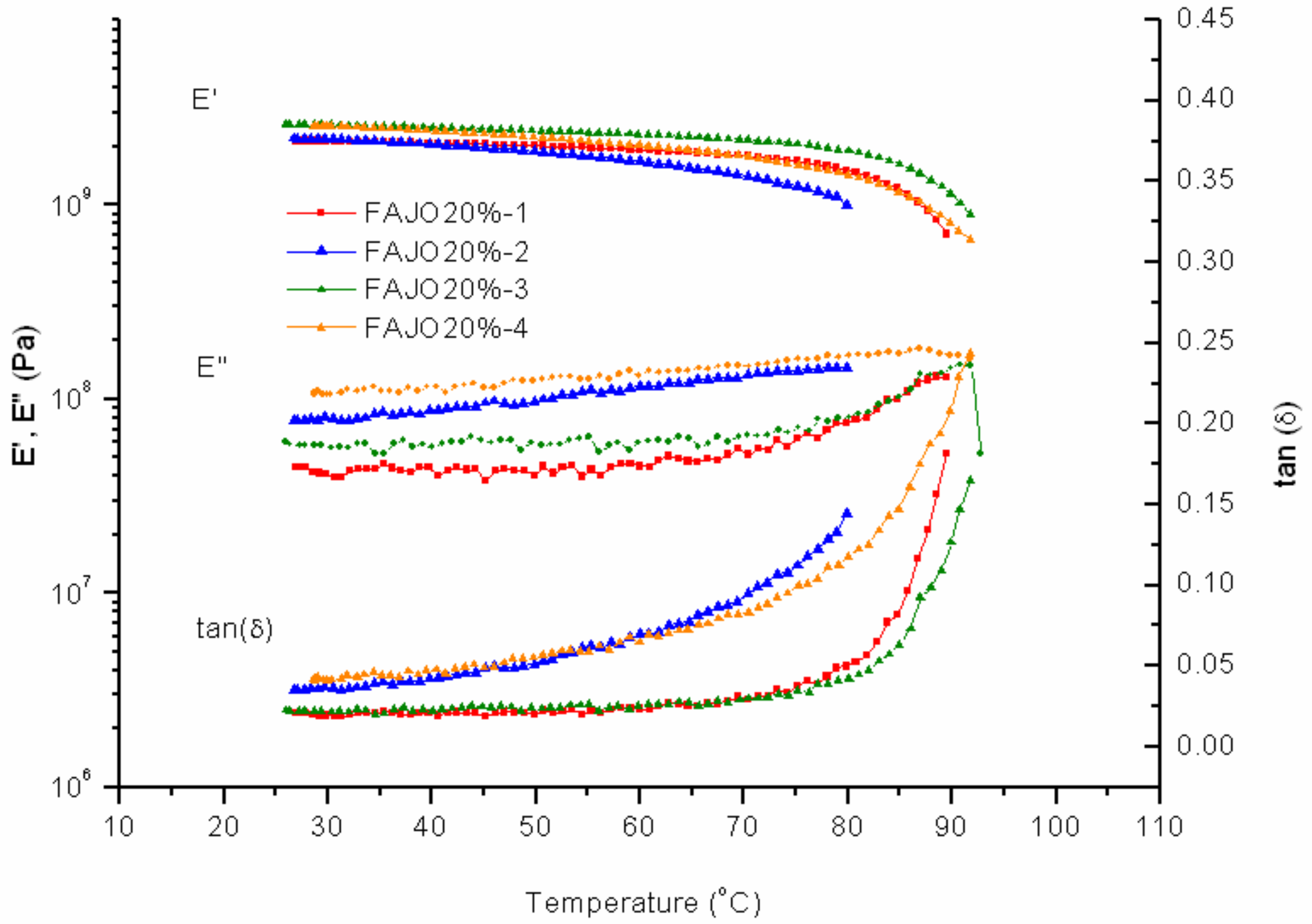
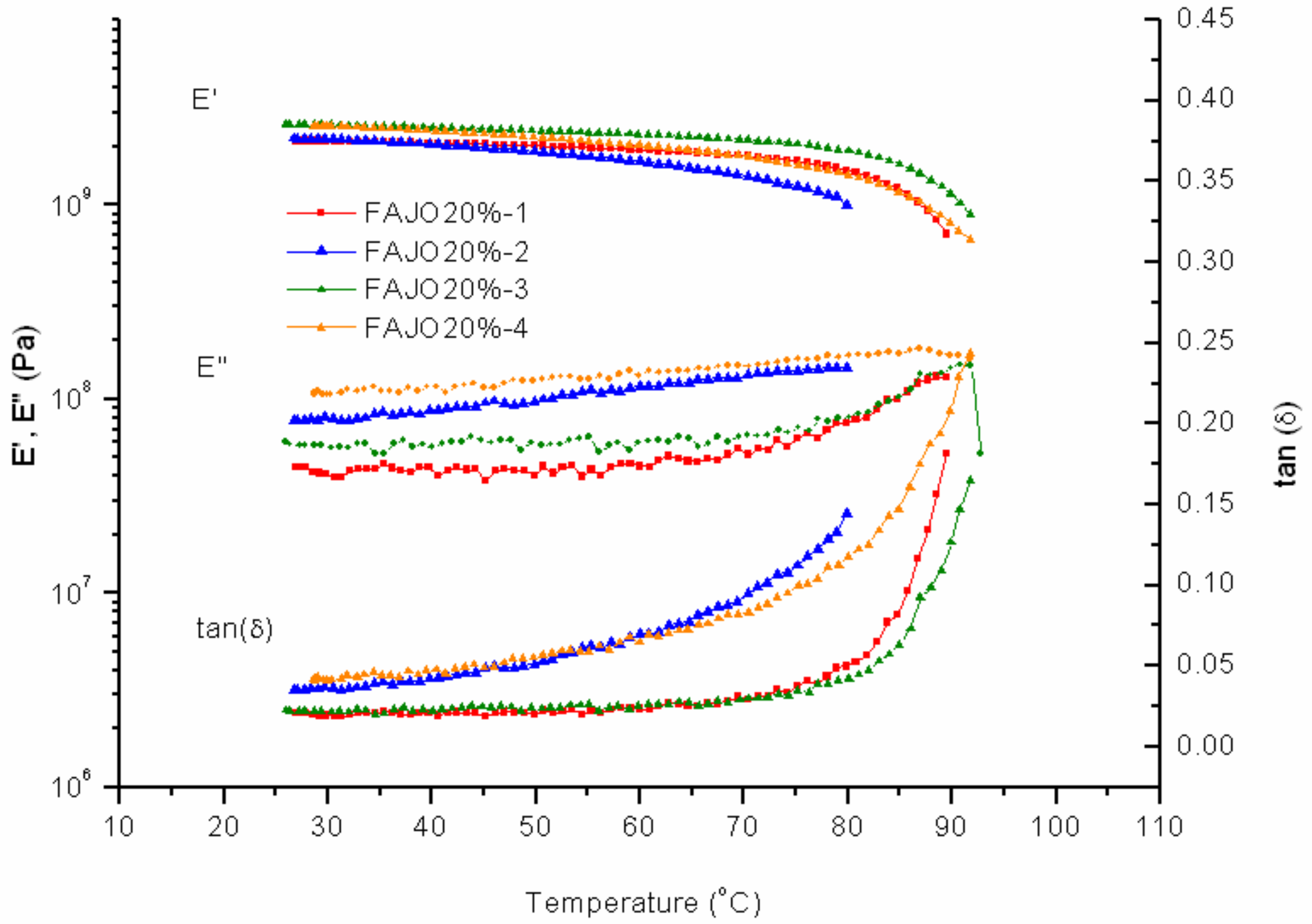
Increasing the amount of FAJO up to 20% gives a more resulting fragile material. At low temperature measurement of the first two cycles, there is no significant resistance on the sample (
Figure 11). The first cycle shows a peak for the storage modulus at 90 °C, whereas PK-Furan showed that peak at 95 °C. The second cycle shows the sample’s initial state has changed. The third cycle shows similarity with the initial mechanical properties (first cycle). The fourth cycle shows a similar profile to the second cycle with higher measured temperature (i.e., up to 90 °C). The intake of 10% FAJO in PK-Furan does not significantly change the mechanical properties with respect to pure PK-Furan. This result is already considered as an improvement in terms of the (partial) use of renewable resources. However, higher FAJO intake (i.e., 20%) does not lead to better mechanical properties (i.e., brittle material).

 ,
, 



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}