High Temperature, Living Polymerization of Ethylene by a Sterically-Demanding Nickel(II) α-Diimine Catalyst



Abstract
:1. Introduction
2. Materials and Methods
3. Results and Discussion
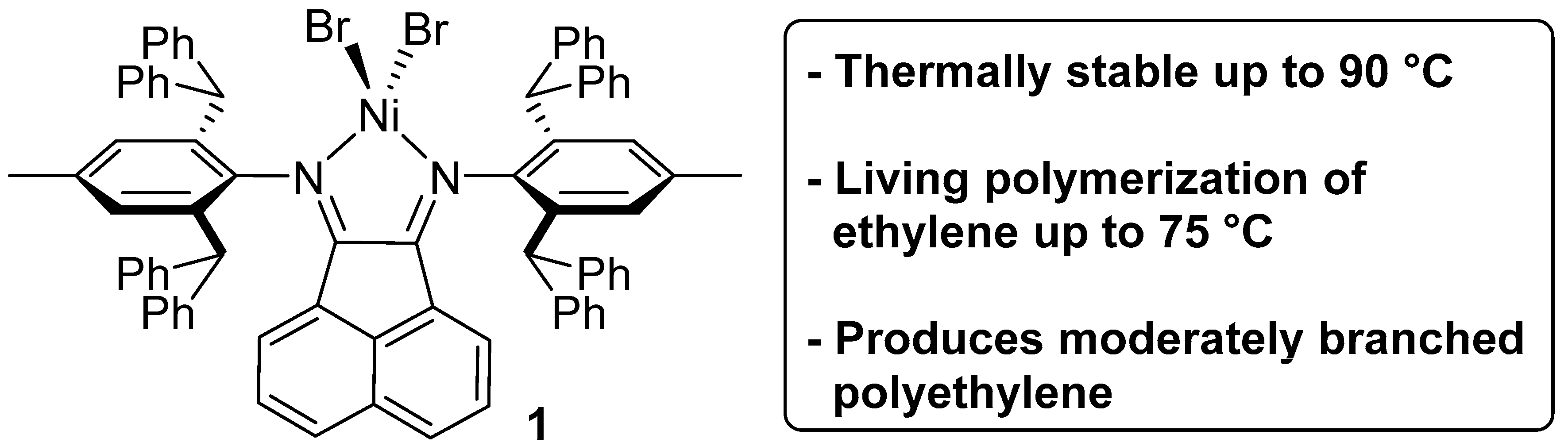
3.1. Synthesis of High Purity Catalyst 1
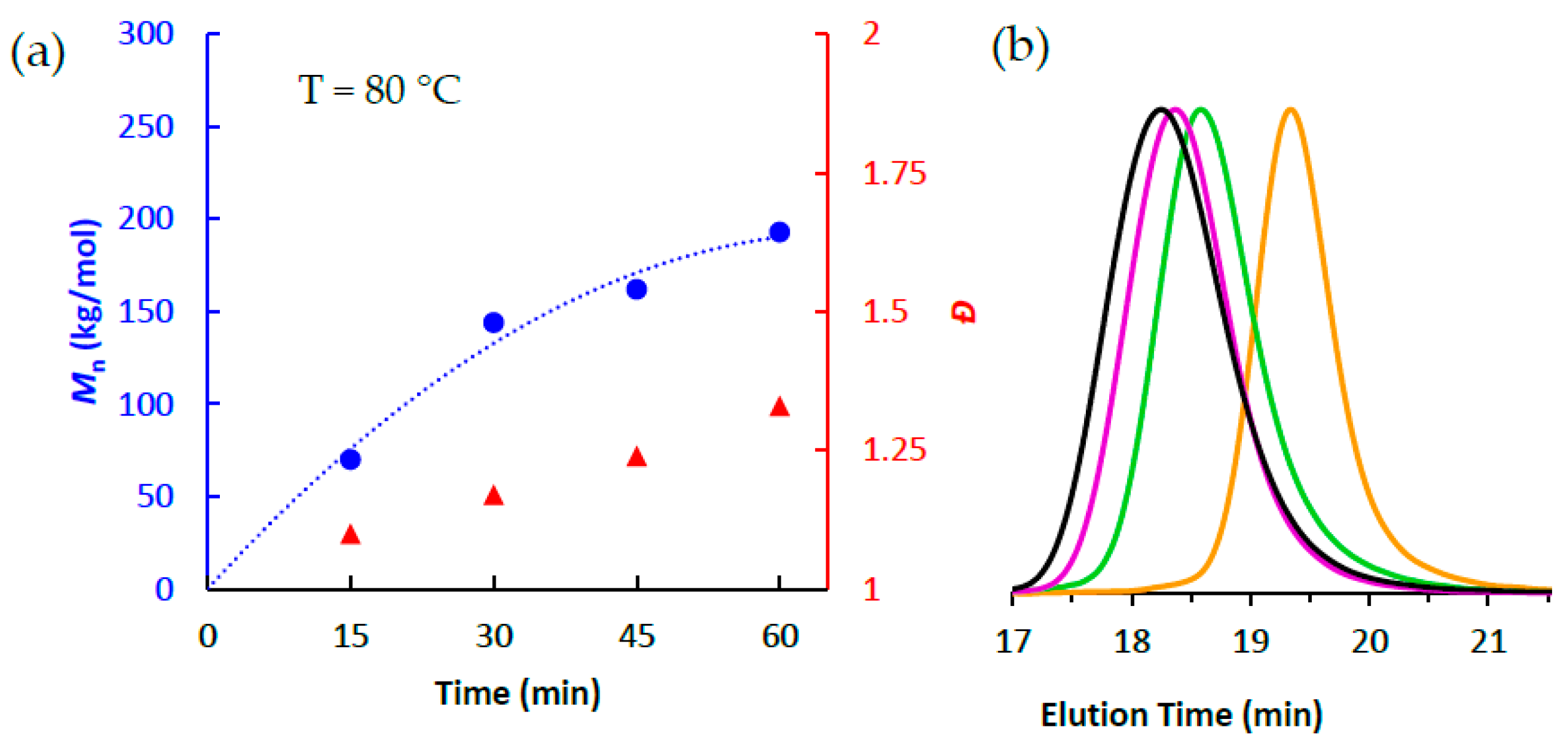
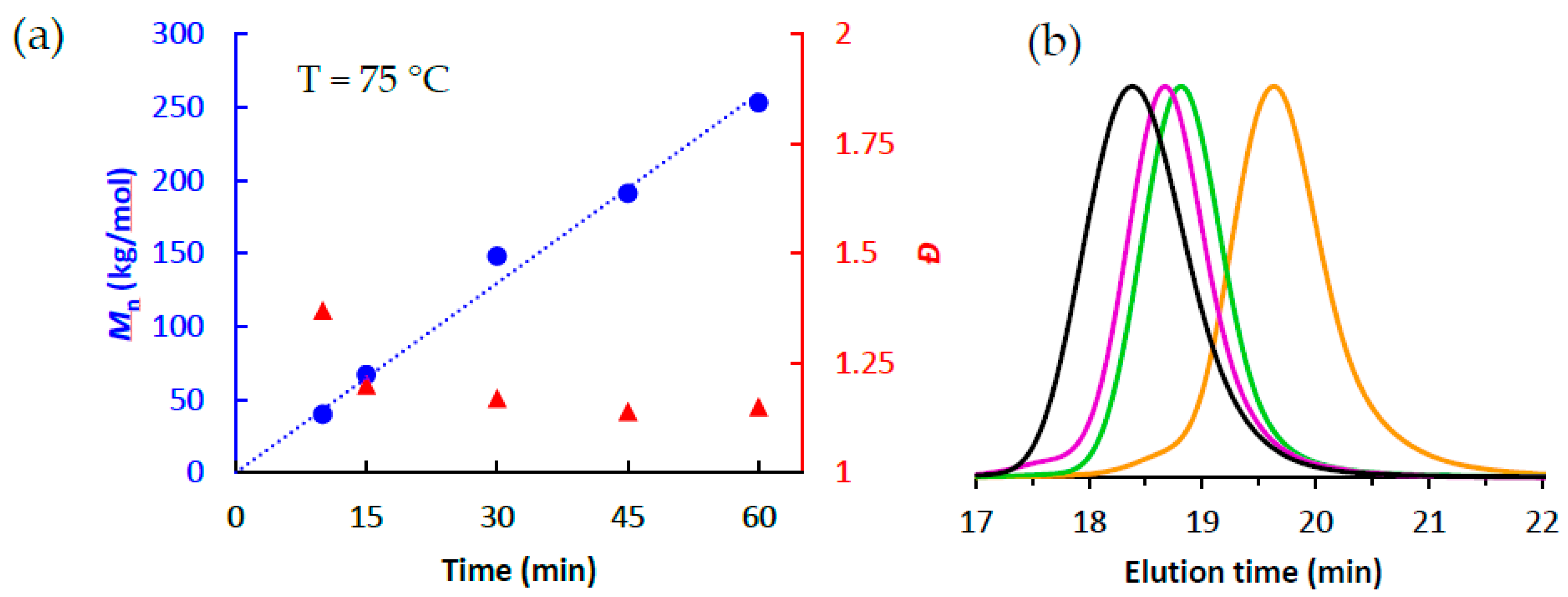
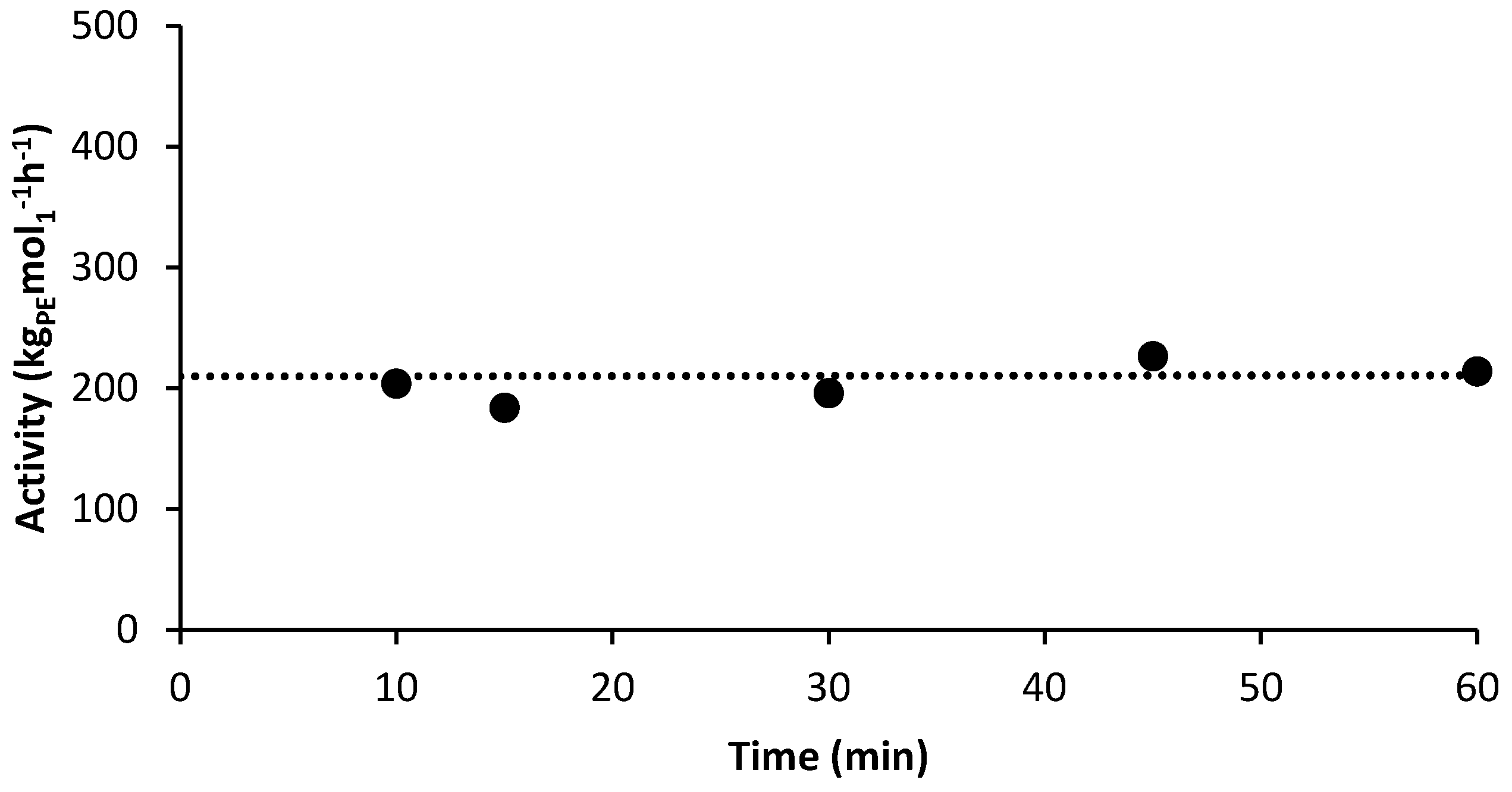
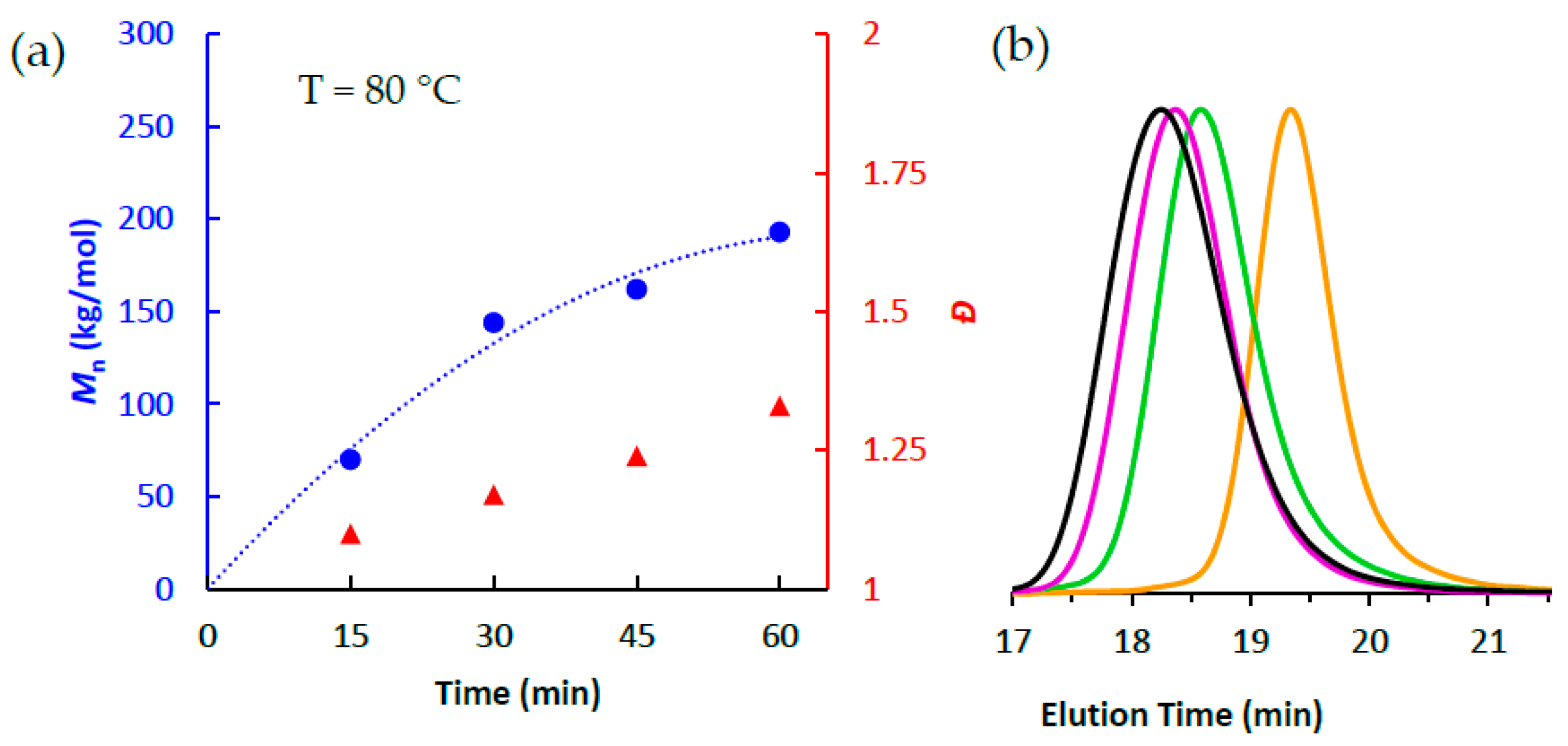
3.2. Ethlene Polymerizations using High-Purity 1/PMAO-IP at Elevated Temperatures
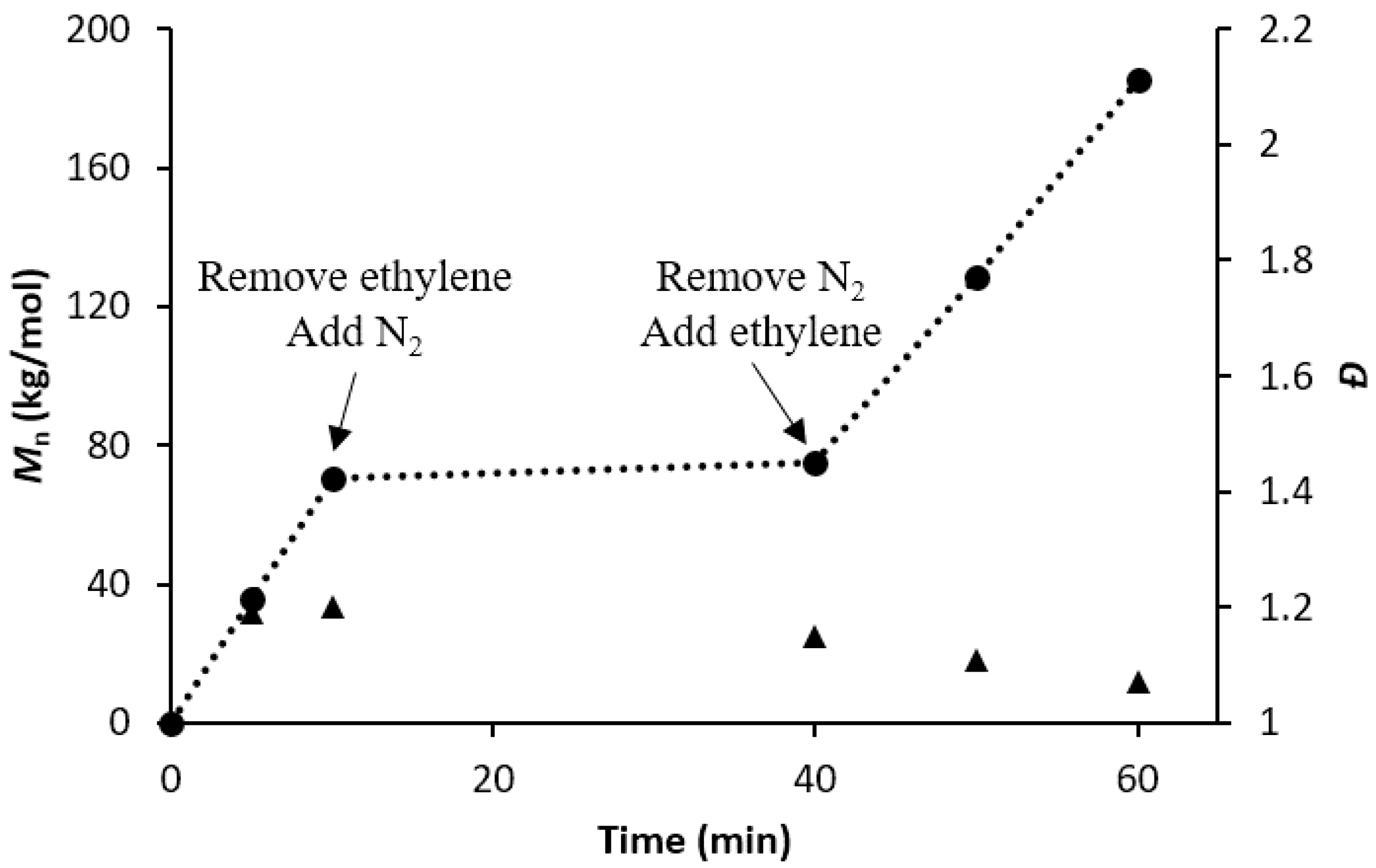
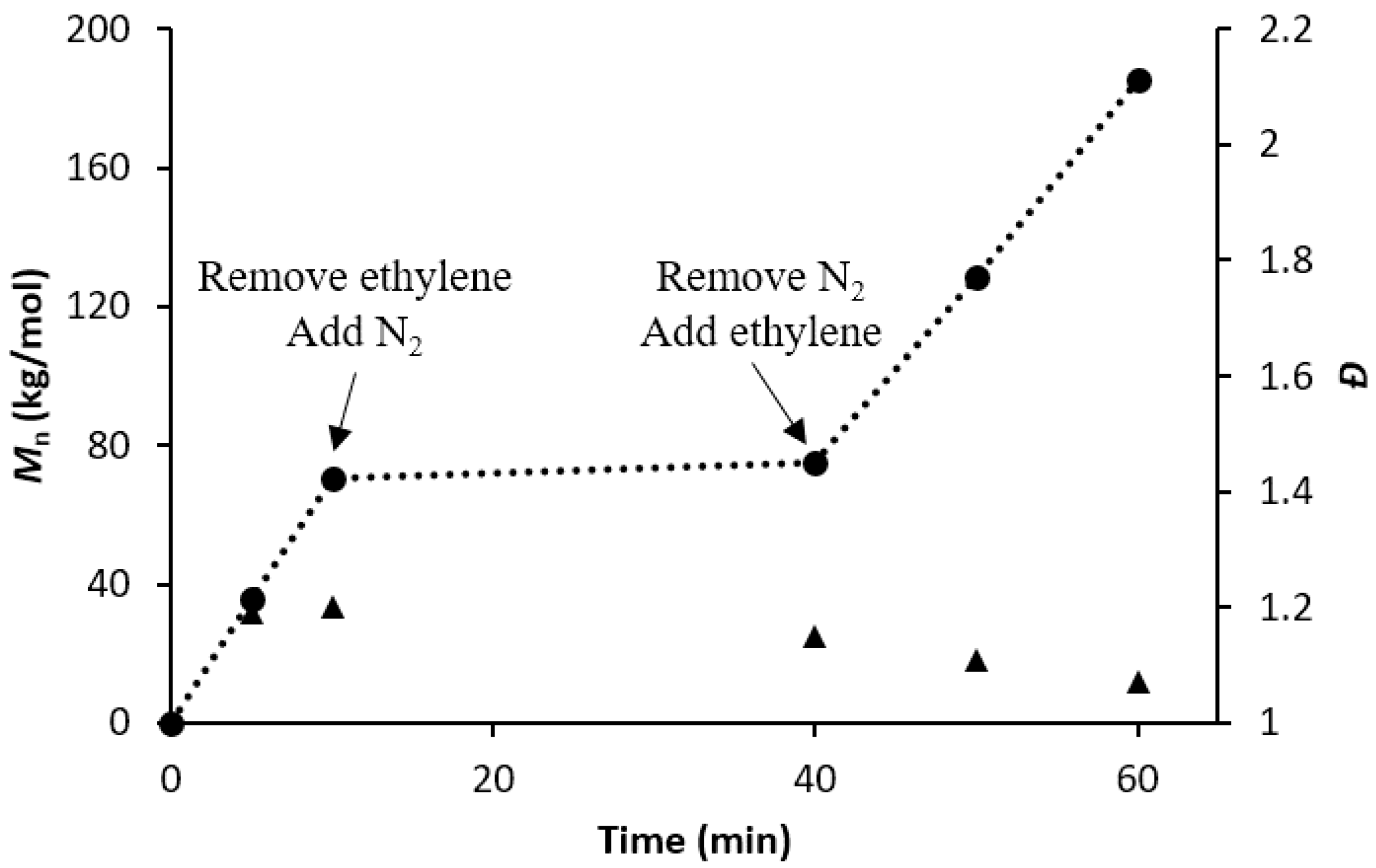
3.3. Evaluating the Livingness of Catalyst 1/PMAO-IP in the Absence of Monomer
3.4. Synthesis of Block Copolyethylenes by 1/PMAO-IP
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Cherian, A.E.; Rose, J.M.; Lobkovsky, E.B.; Coates, G.W. A C2-symmetric, living α-diimine Ni(II) catalyst: Regioblock copolymers from propylene. J. Am. Chem. Soc. 2005, 127, 13770–13771. [Google Scholar] [CrossRef] [PubMed]
- Domski, G.J.; Rose, J.M.; Coates, G.W.; Bolig, A.D.; Brookhart, M. Living alkene polymerization: New methods for the precision synthesis of polyolefins. Prog. Polym. Sci. 2007, 32, 30–92. [Google Scholar] [CrossRef]
- Azoulay, J.D.; Schneider, Y.; Galland, G.B.; Bazan, G.C. Living polymerization of ethylene and α-olefins using a nickel α-keto-β-diimine initiator. Chem. Commun. 2009, 6177–6179. [Google Scholar] [CrossRef] [PubMed]
- Azoulay, J.D.; Bazan, G.C.; Galland, G.B. Microstructural Characterization of Poly(1-hexene) Obtained Using a Nickel α-Keto-β-diimine Initiator. Macromolecules 2010, 43, 2794–2800. [Google Scholar] [CrossRef]
- Ittel, S.D.; Johnson, L.K.; Brookhart, M. Late-metal catalysts for ethylene homo- and copolymerization. Chem. Rev. 2000, 100, 1169–1203. [Google Scholar] [CrossRef] [PubMed]
- Harney, M.B.; Keaton, R.J.; Fettinger, J.C.; Sita, L.R. Living Ziegler-Natta polymerization by early transition metals: Synthesis and evaluation of cationic zirconium alkyl complexes bearing β-hydrogens as models for propagating centers. J. Am. Chem. Soc. 2006, 128, 3420–3432. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Wei, J.; Sita, L.R. Living Coordinative Chain-Transfer Polymerization and Copolymerization of Ethene, α-Olefins, and α,ω-Nonconjugated Dienes Using Dialkylzinc as “Surrogate” Chain-Growth Sites. Macromolecules 2008, 41, 7829–7833. [Google Scholar] [CrossRef]
- Webster, O.W. Living Polymerization Methods. Science 1991, 251, 887–893. [Google Scholar] [CrossRef] [PubMed]
- Killian, C.M.; Tempel, D.J.; Johnson, L.K.; Brookhart, M. Living polymerization of α-olefins using Ni-II-α-diimine catalysts. Synthesis of new block polymers based on α-olefins. J. Am. Chem. Soc. 1996, 118, 11664–11665. [Google Scholar] [CrossRef]
- Coates, G.W.; Hustad, P.D.; Reinartz, S. Reinartz. Catalysts for the living insertion polymerization of alkenes: Access to new polyolefin architectures using Ziegler-Natta chemistry. Angew. Chem. Int. Ed. 2002, 41, 2236–2257. [Google Scholar]
- Reinartz, S.; Mason, A.F.; Lobkovsky, E.B.; Coates, G.W. Titanium Catalysts with Ancillary Phenoxyketimine Ligands for Living Ethylene Polymerization. Organometallics 2003, 22, 2542–2544. [Google Scholar] [CrossRef]
- Makio, H.; Terao, H.; Iwashita, A.; Fujita, T. FI catalysts for olefin polymerization—A comprehensive treatment. Chem. Rev. 2011, 111, 2363–2449. [Google Scholar] [CrossRef] [PubMed]
- Long, B.K.; Eagan, J.M.; Mulzer, M.; Coates, G.W. Semi-Crystalline Polar Polyethylene: Ester-Functionalized Linear Polyolefins Enabled by a Functional-Group-Tolerant, Cationic Nickel Catalyst. Angew. Chem. Int. Ed. 2016, 55, 7106–7110. [Google Scholar] [CrossRef] [PubMed]
- Yu, S.M.; Mecking, S. Extremely narrow-dispersed high molecular weight polyethylene from living polymerization at elevated temperatures with o-F substituted Ti enolatoimines. J. Am. Chem. Soc. 2008, 130, 13204–13205. [Google Scholar] [CrossRef] [PubMed]
- Xie, T.Y.; Mcauley, K.B.; Hsu, J.C.C.; Bacon, D.W. Gas-Phase Ethylene Polymerization—Production Processes, Polymer Properties, and Reactor Modeling. Ind. Eng. Chem. Res. 1994, 33, 449–479. [Google Scholar] [CrossRef]
- Ali, E.M.; Abasaeed, A.E.; Al-Zahrani, S.M. Optimization and control of industrial gas-phase ethylene polymerization reactors. Ind. Eng. Chem. Res. 1998, 37, 3414–3423. [Google Scholar] [CrossRef]
- Camacho, D.H.; Guan, Z. Living Polymerization of α-Olefins at Elevated Temperatures Catalyzed by a Highly Active and Robust Cyclophane-Based Nickel Catalyst. Macromolecules 2005, 38, 2544–2546. [Google Scholar] [CrossRef]
- Zai, S.; Liu, F.; Gao, H.; Li, C.; Zhou, G.; Cheng, S.; Guo, L.; Zhang, L.; Zhu, F.; Wu, Q. Longstanding living polymerization of ethylene: Substituent effect on bridging carbon of 2-pyridinemethanamine nickel catalysts. Chem. Commun. (Camb.) 2010, 46, 4321–4323. [Google Scholar] [CrossRef] [PubMed]
- Gao, H.; Hu, H.; Zhu, F.; Wu, Q. A thermally robust amine-imine nickel catalyst precursor for living polymerization of ethylene above room temperature. Chem. Commun. 2012, 48, 3312–3314. [Google Scholar] [CrossRef] [PubMed]
- Hu, H.; Zhang, L.; Gao, H.; Zhu, F.; Wu, Q. Design of Thermally Stable Amine–Imine Nickel Catalyst Precursors for Living Polymerization of Ethylene: Effect of Ligand Substituents on Catalytic Behavior and Polymer Properties. Chem. Eur. J. 2014, 20, 3225–3233. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Chen, D.; Wu, H.; Xiao, Z.; Gao, H.; Zhu, F.; Wu, Q. Polymerization of α-Olefins Using a Camphyl α-Diimine Nickel Catalyst at Elevated Temperature. Macromolecules 2014, 47, 3325–3331. [Google Scholar] [CrossRef]
- Hu, H.; Chen, D.; Gao, H.; Zhong, L.; Wu, Q. Amine-imine palladium catalysts for living polymerization of ethylene and copolymerization of ethylene with methyl acrylate: Incorporation of acrylate units into the main chain and branch end. Polym. Chem. 2016, 7, 529–537. [Google Scholar] [CrossRef]
- Rhinehart, J.L.; Mitchell, N.E.; Long, B.K. Enhancing α-Diimine Catalysts for High-Temperature Ethylene Polymerization. ACS Catal. 2014, 4, 2501–2504. [Google Scholar] [CrossRef]
- Hicks, F.A.; Jenkins, J.C.; Brookhart, M. Synthesis and ethylene polymerization activity of a series of 2-anilinotropone-based neutral nickel(II) catalysts. Organometallics 2003, 22, 3533–3545. [Google Scholar] [CrossRef]
- Rhinehart, J.L.; Brown, L.A.; Long, B.K. A Robust Ni(II) α-Diimine Catalyst for High Temperature Ethylene Polymerization. J. Am. Chem. Soc. 2013, 135, 16316–16319. [Google Scholar] [CrossRef] [PubMed]
- Dai, S.; Chen, C. Direct Synthesis of Functionalized High-Molecular-Weight Polyethylene by Copolymerization of Ethylene with Polar Monomers. Angew. Chem. Int. Ed. 2016, 55, 13281–13285. [Google Scholar] [CrossRef] [PubMed]
- Dai, S.; Sui, X.; Chen, C. Highly Robust Palladium(II) α-Diimine Catalysts for Slow-Chain-Walking Polymerization of Ethylene and Copolymerization with Methyl Acrylate. Angew. Chem. Int. Ed. 2015, 54, 9948–9953. [Google Scholar] [CrossRef] [PubMed]
- Guo, L.; Dai, S.; Sui, X.; Chen, C. Palladium and Nickel Catalyzed Chain Walking Olefin Polymerization and Copolymerization. ACS Catal. 2016, 6, 428–441. [Google Scholar] [CrossRef]
- Fan, L.; Yue, E.; Du, S.; Guo, C.Y.; Hao, X.; Sun, W.H. Enhancing thermo-stability to ethylene polymerization: Synthesis, characterization and the catalytic behavior of 1-(2,4-dibenzhydryl-6-chlorophenylimino)-2-aryliminoacenaphthylnickel halides. RSC Adv. 2015, 5, 93274–93282. [Google Scholar] [CrossRef]
- Guan, Z.B.; Cotts, P.M.; McCord, E.F.; McLain, S.J. Chain walking: A new strategy to control polymer topology. Science 1999, 283, 2059–2062. [Google Scholar] [CrossRef] [PubMed]
- Gates, D.P.; Svejda, S.K.; Onate, E.; Killian, C.M.; Johnson, L.K.; White, P.S.; Brookhart, M. Synthesis of branched polyethylene using (α-diimine)nickel(II) catalysts: Influence of temperature, ethylene pressure, and ligand structure on polymer properties. Macromolecules 2000, 33, 2320–2334. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Entry | Time (min) | Trxn (°C) | Yield (g) | TOF b | Mn c (kg/mol) | Đ c | B d |
|---|---|---|---|---|---|---|---|
| 1 | 10 | 70 | 0.16 | 6900 | 44 | 1.35 | 47 |
| 2 | 15 | 70 | 0.22 | 6300 | 50 | 1.22 | 48 |
| 3 | 30 | 70 | 0.49 | 7000 | 128 | 1.14 | 48 |
| 4 | 45 | 70 | 0.77 | 7300 | 164 | 1.18 | 47 |
| 5 | 60 | 70 | 0.96 | 6900 | 244 | 1.17 | 48 |
| 6 | 10 | 75 | 0.17 | 7300 | 40 | 1.37 | 47 |
| 7 | 15 | 75 | 0.23 | 6600 | 67 | 1.20 | 49 |
| 8 | 30 | 75 | 0.49 | 7000 | 155 | 1.17 | 47 |
| 9 | 45 | 75 | 0.85 | 8100 | 191 | 1.14 | 50 |
| 10 | 60 | 75 | 1.07 | 7600 | 253 | 1.15 | 46 |
| 11 | 15 | 80 | 0.30 | 8600 | 70 | 1.10 | 48 |
| 12 | 30 | 80 | 0.61 | 8700 | 114 | 1.17 | 49 |
| 13 | 45 | 80 | 0.72 | 6900 | 162 | 1.24 | 50 |
| 14 | 60 | 80 | 0.93 | 6600 | 193 | 1.33 | 45 |
| Entry | t1 (min)/T1 (°C) | t2 (min)/T2 (°C) | t3 (min)/T3 (°C) | Mn b, Tot (kg/mol) | Đ b | Tm c (°C) | E d (MPa) |
|---|---|---|---|---|---|---|---|
| 1 | 25/−40 | - | - | 52 | 1.22 | 128.5 | 120.5 ± 3.8 |
| 2 | 45/75 | - | - | 191 | 1.14 | 62.4 | 13.5 ± 5.9 |
| 3 | 25/−40 | 45/75 | - | 279 | 1.26 | 61.3, 113.5 | 40.4 ± 3.0 |
| 4 | 25/−40 | 45/75 | 25/−40 | 352 | 1.23 | 66.2, 111.0 | 40.7 ± 8.2 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Brown, L.A.; Anderson, W.C.; Mitchell, N.E.; Gmernicki, K.R.; Long, B.K. High Temperature, Living Polymerization of Ethylene by a Sterically-Demanding Nickel(II) α-Diimine Catalyst. Polymers 2018, 10, 41. https://doi.org/10.3390/polym10010041
Brown LA, Anderson WC, Mitchell NE, Gmernicki KR, Long BK. High Temperature, Living Polymerization of Ethylene by a Sterically-Demanding Nickel(II) α-Diimine Catalyst. Polymers. 2018; 10(1):41. https://doi.org/10.3390/polym10010041
Chicago/Turabian StyleBrown, Lauren A., W. Curtis Anderson, Nolan E. Mitchell, Kevin R. Gmernicki, and Brian K. Long. 2018. "High Temperature, Living Polymerization of Ethylene by a Sterically-Demanding Nickel(II) α-Diimine Catalyst" Polymers 10, no. 1: 41. https://doi.org/10.3390/polym10010041
APA StyleBrown, L. A., Anderson, W. C., Mitchell, N. E., Gmernicki, K. R., & Long, B. K. (2018). High Temperature, Living Polymerization of Ethylene by a Sterically-Demanding Nickel(II) α-Diimine Catalyst. Polymers, 10(1), 41. https://doi.org/10.3390/polym10010041