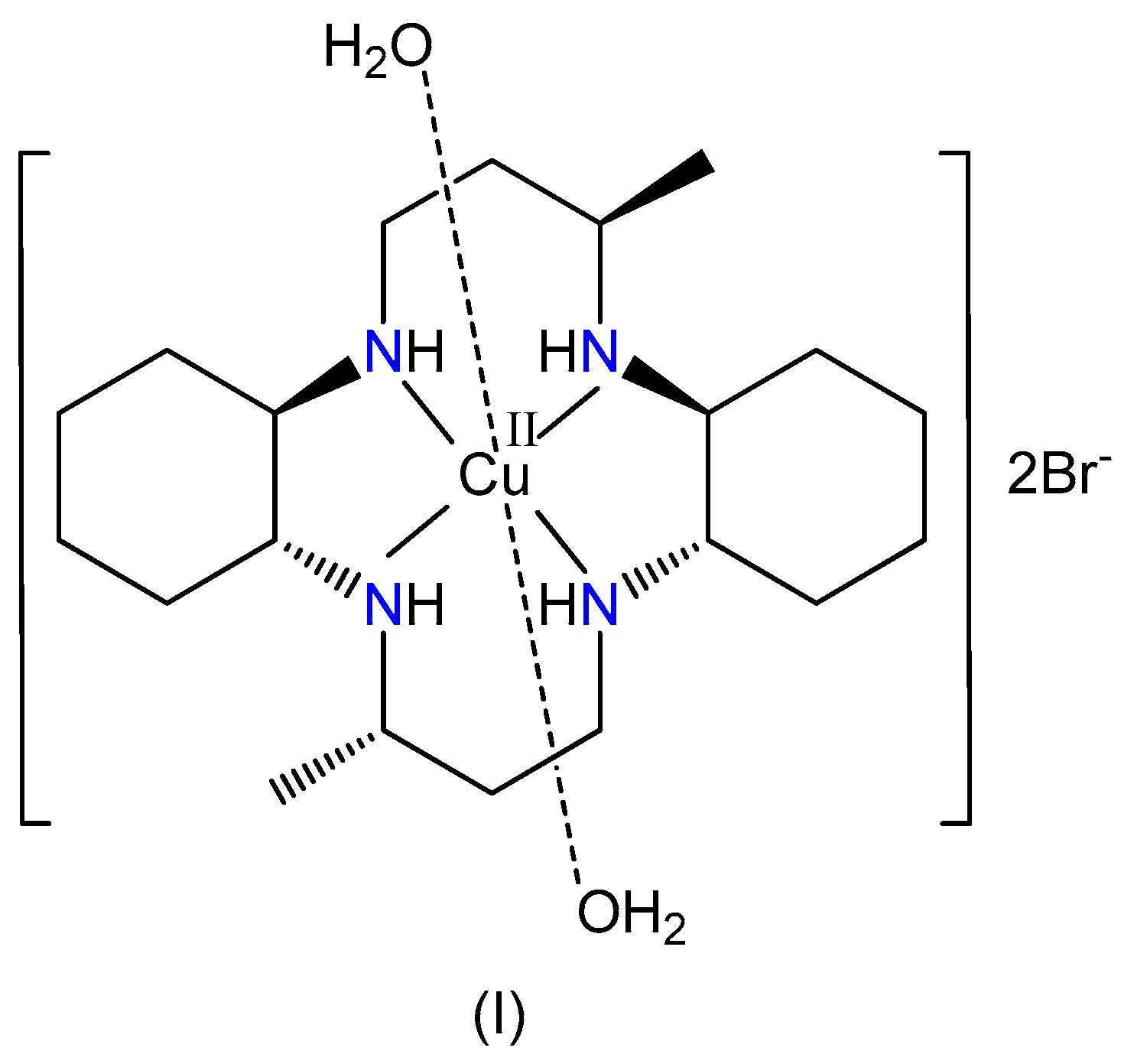
Synthesis, Crystal Structure, Spectroscopic Properties, and Hirshfeld Surface Analysis of Diaqua [3,14-dimethyl-2,6,13,17 tetraazatricyclo(16.4.0.07,12)docosane]copper(II) Dibromide



Abstract
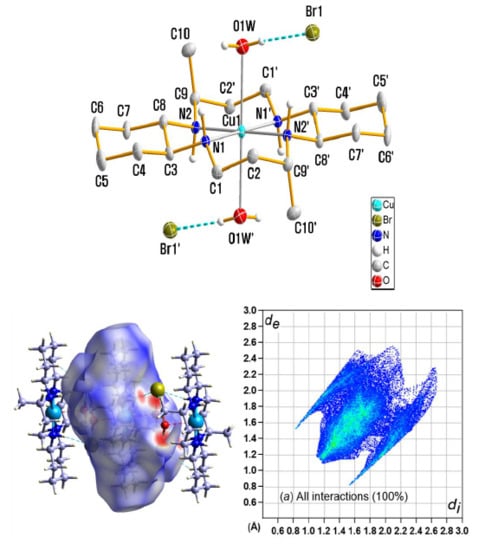
:
1. Introduction
2. Experimental Section
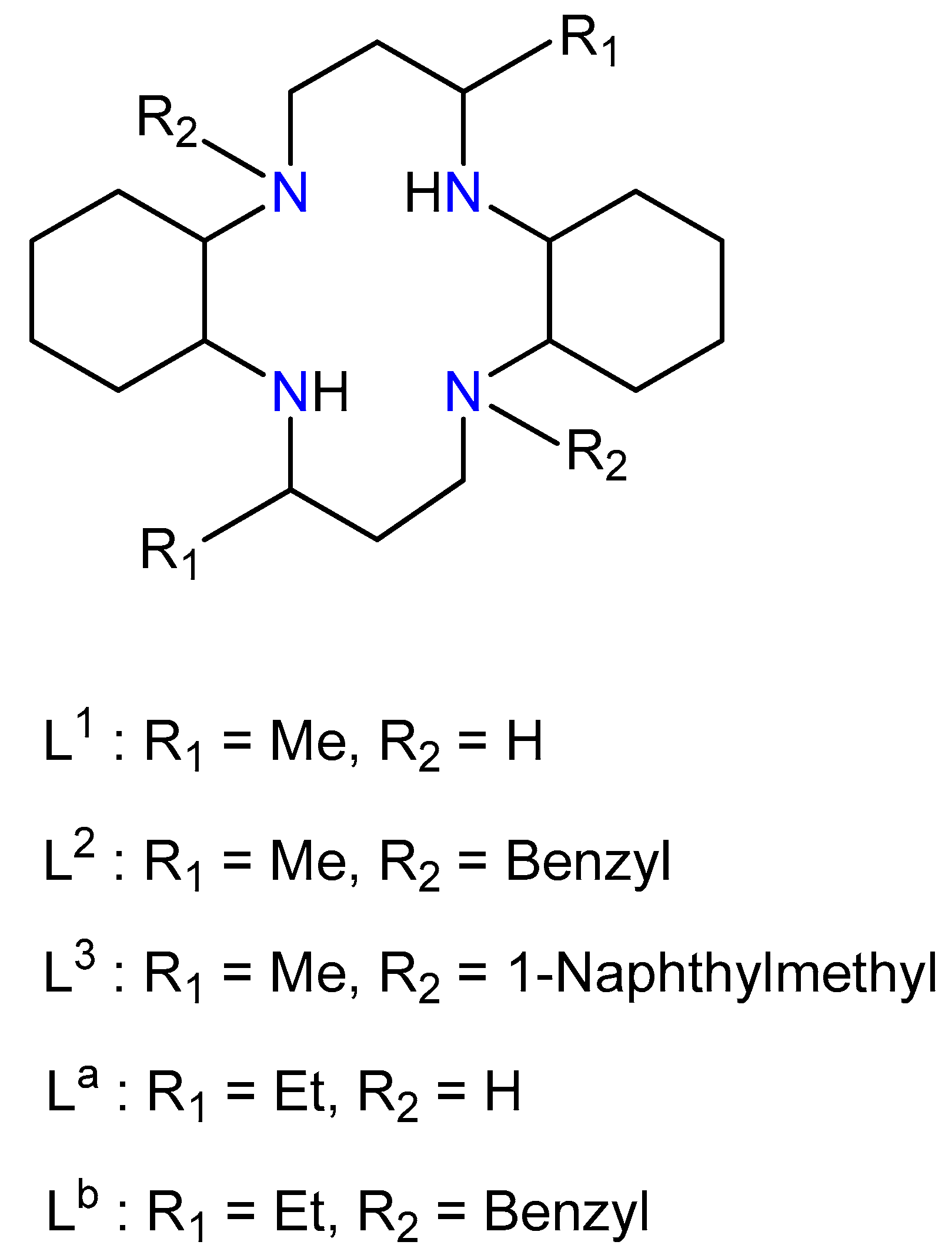
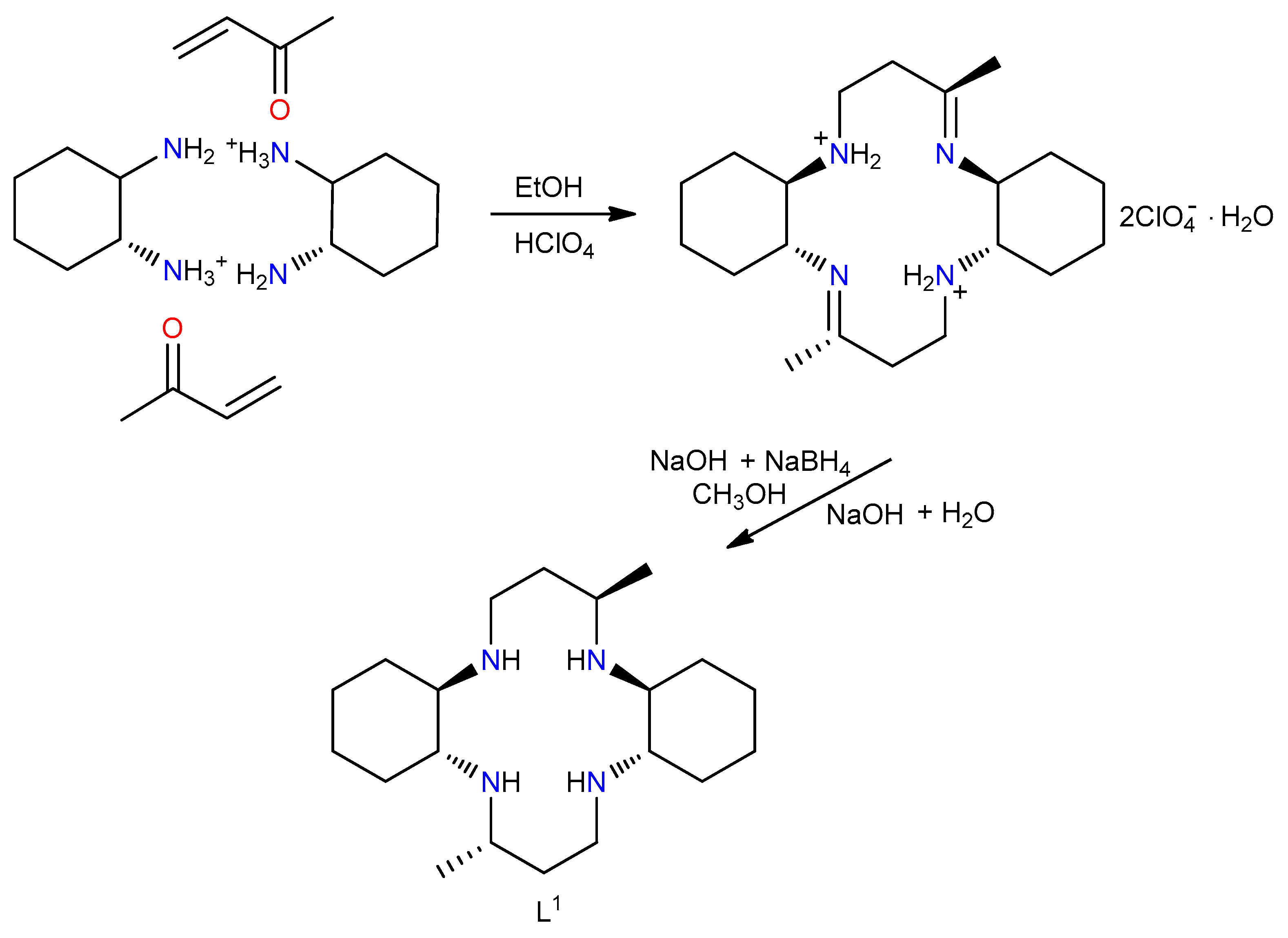
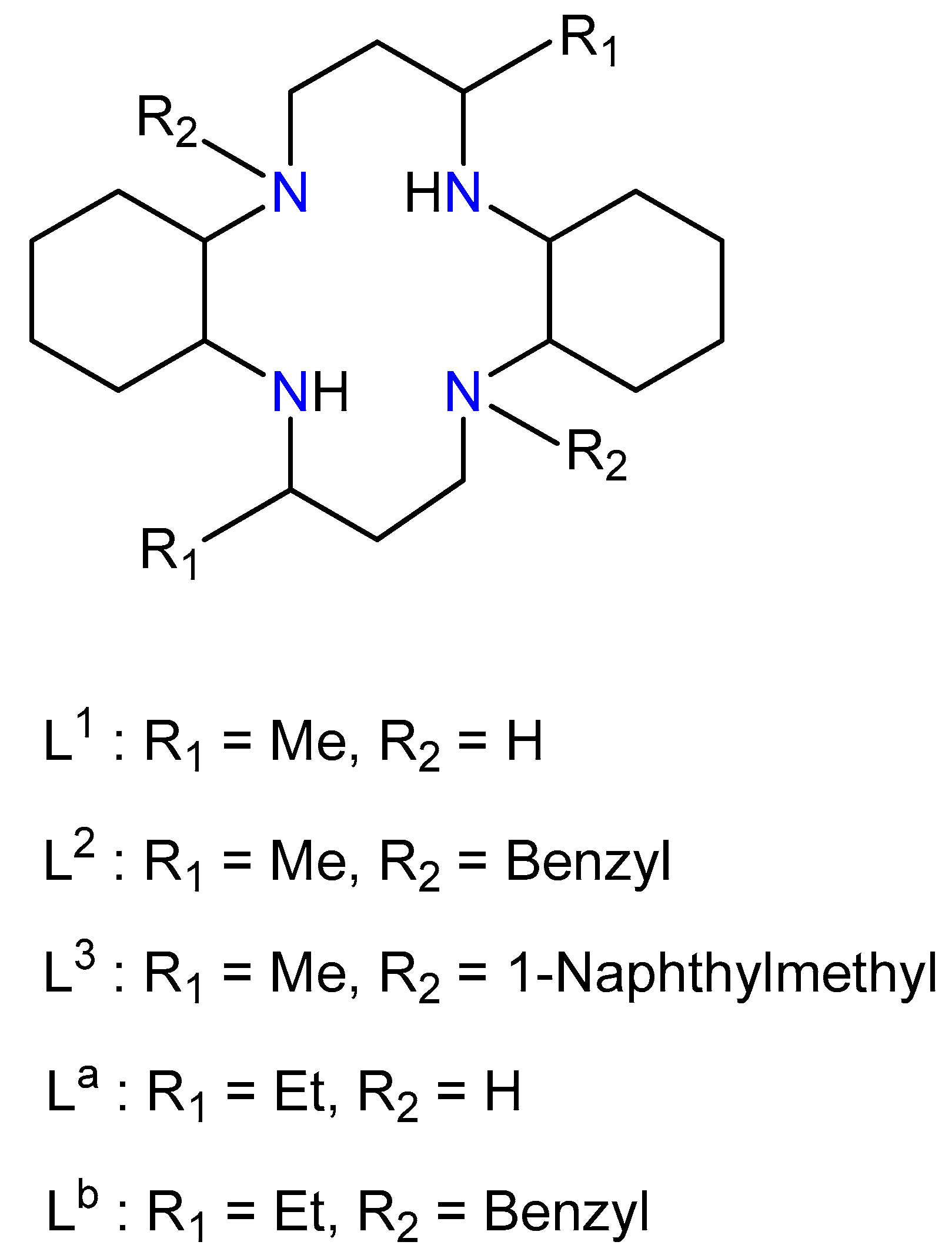
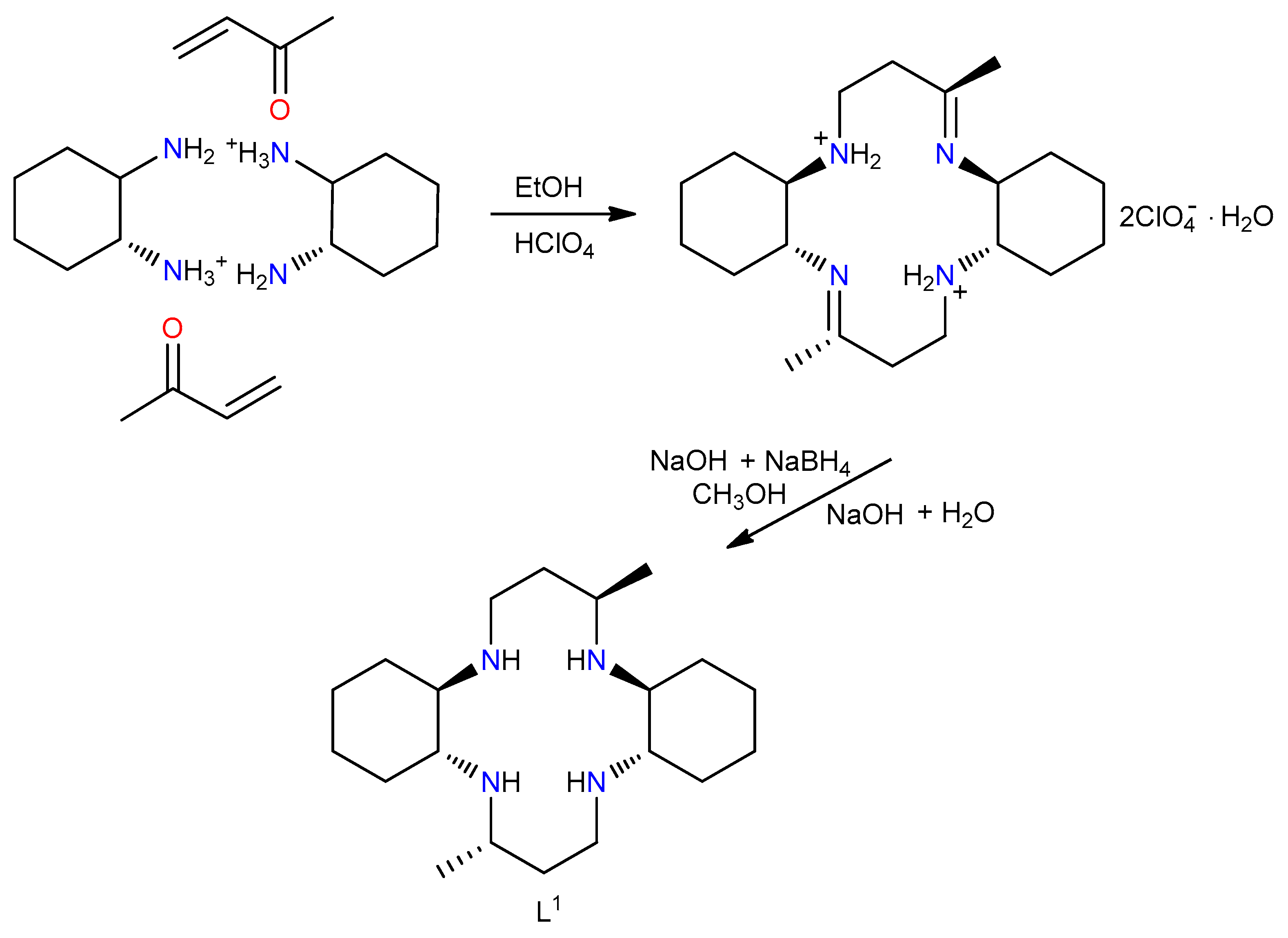
2.1. Synthesis of L1
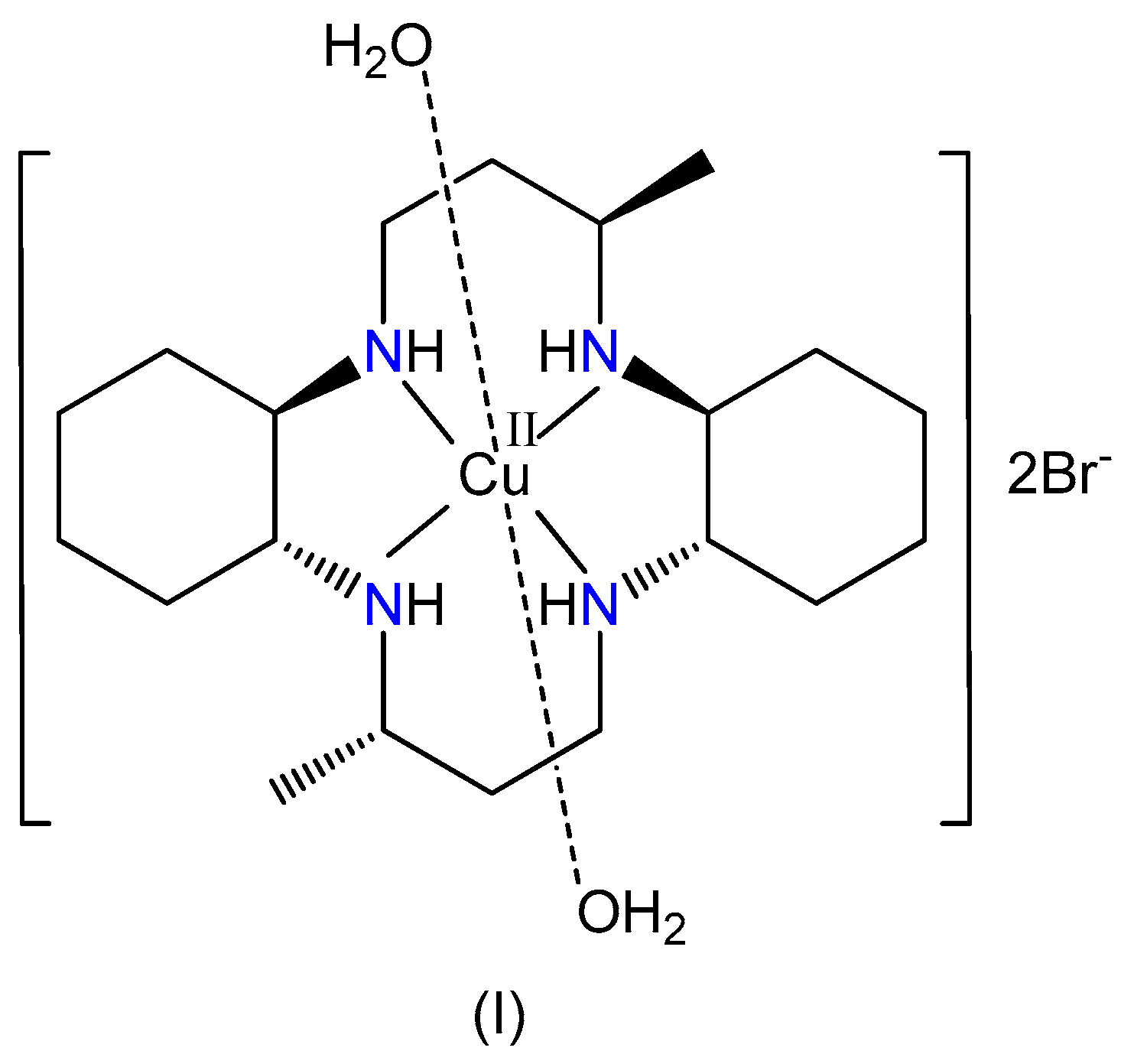
2.2. Synthesis of [Cu(L1)(H2O)2]Br2, (I)
2.3. Physical Measurements
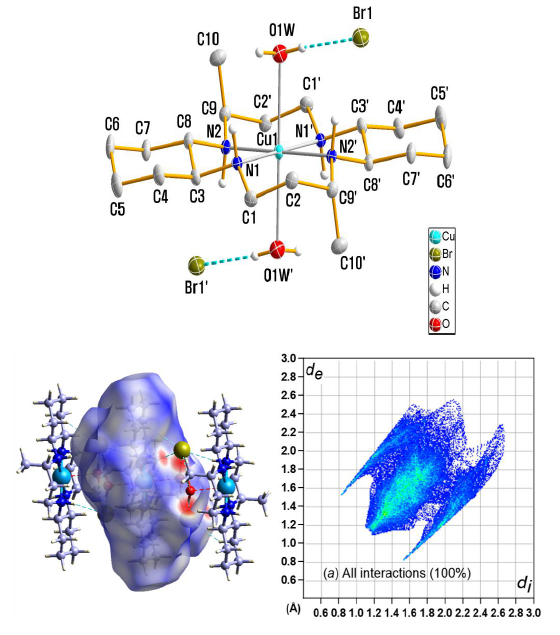
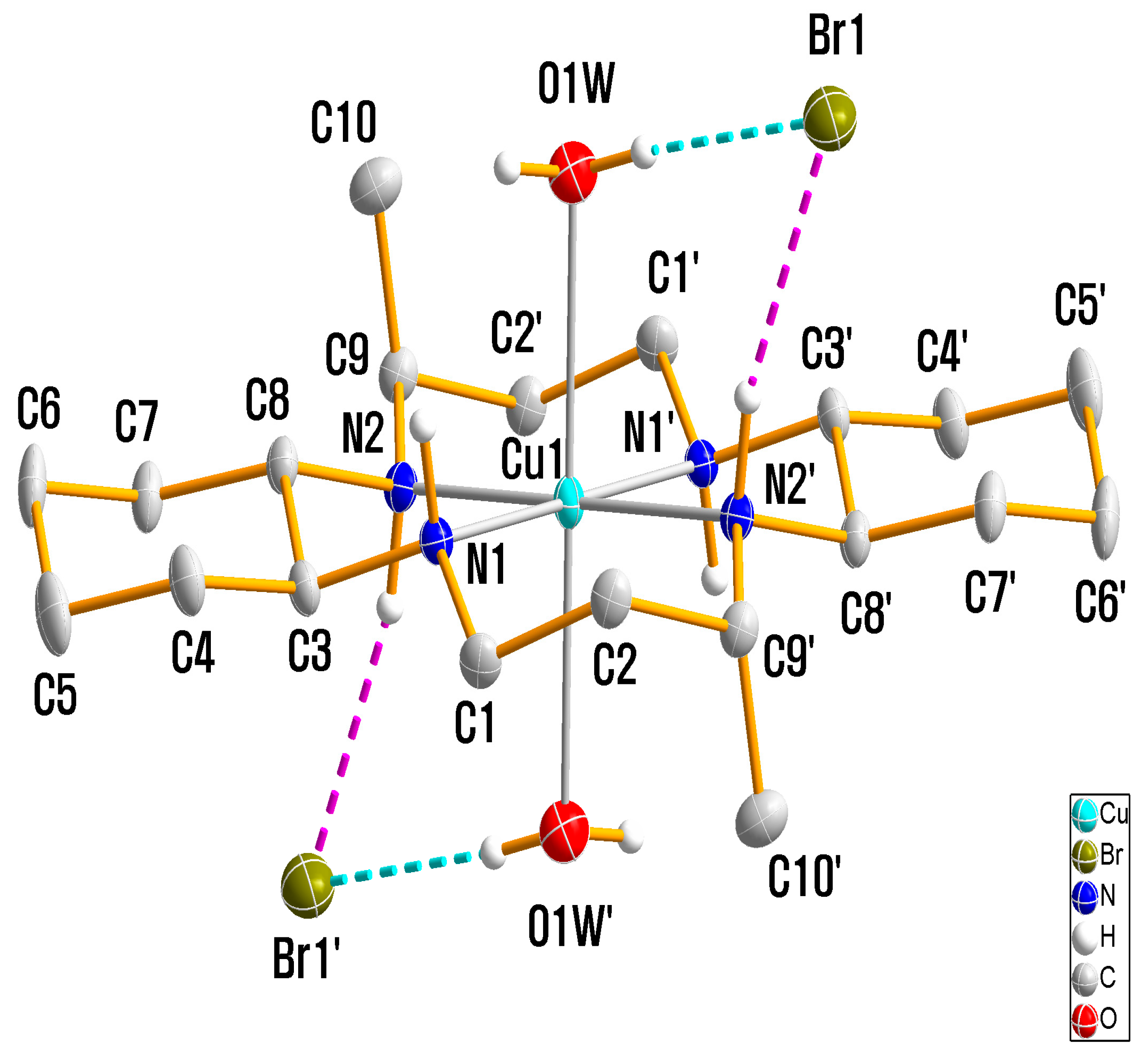
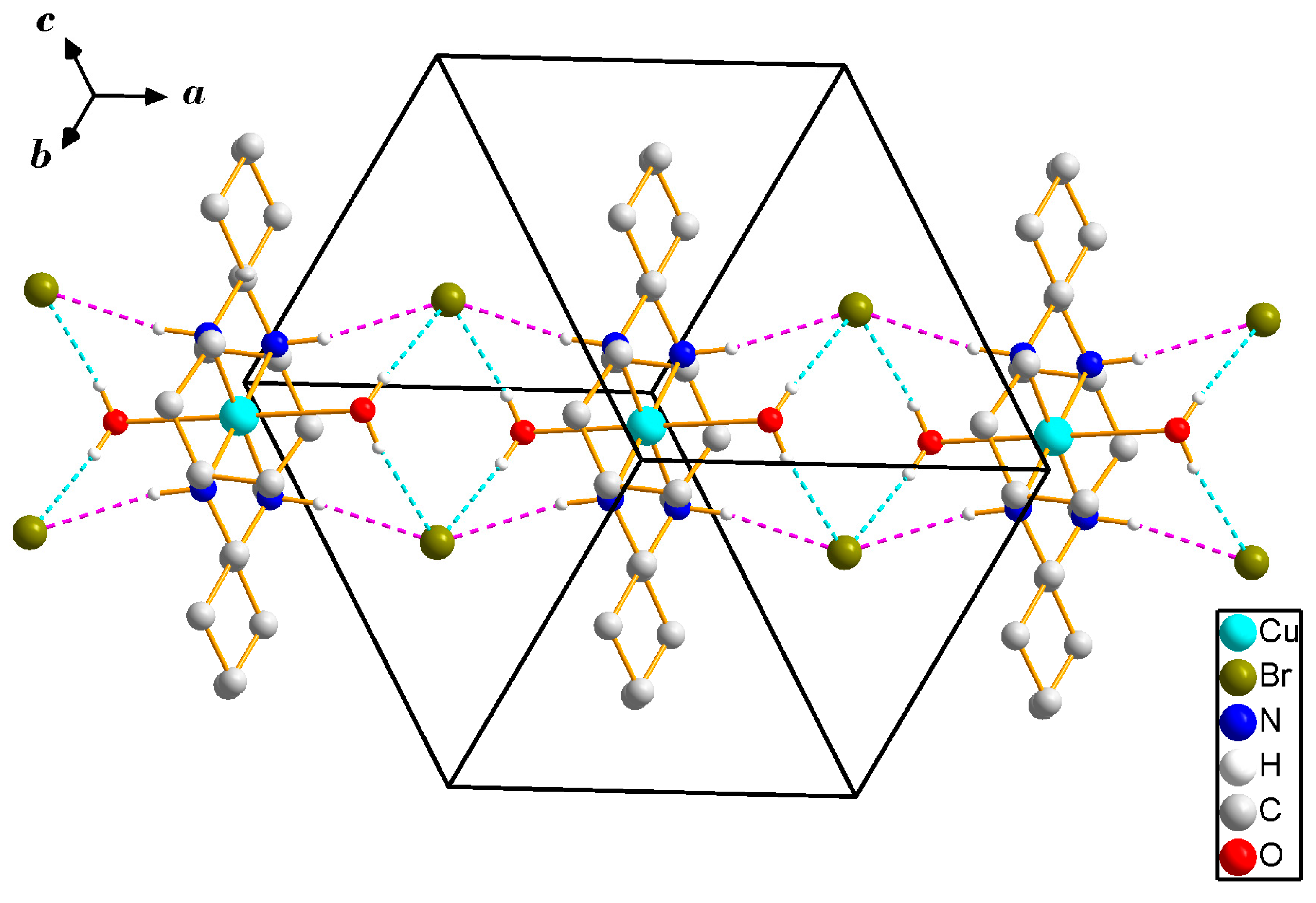
2.4. Crystal Structure Analysis
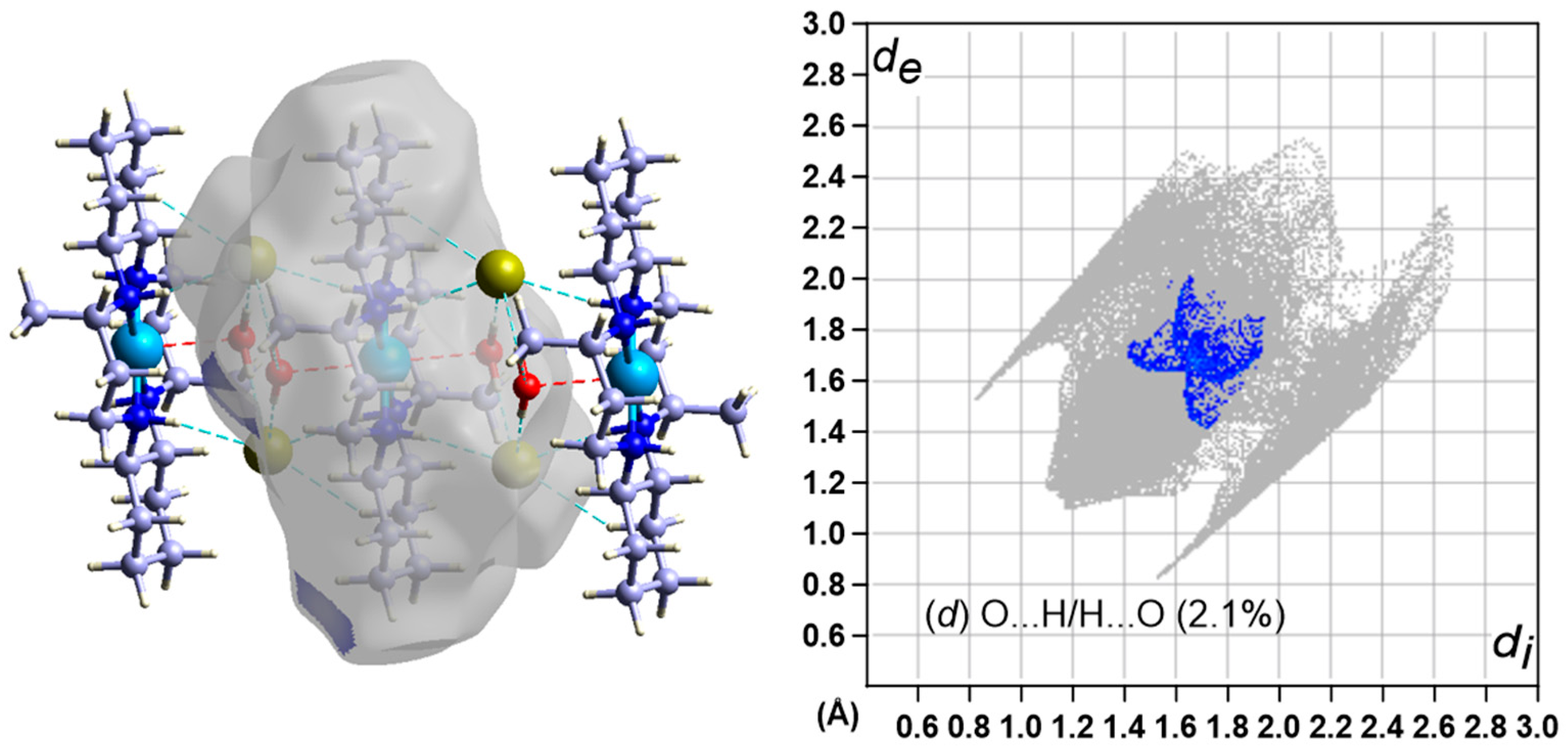
2.5. Hirshfeld Surface Analysis
3. Results and Discussion
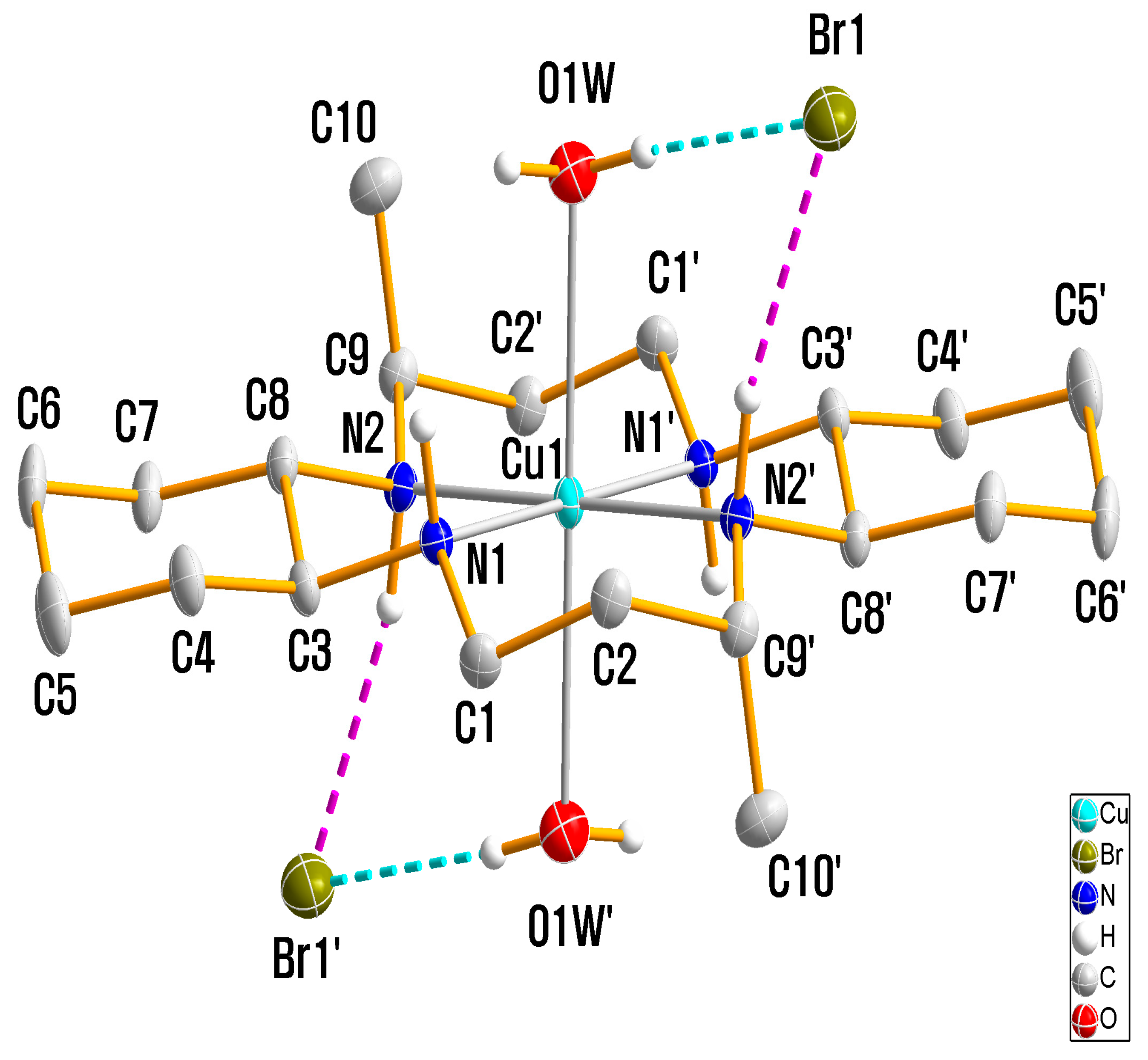
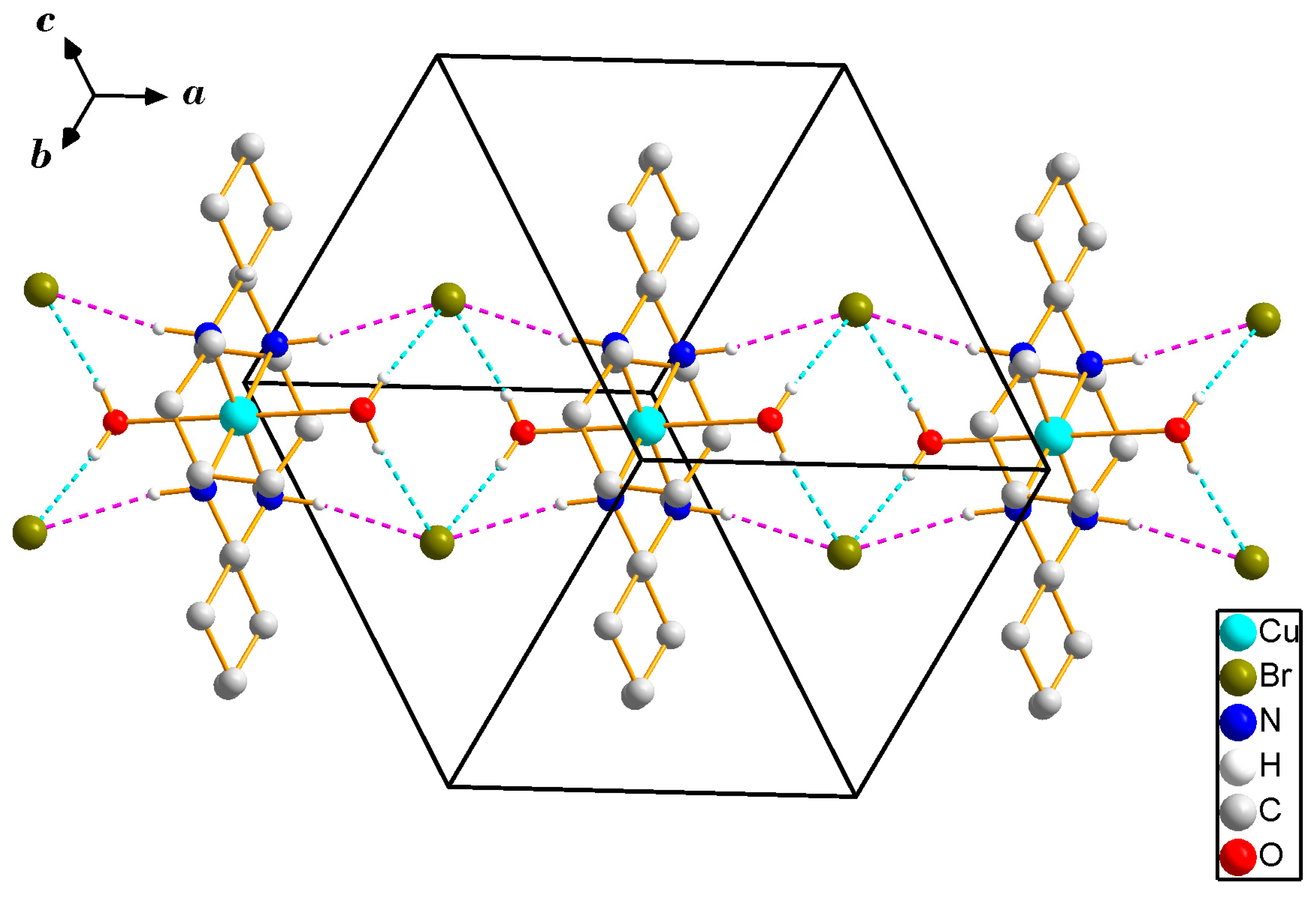
3.1. X-ray Crystallography
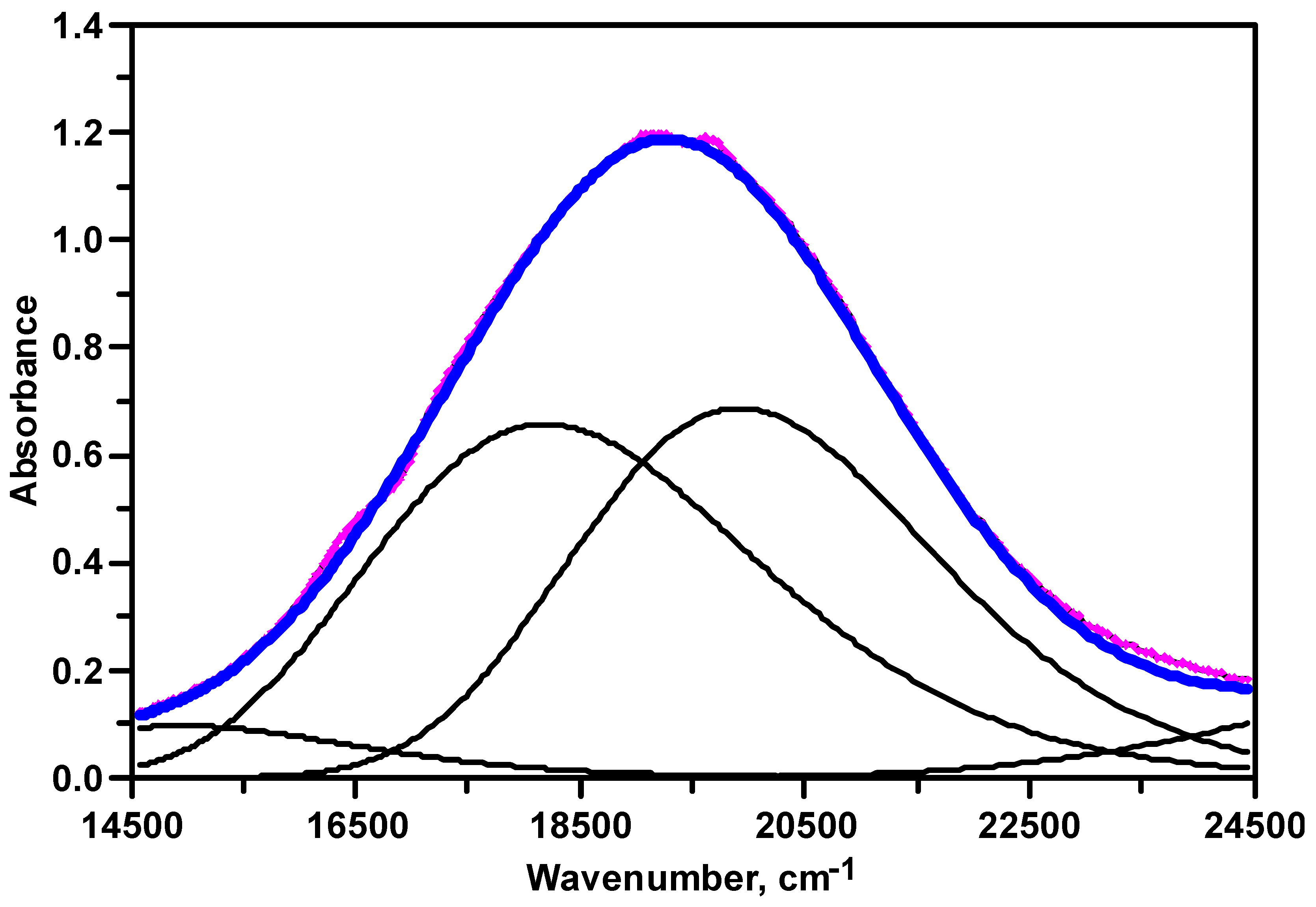
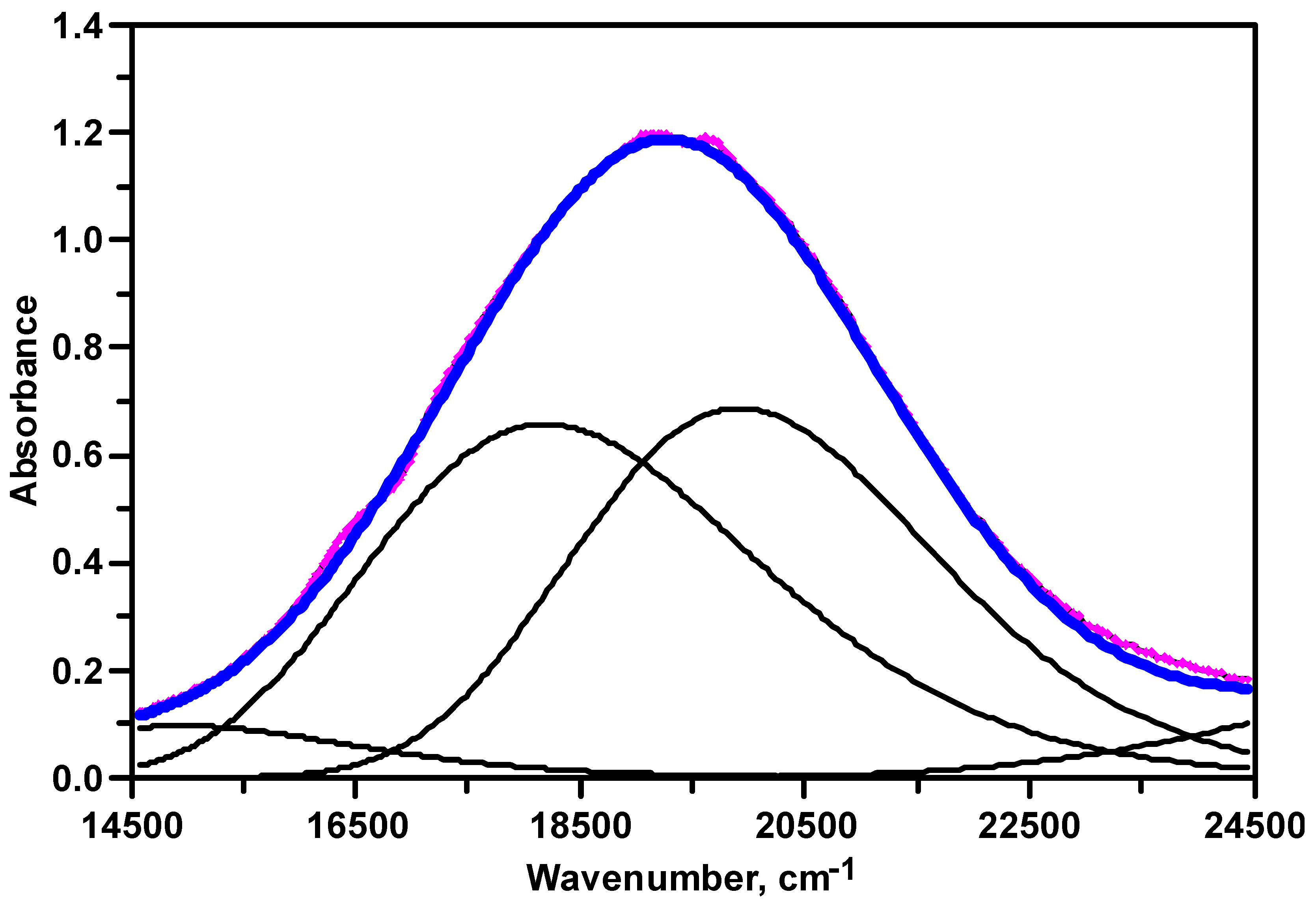
3.2. UV–Visible Spectroscopy
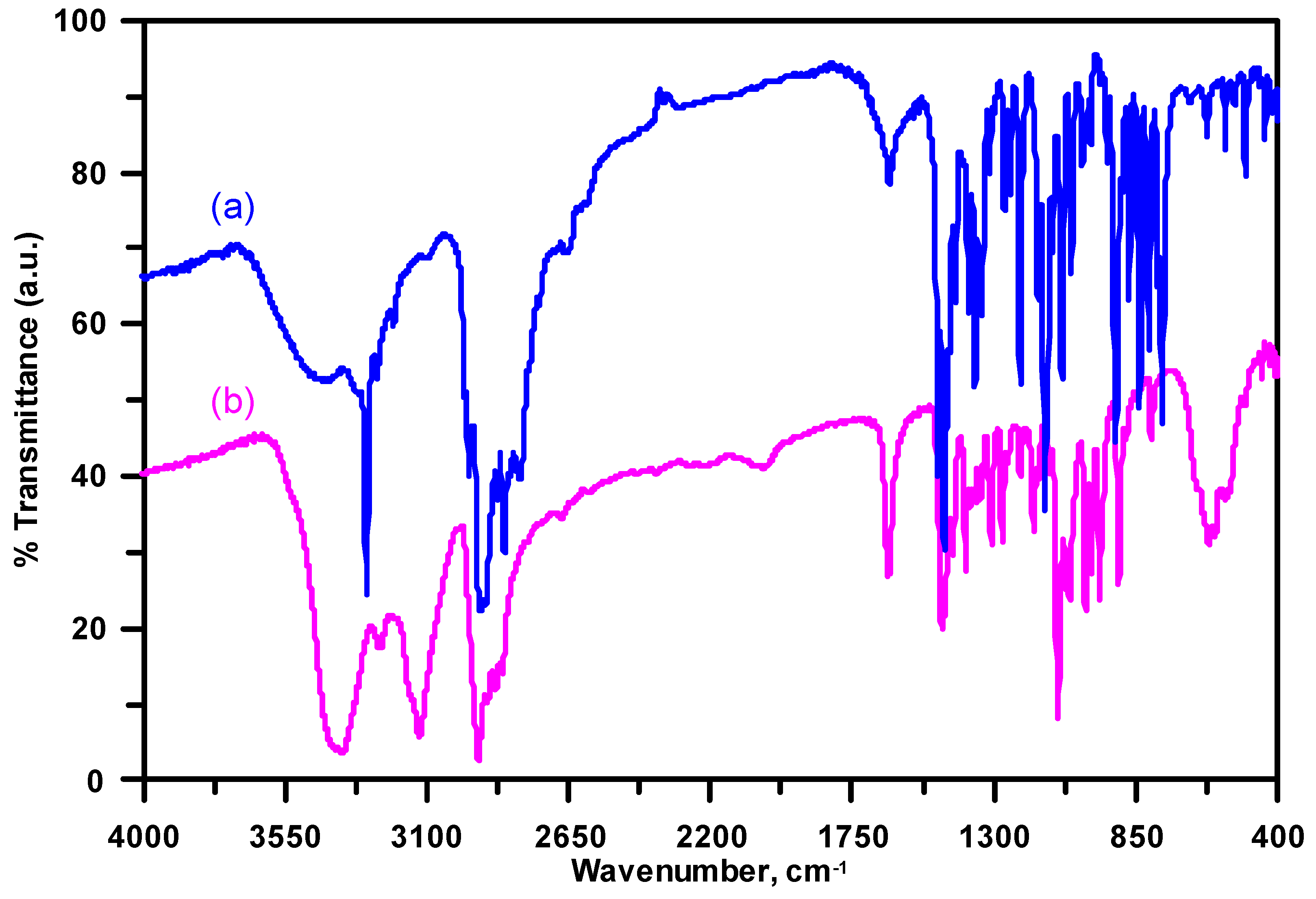
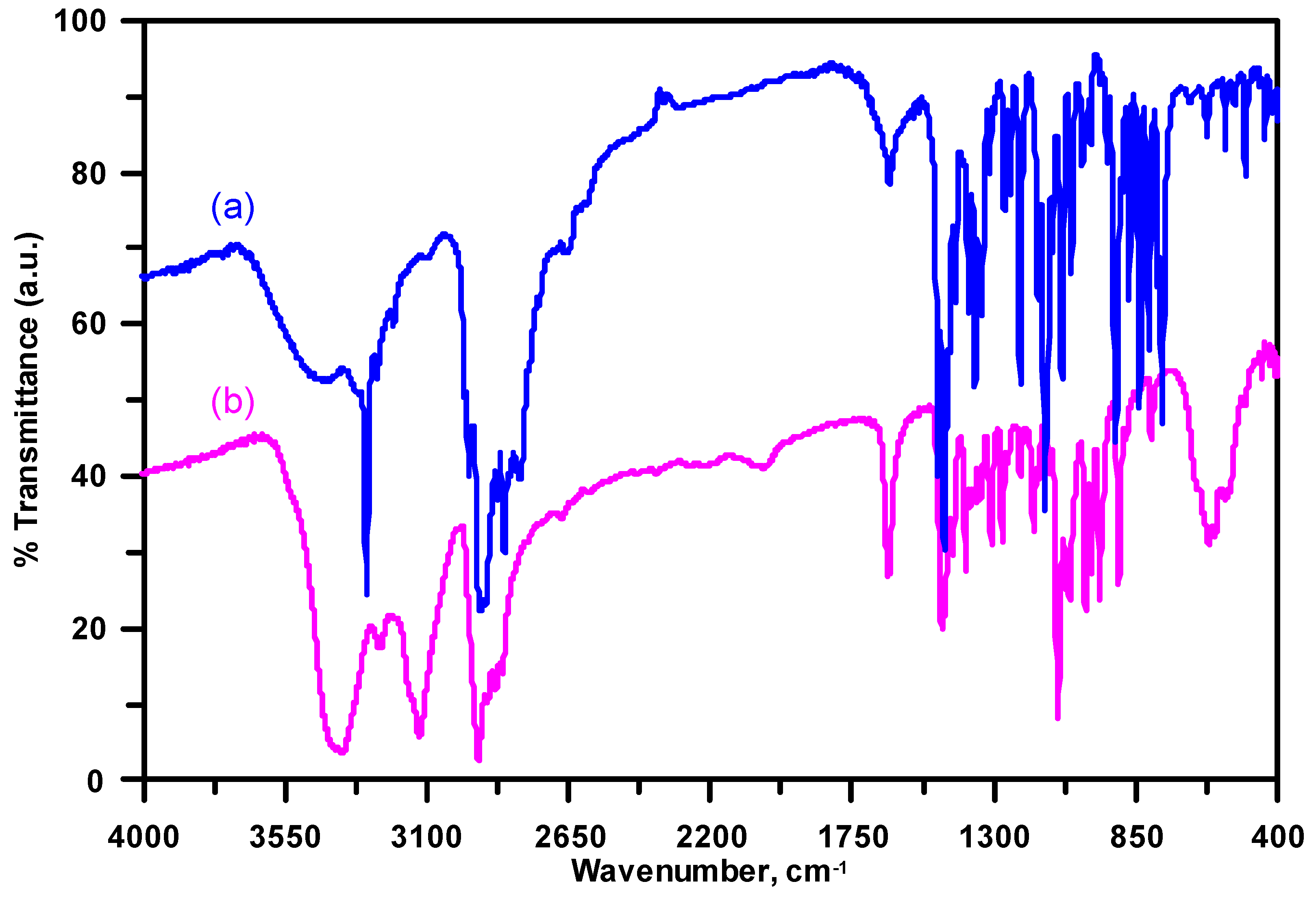
3.3. Infrared Spectroscopy
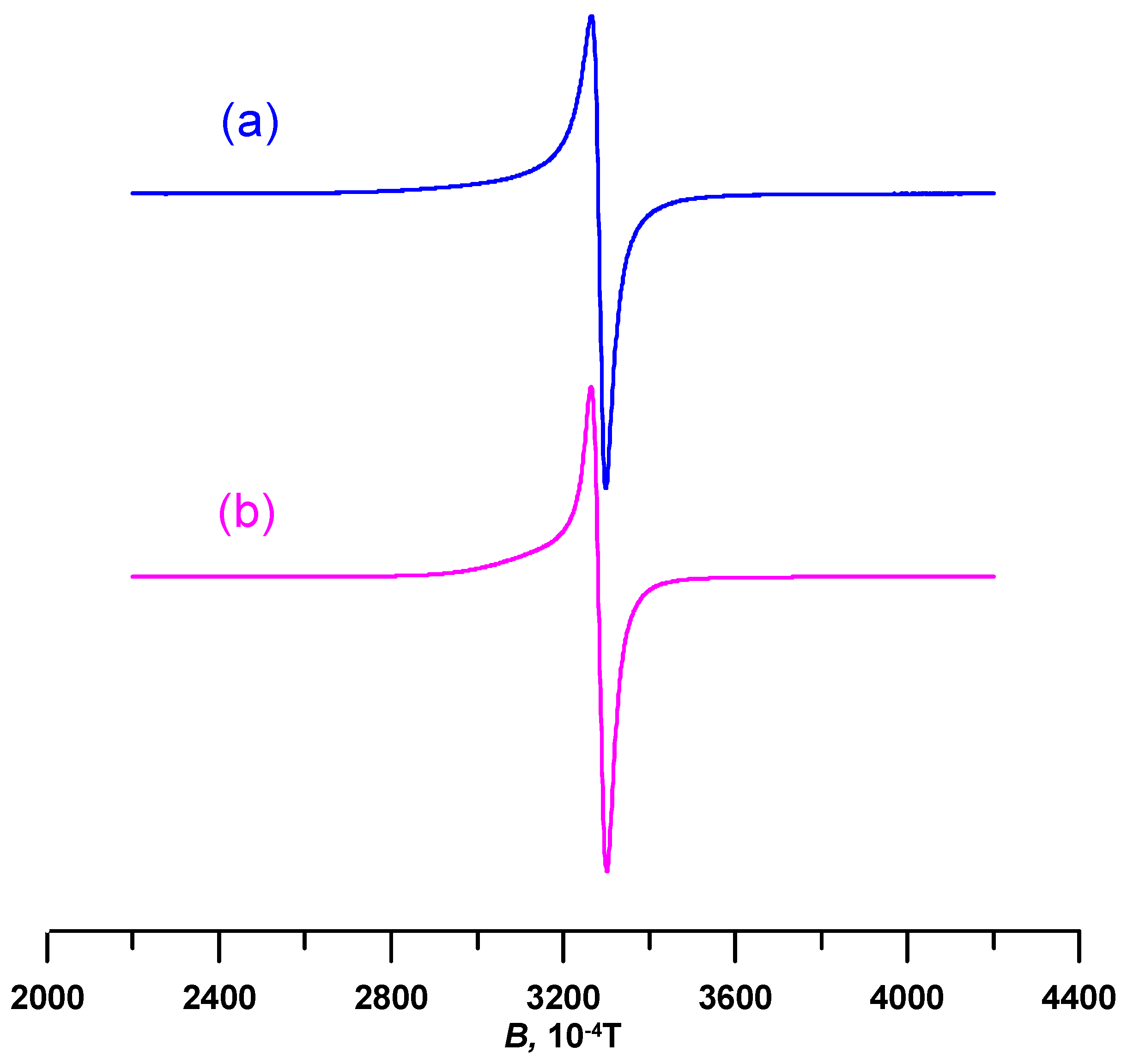
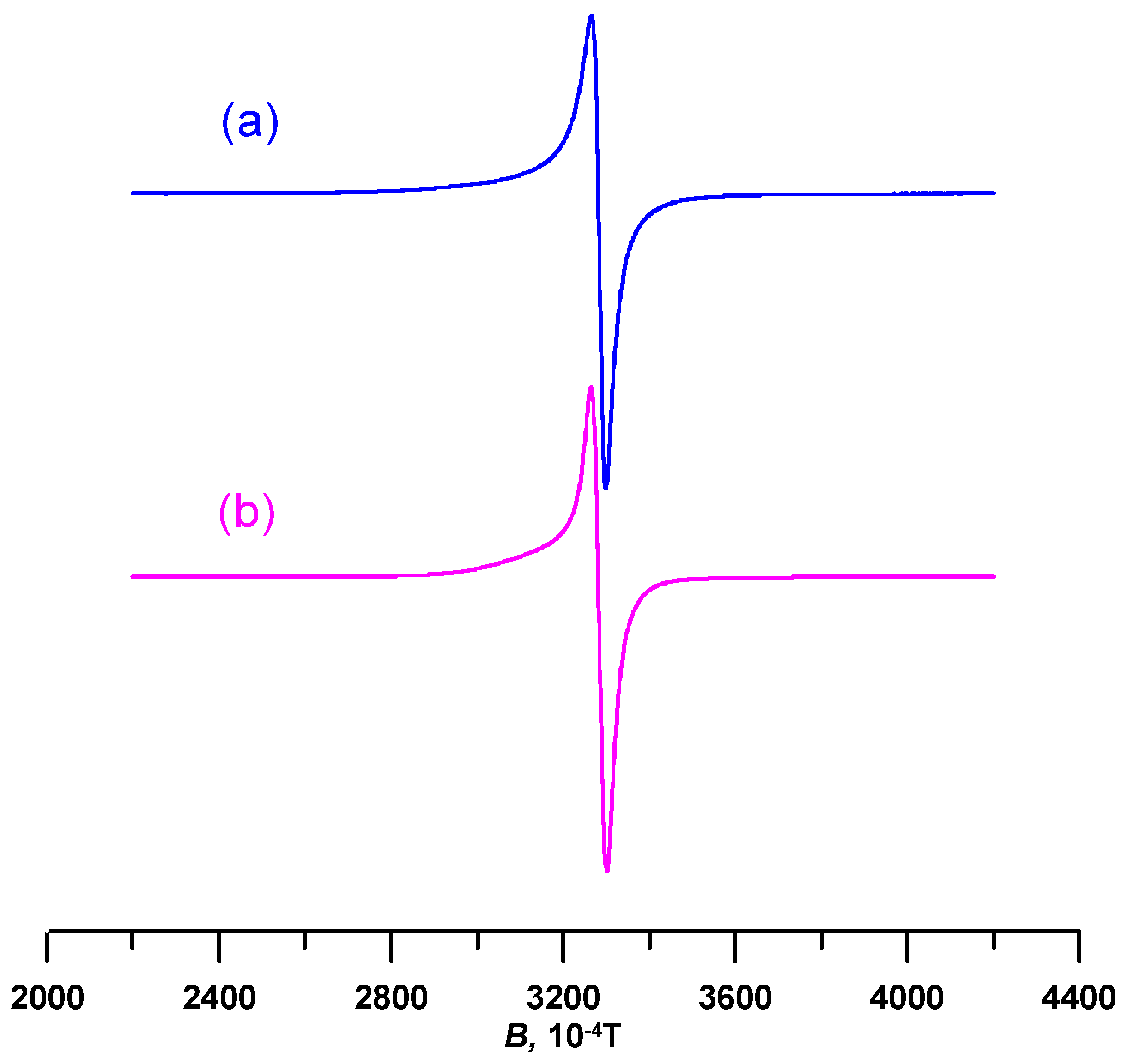
3.4. Electron Paramagnetic Resonace (EPR) Spectroscopy
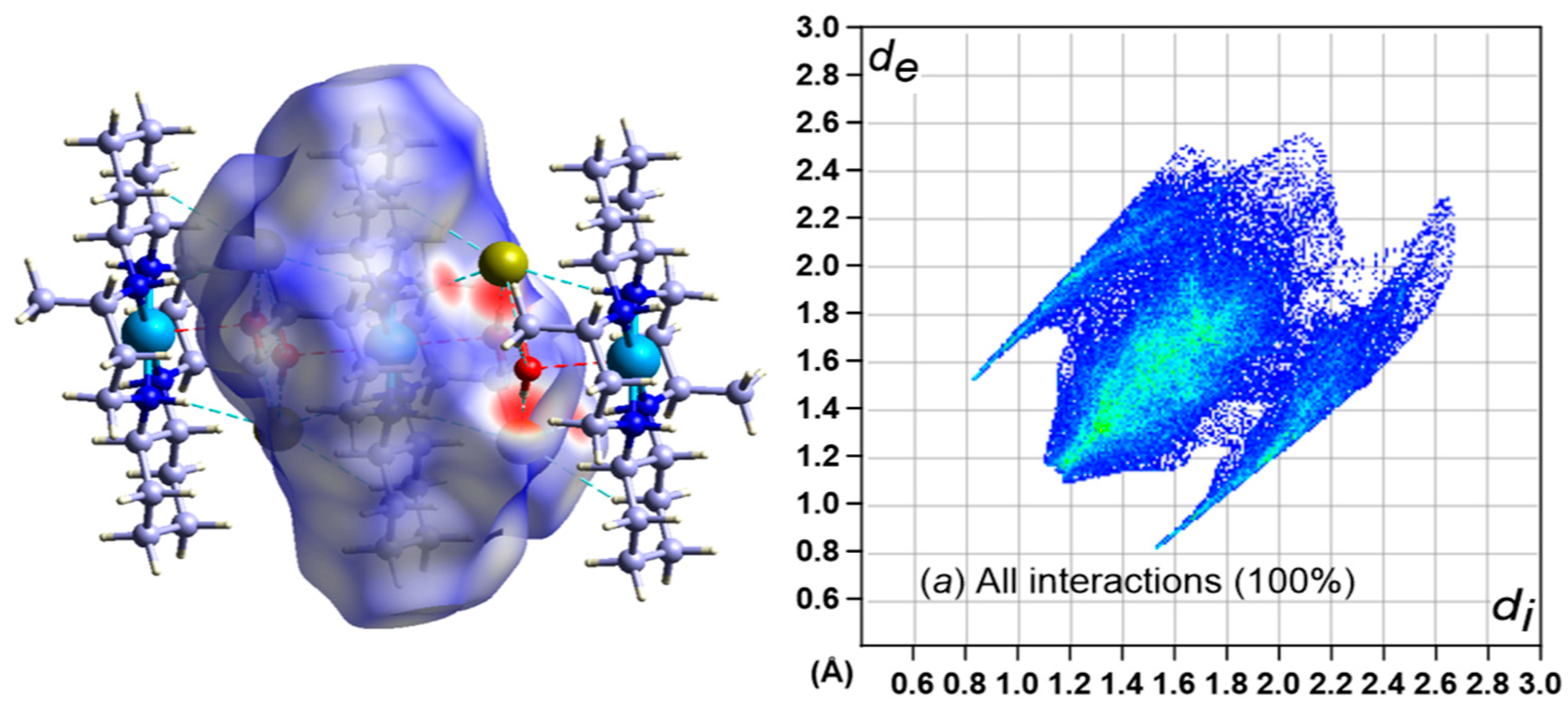
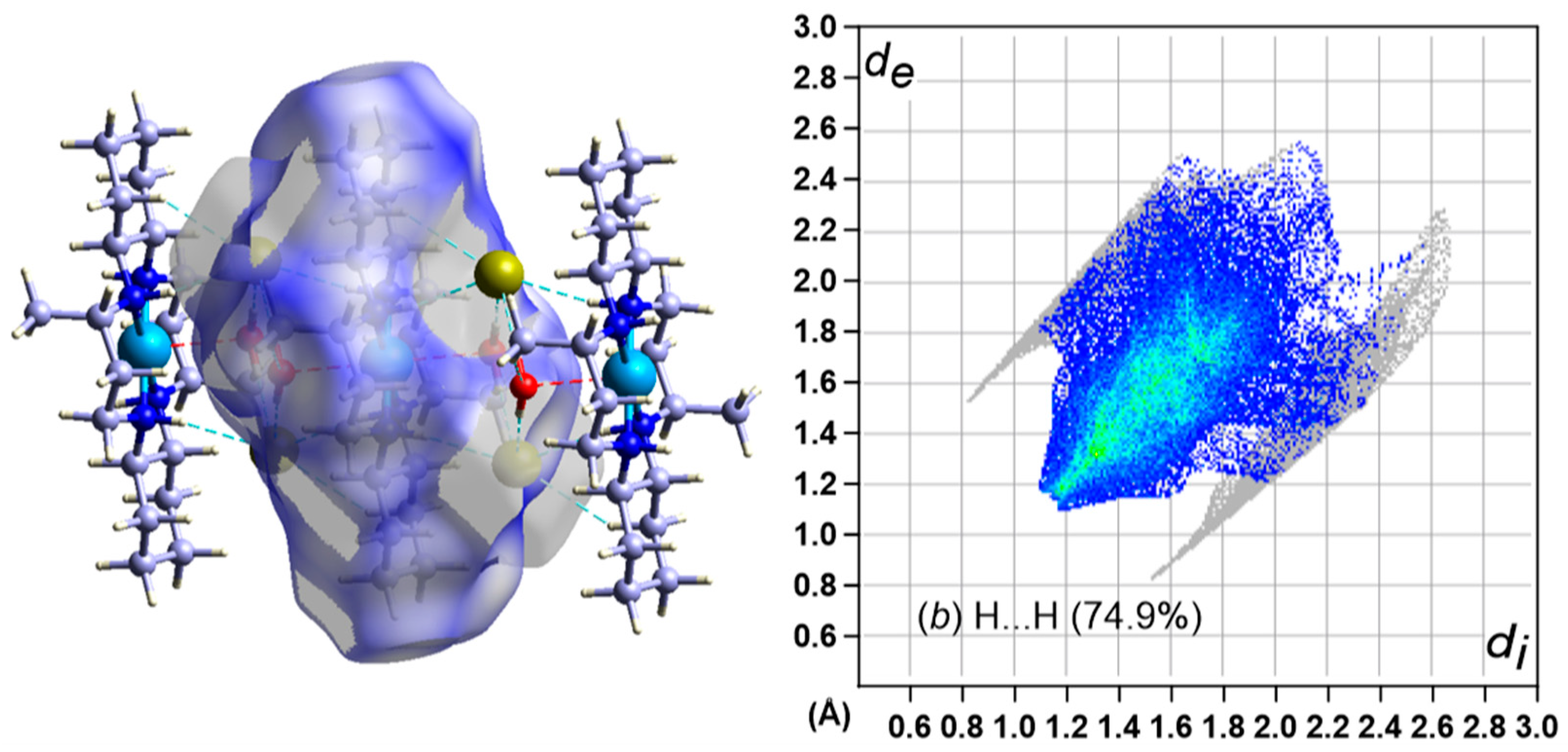
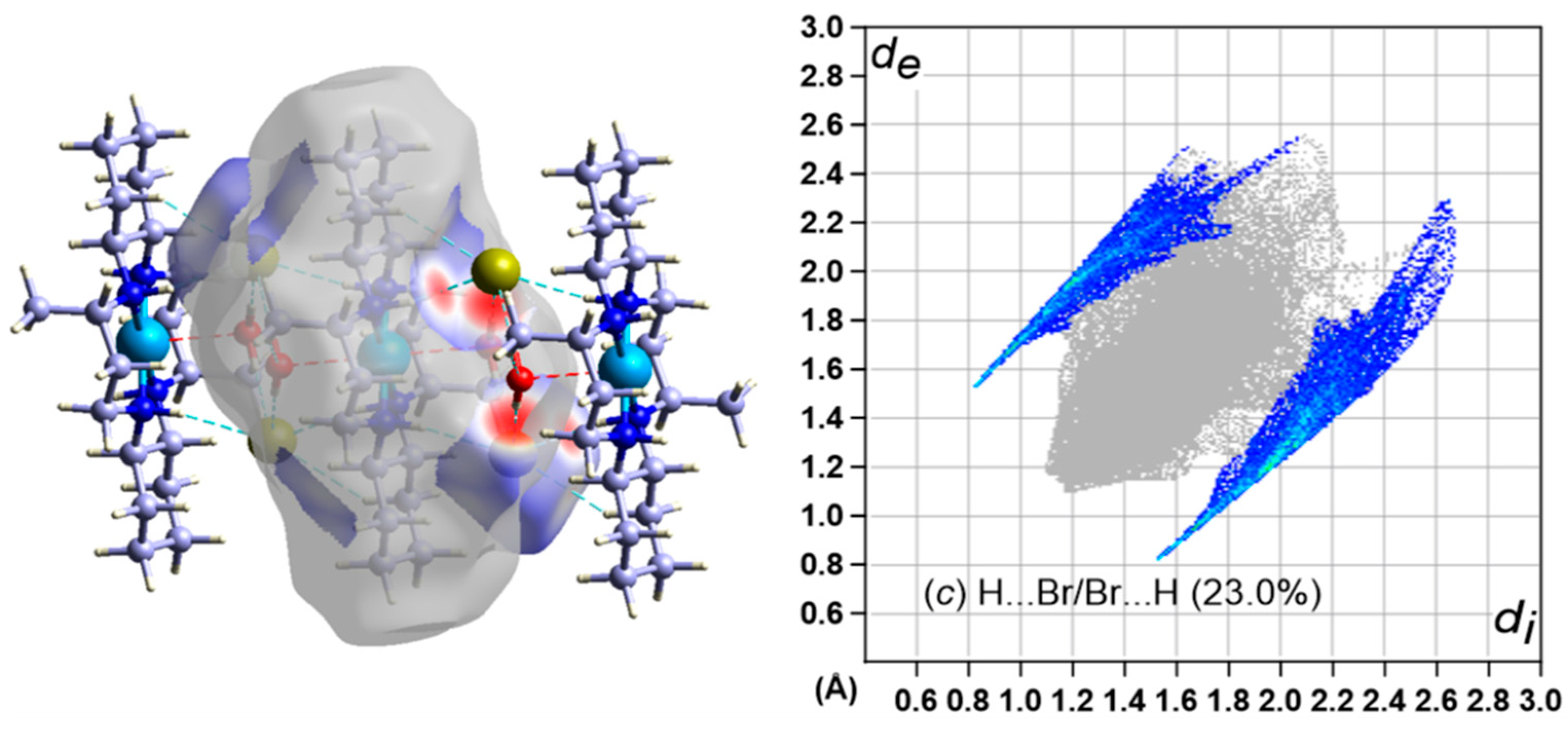
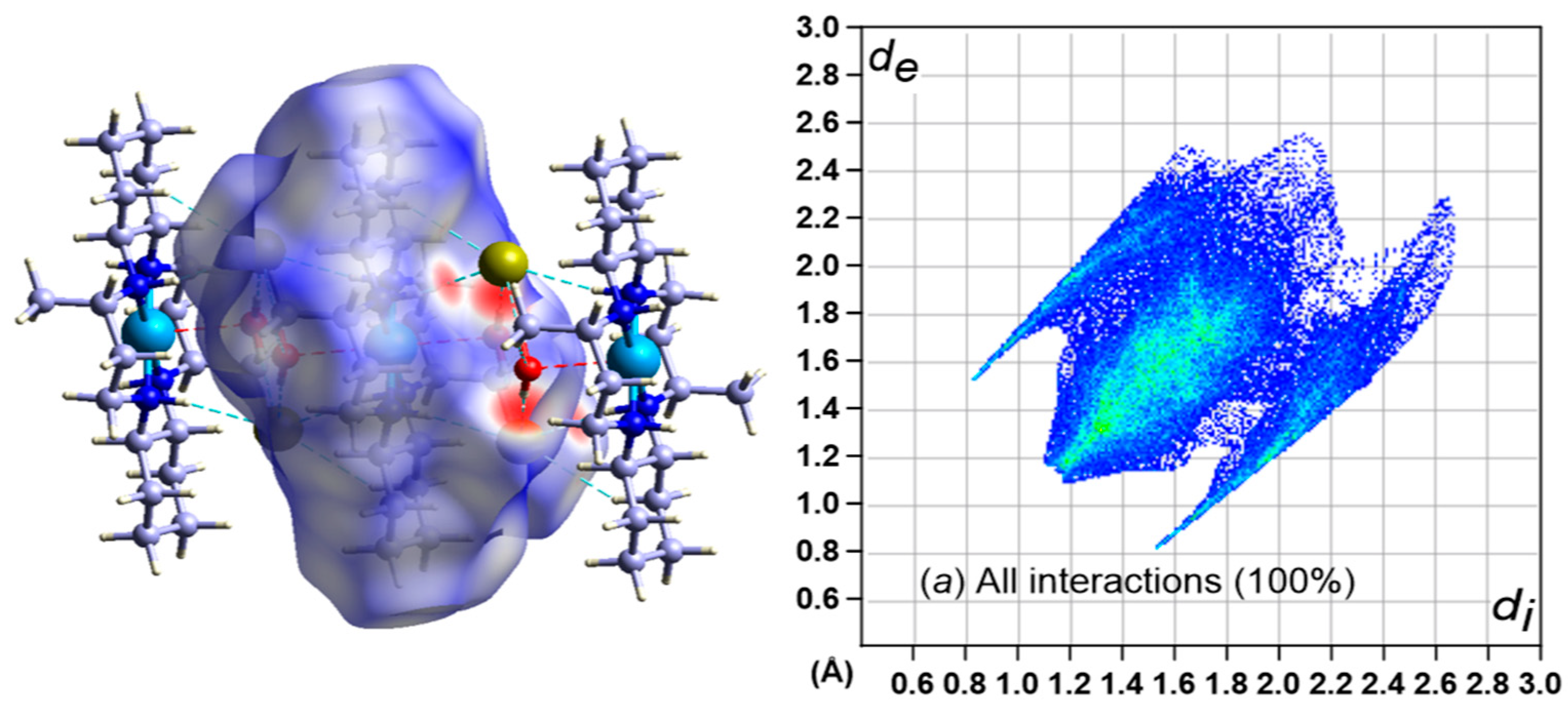
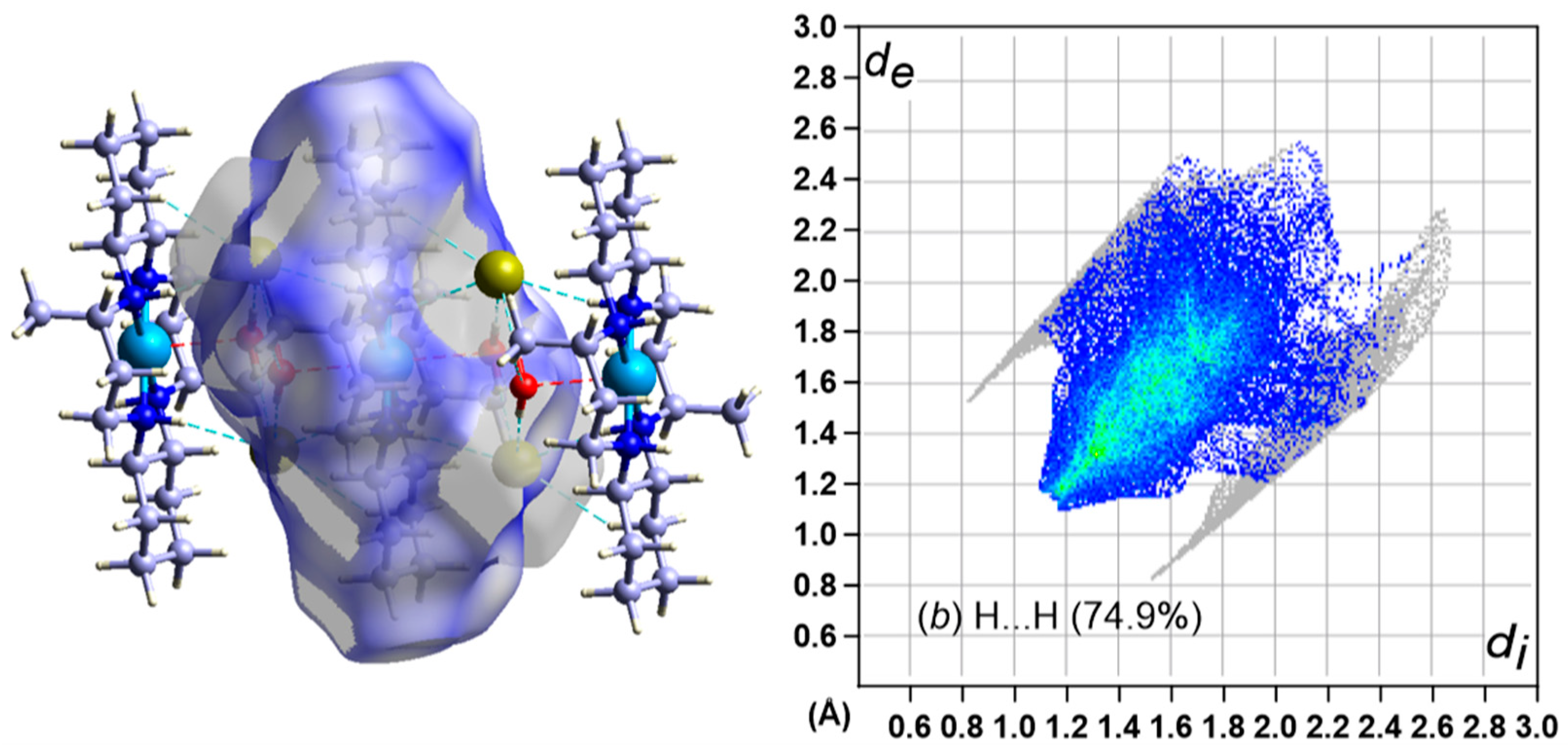
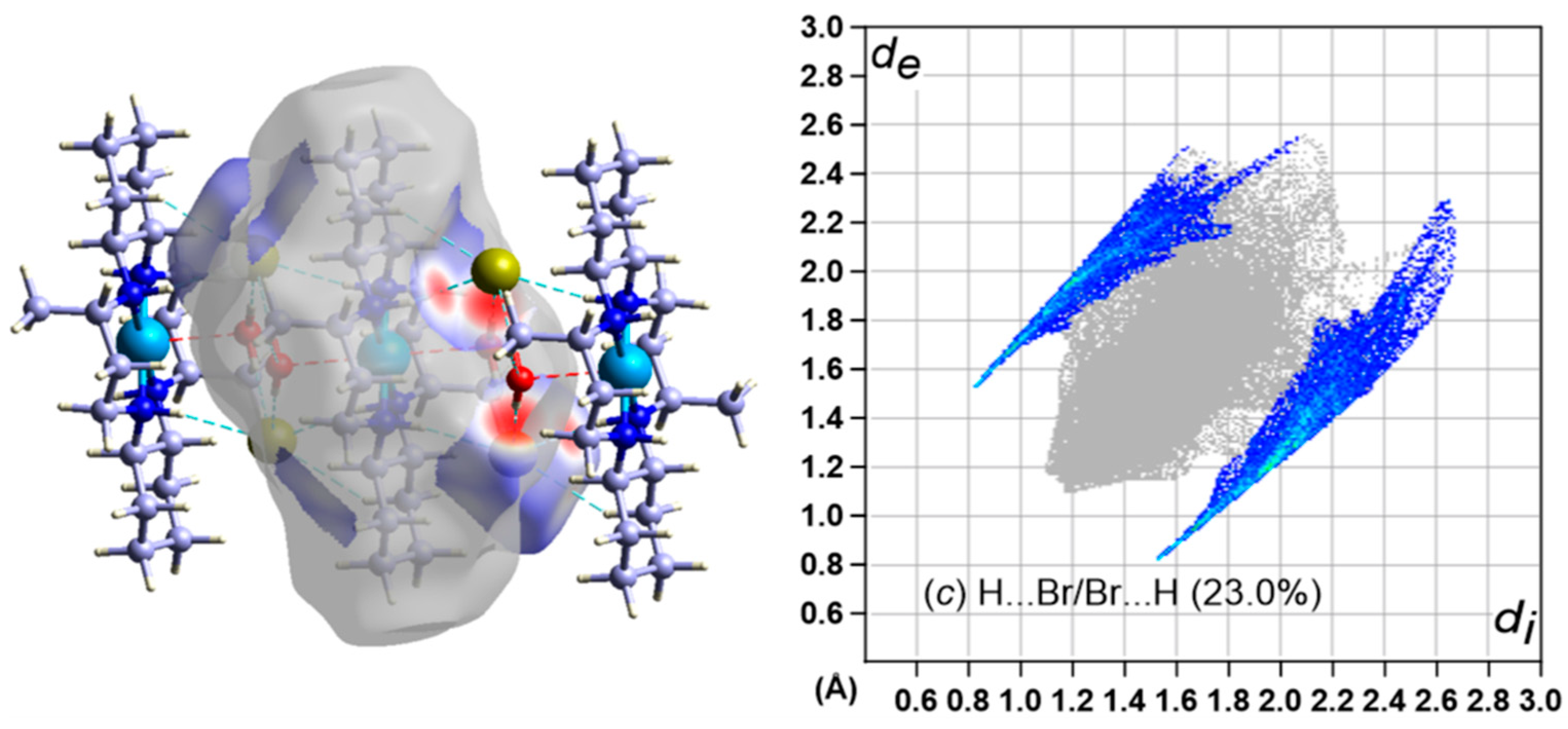
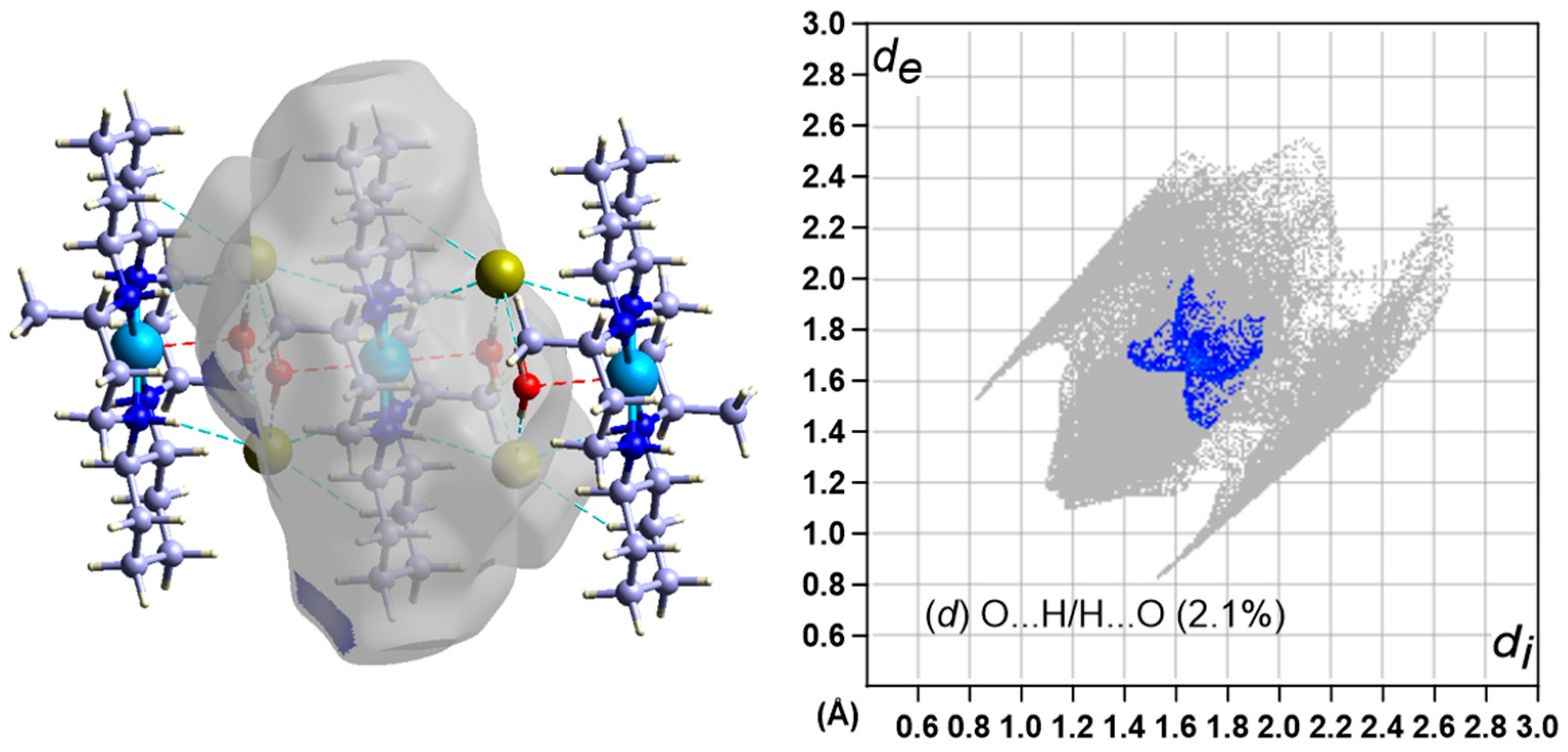
3.5. Hirshfeld Surface Analysis
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Valks, G.C.; McRobbie, G.; Lewis, E.A.; Hubin, T.J.; Hunter, T.M.; Sadler, P.J.; Pannecouque, C.; De Clercq, E.; Archibald, S.J. Configurationally restricted bismacrocyclic CXCR4 receptor antagonists. J. Med. Chem. 2006, 49, 6162–6165. [Google Scholar] [CrossRef] [PubMed]
- Ronconi, L.; Sadler, P. Using coordination chemistry to design new medicines. Coord. Chem. Rev. 2007, 251, 1633–1648. [Google Scholar] [CrossRef]
- De Clercq, E. Highlights in the discovery of antiviral drugs: A personal retrospective. J. Med. Chem. 2010, 53, 1438–1450. [Google Scholar] [CrossRef] [PubMed]
- Ross, A.; Choi, J.-H.; Hunter, T.M.; Pannecouque, C.; Moggach, S.A.; Parsons, S.; De Clercq, E.; Sadler, P. Zinc(II) Complexes of Constrained Antiviral Macrocycles. Dalton Trans. 2012, 41, 6408–6418. [Google Scholar] [CrossRef] [PubMed]
- Choi, J.-H. Crystal structure and spectroscopic properties of trans-bis(nicotinato)(1,4,8,11-tetraazacyclotetradecane)chromium(III) perchlorate. Inorg. Chim. Acta 2009, 362, 4231–4236. [Google Scholar] [CrossRef]
- Subhan, M.A.; Choi, J.-H.; Ng, S.W. Synthesis, crystal structure and spectroscopic properties of (acetylacetonato)(1,4,8,11-tetraazacyclotetradecane)chromium(III) diperchlorate. Z. Anorg. Allg. Chem. 2011, 637, 2193–2197. [Google Scholar] [CrossRef]
- Choi, J.-H.; Tanmaya, T.; Spiccia, L. Syntheses, structural and spectroscopic properties of the copper(II) complexes of constrained macrocyclic ligands. Z. Anorg. Allg. Chem. 2012, 638, 146–151. [Google Scholar] [CrossRef]
- Choi, K.-Y.; Kim, J.C.; Jensen, W.P.; Suh, I.H.; Choi, S.S. Copper(II) and nickel(II) complexes with 3,14-dimethyl-2,6,13,17-tetraazatricyclo[16.4.0.07,12]docosane, [M(C20H40N4)]Cl2·2H2O (M = Cu II and Ni II). Acta Crystallogr. 1996, C52, 2166–2168. [Google Scholar]
- Choi, J.-H.; Ryoo, K.S.; Park, K.M. [3,14-Dimethyl-2,6,13,17-tetraazatricyclo(16.4.0.07,12)docosane-κ4N]bis(perchlorato-κO)copper(II). Acta Crystallogr. 2007, E63, m2674–m2675. [Google Scholar]
- Choi, J.-H.; Subhan, M.A.; Ng, S.W. [3,14-Dimethyl-2,6,13,17-tetraazatricyclo(16.4.0.07,12)docosane-κ4N]bis(nitrato-κO)copper(II). Acta Crystallogr. 2012, E68, m190. [Google Scholar]
- Choi, J.-H.; Suzuki, T.; Kaizaki, S. [3,14-Dimethyl-2,6,13,17-tetraazatricyclo(16.4.0.07,12)docosane-κ4N]dinitratocopper(II) trihydrate. Acta Crystallogr. 2006, E62, m2383–m2385. [Google Scholar]
- Subhan, M.A.; Ryoo, K.S.; Choi, J.-H. Crystal structure and spectroscopic properties of [3,14-dimethyl-2,6,13,17-tetraazatricyclo(16.4.0.07,12)docosane]copper(II) dithiocyanate dihydrate. Int. J. Chem. Sci. 2015, 13, 593–604. [Google Scholar]
- Subhan, M.A.; Ng, S.W.; Lee, C.S.; Choi, J.-H. Synthesis, crystal structure, and spectroscopic properties of isothiocyanato[3,14-dimethyl-2,6,13,17-tetraazatricyclo(16.4.0.07,12)docosane]copper(II) thiocyanate. J. Struct. Chem. 2017, 58, 742–749. [Google Scholar] [CrossRef]
- Choi, K.-Y. Synthesis and crystal structure of a macrocyclic azidotetraamine copper(II) complex. J. Chem. Crystallogr. 1998, 28, 875–878. [Google Scholar] [CrossRef]
- Choi, J.-H.; Clegg, W.; Harrington, R.W. Crystal structure of [2,13-dibenzyl-5,16-dimethyl-2,6,13,17-tetraazatricyclo(14,4,0,07.12)docosane]copper(II) diperchlorate. J. Chem. Crystallogr. 2010, 40, 80–84. [Google Scholar] [CrossRef]
- Choi, J.-H.; Clegg, W.; Gary, G.S. Synthesis, crystal structure and spectroscopic properties of [2,13-bis(1-naphthalenylmethyl)-5,16-dimethyl-2,6,13,17-tetraazatricyclo(14,4,0,07.12)docosane]copper(II) diperchlorate acetonitrile disolvate. Z. Anorg. Allg. Chem. 2010, 636, 1612–1616. [Google Scholar] [CrossRef]
- Lim, J.H.; Kang, J.S.; Kim, H.C.; Koh, E.K.; Hong, C.S. Synthesis, crystal structures, and magnetic properties of cyano-bridged honeycomb like layers MV-CuII (M=Mo, W) chelated by a macrocyclic ligand. Inorg. Chem. 2006, 45, 7821–7827. [Google Scholar] [CrossRef] [PubMed]
- Moncol, J.; Mazúr, M.; Valko, M.; Choi, J.-H. Synthesis, structural characterization, EPR spectroscopy and Hirshfeld surface analysis of novel Cu2+ doped 3,14-diethyl-2,13-diaza-6,17-diazoniatricyclo(16.4.0.07,12)docosane diperchlorate. Acta Crystallogr. 2019, C75, 612–622. [Google Scholar] [CrossRef] [PubMed]
- Choi, J.-H.; Subhan, M.A.; Ng, S.W. Syntheses, crystal structures and spectroscopic properties of [Cu(L)(NO3)2] and [Cu(L)(H2O)2](SCN)2 with 3,14-diethyl-2,6,13,17-tetraazatricyclo(16.4.0.07,12)docosane (L). J. Coord. Chem. 2012, 65, 3481–3491. [Google Scholar] [CrossRef]
- Subhan, M.A.; Choi, J.-H. X-ray structure and spectroscopy of novel trans-[Ni(L)(NO3)2] and [Ni(L)](ClO4)2·2H2O complexes. Spectrochim. Acta Part A 2014, 123, 410–415. [Google Scholar] [CrossRef] [PubMed]
- Lim, I.-T.; Kim, C.-H.; Choi, K.-Y. Synthesis and structural characterization of nickel(II) complexes of 3,14-diethyl-2,6,13,17-tetraazatricyclo(14.4.0.07,12)docosane with inorganic salts. Polyhedron 2015, 100, 43–48. [Google Scholar]
- Subhan, M.A.; Moon, D.; Choi, J.-H. Synthesis, crystal structure determination, and spectroscopic characterization of [2,13-dibenzyl-5,16-diethyl-2,6,13,17-tetraazatricyclo(16.4.0.07,12)docosane]copper(II) dinitrate. Main Group Chem. 2017, 16, 27–36. [Google Scholar] [CrossRef]
- Jimenez, D.; Martinez-Manez, R.; Sancenon, F.; Ros-Lis, J.V.; Soto, J. A new chromo-chemodosimeter selective for sulfide anion. J. Am. Chem. Soc. 2003, 125, 9000–9001. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Manez, R.; Sancenon, F. Fluorogenic and chromogenic chemosensors and reagents for anions. Chem. Rev. 2003, 103, 4419–4476. [Google Scholar]
- Kang, S.G.; Kweon, J.K.; Jung, S.K. Synthesis of new tetraaza macrocyclic ligands with cyclohexane ring and their Ni(II) and Cu(II) complexes. Bull. Korean Chem. Soc. 1991, 12, 483–487. [Google Scholar]
- Moon, D.; Choi, J.-H. Crystal structure of 3,14-dimethyl-2,6,13,17-tetraazoniatricyclo(16.4.0.07,12)docosane tetrachloride tetrahydrate from synchrotron X-ray data. Acta Crystallogr. 2018, E74, 1039–1041. [Google Scholar]
- Mazur, M.; Valko, M.; Klement, R.; Morris, H. Quantitative EPR spectroscopy with a TE104 double rectangular cavity. Part 1. A simple alignment procedure for the precision positioning of the sample. Anal. Chim. Acta 1996, 333, 249–252. [Google Scholar] [CrossRef]
- Thiele, H.; Etstling, J.; Such, P.; Hoefer, P. WINEPR; Bruker Analytic Gmb: Rheinstetten, Germany, 1992. [Google Scholar]
- Weber, R.T. WINEPR SimFonia; EPR Division, Bruker Instr. Inc.: Billerica, MA, USA, 1995. [Google Scholar]
- Sheldrick, G.M. SHELXT-Integrated space-group and crystal-structure determination. Acta Crystallogr. 2015, A71, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Crystallogr. 2015, C71, 3–8. [Google Scholar]
- Koziskova, J.; Hahn, F.; Richter, J.; Kozisek, J. Comparison of different absorption corrections on the model structure of tetrakis(μ2-acetato)diaquadicopper(II). Acta Chim. Slovaca 2016, 9, 136–140. [Google Scholar] [CrossRef]
- Putz, H.; Brandenburg, K. DIAMOND-3; University of Bonn: Bonn, Germany, 2014. [Google Scholar]
- Turner, M.J.; McKinnon, J.J.; Wolff, S.K.; Grimwood, D.J.; Spackman, P.R.; Jayatilaka, D.; Spackman, M.A. CrystalExplorer17.5. The University of Western Australia. 2017. Available online: http://crystalexplorer.scb.uwa.edu.au/ (accessed on 27 June 2019).
- McKinnon, J.J.; Spackman, M.A.; Mitchell, A.S. Novel tools for visualizing and exploring intermolecular interactions in molecular crystals. Acta Crystallogr. 2004, B60, 627–668. [Google Scholar] [CrossRef]
- McKinnon, J.J.; Jayatilaka, D.; Spackman, M.A. Towards quantitative analysis of intermolecular interactions with Hirshfeld surfaces. Chem. Commun. 2007, 3814–3816. [Google Scholar] [CrossRef]
- Hathaway, B.J.; Billing, D.E. The electronic properties and stereochemistry of mononuclear complexes of copper(II) ion. Coord. Chem. Rev. 1970, 5, 43–207. [Google Scholar] [CrossRef]
- Hathaway, B.J.; Hodgson, P.G. Copper-ligand bond-lengths in axial complexes of the copper(II) ion. J. Inorg. Nucl. Chem. 1973, 35, 4071–4081. [Google Scholar] [CrossRef]
- Lever, A.B.P. Inorganic Electronic Spectroscopy, 2nd ed.; Elsevier: Amsterdam, The Netherlands, 1984. [Google Scholar]
- Nakamoto, K. Infrared and Raman Spectra of Inorganic and Coordination Compounds, Part A: Theory and Applications in Inorganic Chemistry; Part B: Applications in Coordination, Organometallic, and Bioinorganic Chemistry, 6th ed.; John Wiley & Sons: New York, NY, USA, 2009. [Google Scholar]
- Choi, J.-H.; Oh, I.-G.; Suzuki, T.; Kaizaki, S. Crystal structure and spectroscopic properties of oxalato(1,4,8,11-tetraazacyclotetradecane)chromium(III) perchlorate. J. Mol. Struct. 2004, 694, 39–44. [Google Scholar] [CrossRef]
- Choi, J.-H. Spectral properties and ligand field analysis of cis-dinitrito(1,4,8,11-tetraazacyclotetradecane)chromium(III) nitrate. Chem. Phys. 2000, 256, 29–35. [Google Scholar] [CrossRef]
- Choi, J.-H. Spectroscopic properties and ligand field analysis of cis-diazido(1,4,8,11-tetraazacyclotetradecane)chromium(III) azide. Spectrochim. Acta Part A 2000, 56, 1653–1660. [Google Scholar] [CrossRef]
- Choi, J.-H.; Oh, I.-G.; Linder, R.; Schönherr, T. Electronic spectroscopy and ligand field analysis of cis-carbonato(rac-5,5,7,12,12,14-hexamethyl-1,4,8,11-tetraazacyclotetradecane)chromium(III) chloride. Chem. Phys. 2004, 297, 7–12. [Google Scholar] [CrossRef]
- Hathaway, B.J.; Tomlinson, A.A.G. Copper ammonia complexes. Coord. Chem. Rev. 1970, 5, 1–43. [Google Scholar] [CrossRef]
- Goodman, B.A.; Raynor, J.B. Electron spin resonance of transition metal complexes. Adv. Inorg. Chem. Radiochem. 1970, 13, 135–362. [Google Scholar]
- Spackman, M.A.; Jayatilaka, D. Hirshfeld surface analysis. CrystEngComm 2009, 11, 19–32. [Google Scholar] [CrossRef]
- Moon, D.; Tanaka, S.; Akitsu, T.; Choi, J.-H. Molecular structure, spectroscopic properties, and Hirshfeld surface analysis of chlorobis(N-methyl-1,3-propanediamine)copper(II) tetrafluoroborate and azidobis(2,2-dimethyl-1,3-propanediamine)copper(II) azide. J. Mol. Struct. 2018, 1154, 338–347, Erratum in 2018, 1168, 329. [Google Scholar]
- Hathwar, V.R.; Sist, M.; Jørgensen, M.R.V.; Mamakhel, A.H.; Wang, X.; Hoffmann, C.M.; Sugimoto, K.; Overgaard, J.; Iversen, B.B. Quantitative analysis of intermolecular interactions in orthorhombic rubrene. IUCrJ 2015, 2, 563–574. [Google Scholar] [CrossRef] [PubMed]
- Spackman, M.A.; McKinnon, J.J.; Jayatilaka, D. Electrostatic potentials mapped on Hirshfeld surfaces provide direct insight into intermolecular interactions in crystals. CrystEngComm 2008, 10, 377–388. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Empirical formula | C20H44CuN4O2Br2 |
| Formula weight | 595.95 g mol−1 |
| Temperature | 100(1) K |
| Wavelength | 1.54186 Å |
| Crystal system, space group | Triclinic, Pī |
| Unit cell dimensions | a = 8.0777(4) Å, α = 98.879(4)° |
| b = 8.8129(4) Å, β = 110.204(3)° | |
| c = 10.0932(5) Å, γ = 109.391(3)° | |
| Volume | 650.97(5) Å3 |
| Z | 1 |
| Radiation type | Cu Kα |
| Density (calculated) | 1.633 Mg m−3 |
| Absorption coefficient | 5.31 mm−1 |
| Crystal size | 0.32 × 0.22 × 0.21 mm3 |
| Theta range for data collection | 4.9 to 70.5° |
| Reflections collected | 19,409 |
| Independent reflections | 9304 [Rint = 0.013] |
| Absorption correction | Multi-scan |
| Max. and min. transmission | 0.012 and 0.263 |
| Refinement method | Full-matrix least-squares on F2 |
| Data/restraints/parameters | 2241/0/134 |
| Goodness-of-fit on F2 | 1.11 |
| Final R indices (F2 > 2) | R1 = 0.034, wR2 |
| Extinction coefficient | 0.0050(3) |
| Largest diff. peak and hole (e Å−3) | 0.68 and −0.87 |
| Cu1–N1 | 2.018 (3) | C2–C9i | 1.527 (5) |
| Cu1–N2 | 2.049 (3) | C3–C4 | 1.531 (4) |
| Cu1–O1W | 2.632 (3) | C3–C8 | 1.526 (5) |
| N1–C1 | 1.489 (4) | C4–C5 | 1.528 (5) |
| N1–C3 | 1.488 (4) | C5–C6 | 1.516 (6) |
| N2–C8 | 1.497 (4) | C6–C7 | 1.528 (5) |
| N2–C9 | 1.498 (4) | C7–C8 | 1.525 (4) |
| C1–C2 | 1.522 (5) | C9–C10 | 1.521 (5) |
| N1–Cu1–N1i | 180.0 | N1–C1–C2 | 110.8 (3) |
| N1i–Cu1–N2 | 95.40 (11) | C1–C2–C9i | 116.1 (3) |
| N1–Cu1–N2 | 84.60 (11) | N1–C3–C4 | 113.4 (3) |
| N1–Cu1–O1Wi | 91.78 (10) | N1–C3–C8 | 106.4 (3) |
| N1–Cu1–O1W | 88.22 (10) | C8–C3–C4 | 111.3 (3) |
| N2–Cu1–N2i | 180.0 | C5–C4–C3 | 110.6 (3) |
| N2–Cu1–O1W | 95.89 (10) | C6–C5–C4 | 110.8 (3) |
| N2–Cu1–O1Wi | 84.11 (10) | C5–C6–C7 | 110.7 (3) |
| O1Wi–Cu1—O1W | 180.0 | C8–C7–C6 | 110.4 (3) |
| C1–N1–Cu1 | 116.0 (2) | N2–C8–C3 | 106.3 (3) |
| C3–N1–Cu1 | 108.07 (19) | N2–C8–C7 | 113.4 (3) |
| C3–N1–C1 | 113.4 (3) | C7–C8–C3 | 111.7 (3) |
| C8–N2–Cu1 | 107.20 (19) | N2–C9–C2i | 109.1 (3) |
| C8–N2–C9 | 114.6 (3) | N2—C9–C10 | 111.8 (3) |
| C9–N2–Cu1 | 121.0 (2) | C10–C9–C2i | 113.3 (3) |
| Compound | M–N | M–X | Conformation | Ref. |
|---|---|---|---|---|
| [Cu(L1)(H2O)2]Br2 | 2.018(3)–2.049(3) | 2.632(3) | trans-III | This work |
| [Cu(L1)(H2O)2](BF4)2·2H2O | 2.028(2)–2.028(2) | 2.693(3) | trans-III | [7] |
| [Cu(L1)(H2O)2]Cl2 | 2.017(2)–2.038(2) | 2.649(2) | trans-III | [8] |
| [Cu(L1)(ClO4)2] | 2.005(2)–2.048(2) | 2.623(2) | trans-III | [9] |
| [Cu(L1)(NO3)2] | 2.007(2)–2.044(2) | 2.463(2) | trans-III | [10] |
| [Cu(L1)(NO3)2]·3H2O | 2.021(2)–2.029(2) | 2.746(2) | trans-III | [11] |
| [Cu(L1)](NCS)2·2H2O | 2.020(7)–2.056(7) | 3.037(7) | trans-III | [12] |
| [Cu(L1)(NCS)]SCN | 2.013(2)–2.052(2) | 2.322(3) | trans-III | [13] |
| [Cu(L1)(N3)]ClO4·H2O | 2.030(3)–2.054(3) | 2.254(4) | trans-III | [14] |
| [Cu(L2)](ClO4)2 | 1.990(4)–2.050(4) | 3.496(3) | trans-I | [15] |
| [Cu(L2)](NO3)2 | 1.999(7) –2.095(7) | 2.964(7) | trans-III | [8] |
| [Cu(L3)](ClO4)2·2CH3CN | 2.030(3)–2.081(2) | 3.264(3) | trans-III | [16] |
| [Cu(La)(ClO4)2] | 2.016(2) –2.040(2) | 2.762(2) | trans-III | [17] |
| [Cux(H2(1-x)La)(ClO4)2] (x = 0.69) | 2.015(3) –2.047(3) | 2.795(3) | trans-III | [18] |
| [Cu(La)(NO3)2] | 2.014(2) –2.047(2) | 2.506(2) | trans-III | [19] |
| [Cu(La)(H2O)2](SCN)2 | 2.021(2) –2.046(2) | 2.569(2) | trans-III | [19] |
| [Ni(La)(NO3)2] | 2.051(4) –2.094(4) | 2.198(3) | trans-III | [20] |
| [Ni(La)](ClO4)2·2H2O | 1.996(7) –2.042(7) | 2.973(7) | trans-III | [20] |
| [Ni(La)(N3)2] | 2.070(1) –2.105(1) | 2.176(1) | trans-III | [21] |
| [Cu(Lb)](NO3)2 | 2.023(2) –2.086(2) | 2.936(2) | trans-III | [22] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jeon, S.; Moncol, J.; Mazúr, M.; Valko, M.; Choi, J.-H. Synthesis, Crystal Structure, Spectroscopic Properties, and Hirshfeld Surface Analysis of Diaqua [3,14-dimethyl-2,6,13,17 tetraazatricyclo(16.4.0.07,12)docosane]copper(II) Dibromide. Crystals 2019, 9, 336. https://doi.org/10.3390/cryst9070336
Jeon S, Moncol J, Mazúr M, Valko M, Choi J-H. Synthesis, Crystal Structure, Spectroscopic Properties, and Hirshfeld Surface Analysis of Diaqua [3,14-dimethyl-2,6,13,17 tetraazatricyclo(16.4.0.07,12)docosane]copper(II) Dibromide. Crystals. 2019; 9(7):336. https://doi.org/10.3390/cryst9070336
Chicago/Turabian StyleJeon, Sunghwan, Ján Moncol, Milan Mazúr, Marián Valko, and Jong-Ha Choi. 2019. "Synthesis, Crystal Structure, Spectroscopic Properties, and Hirshfeld Surface Analysis of Diaqua [3,14-dimethyl-2,6,13,17 tetraazatricyclo(16.4.0.07,12)docosane]copper(II) Dibromide" Crystals 9, no. 7: 336. https://doi.org/10.3390/cryst9070336
APA StyleJeon, S., Moncol, J., Mazúr, M., Valko, M., & Choi, J.-H. (2019). Synthesis, Crystal Structure, Spectroscopic Properties, and Hirshfeld Surface Analysis of Diaqua [3,14-dimethyl-2,6,13,17 tetraazatricyclo(16.4.0.07,12)docosane]copper(II) Dibromide. Crystals, 9(7), 336. https://doi.org/10.3390/cryst9070336