Fab Fragment of VHH-Based Antibody Netakimab: Crystal Structure and Modeling Interaction with Cytokine IL-17A

 ,
, 
Abstract
1. Introduction
2. Materials and Methods
2.1. Production and Purification of the Fab Fragment and IL-17A
2.2. Production and Purification of the VHH Domain
2.3. Circular Dichroism Measurements
2.4. Production and Purification of IL-17A
2.5. Analysis of the Interaction of the Fab Fragment and the VHH domain with IL-17A by Surface Plasmon Resonance Approach (SPR)
2.6. Crystallization of the Fab Fragment
2.7. Diffraction Data Collection and Determining the Structure of the Fab Fragment
2.8. Docking and Molecular Dynamics Simulations
3. Results
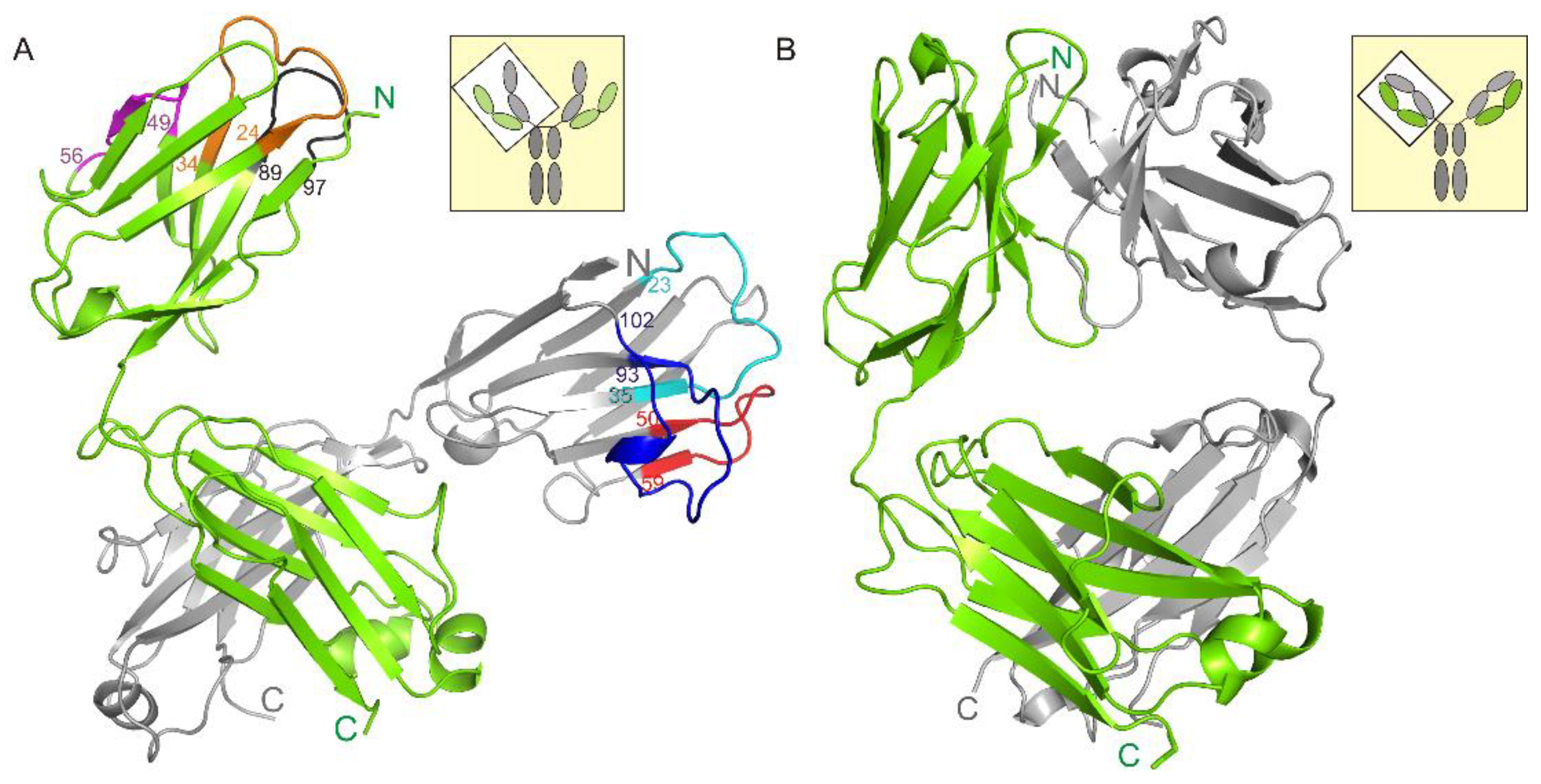
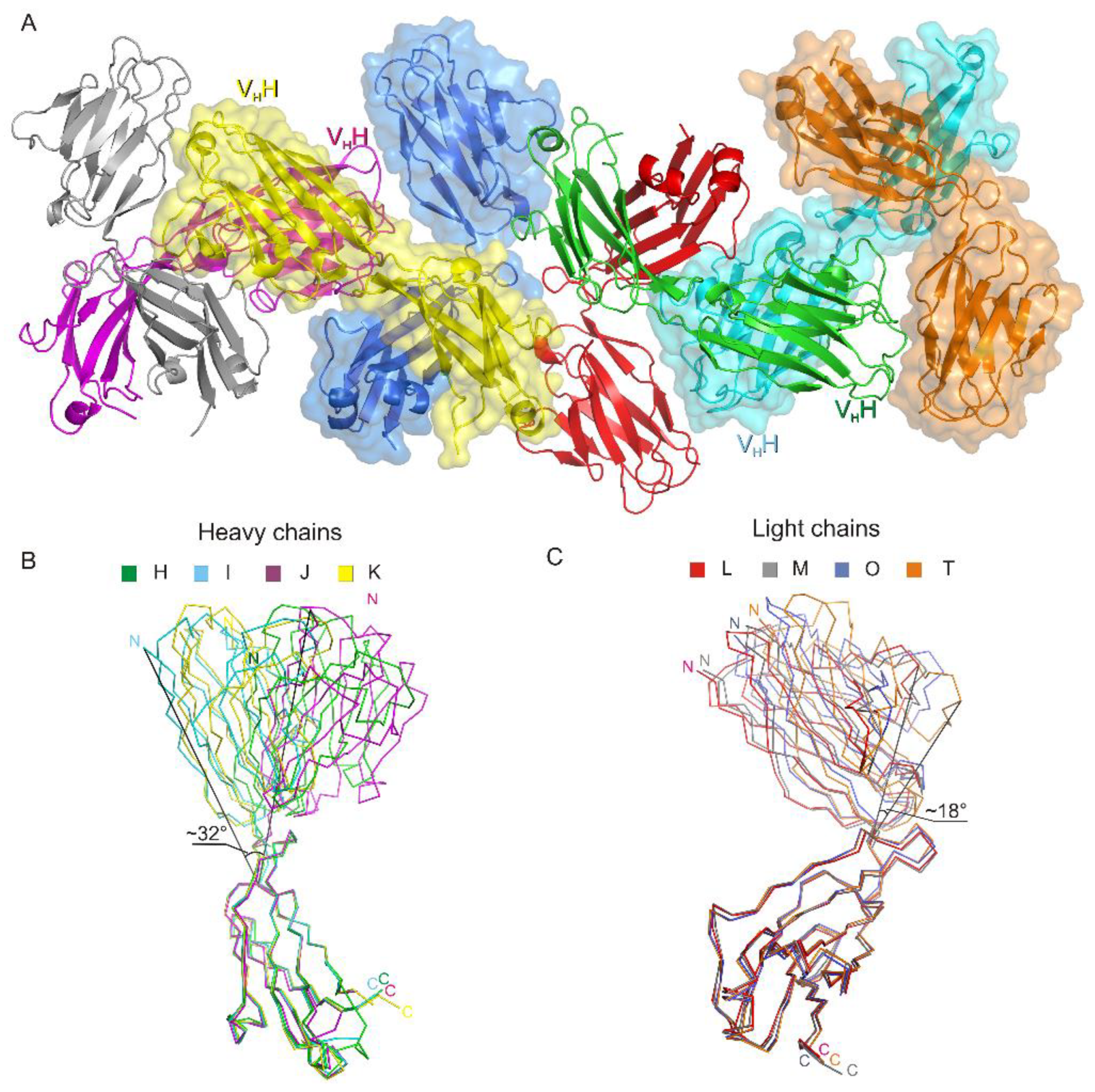
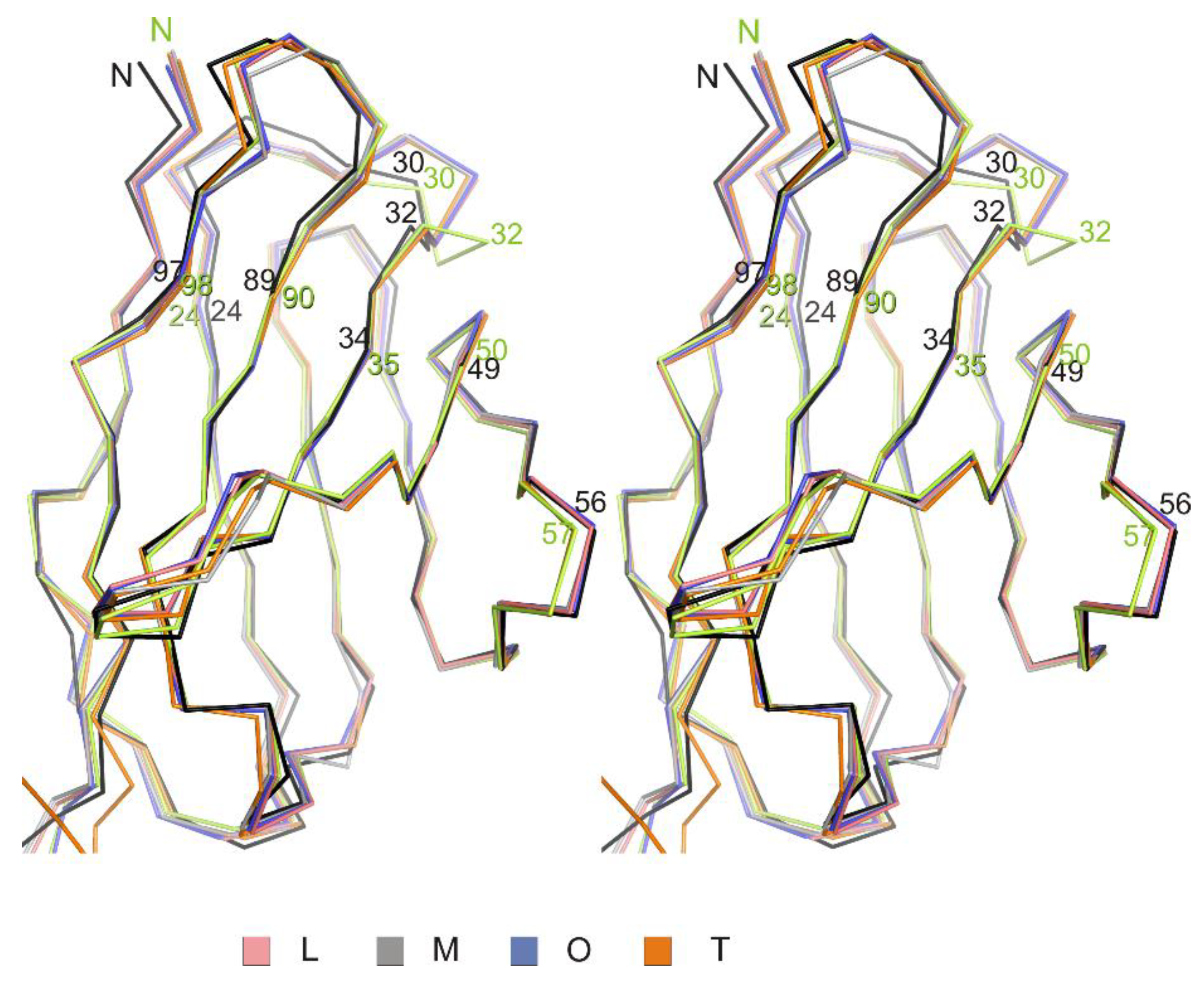
3.1. Crystal Structure of the Fab Fragment of Netakimab at 1.9 Å Resolution
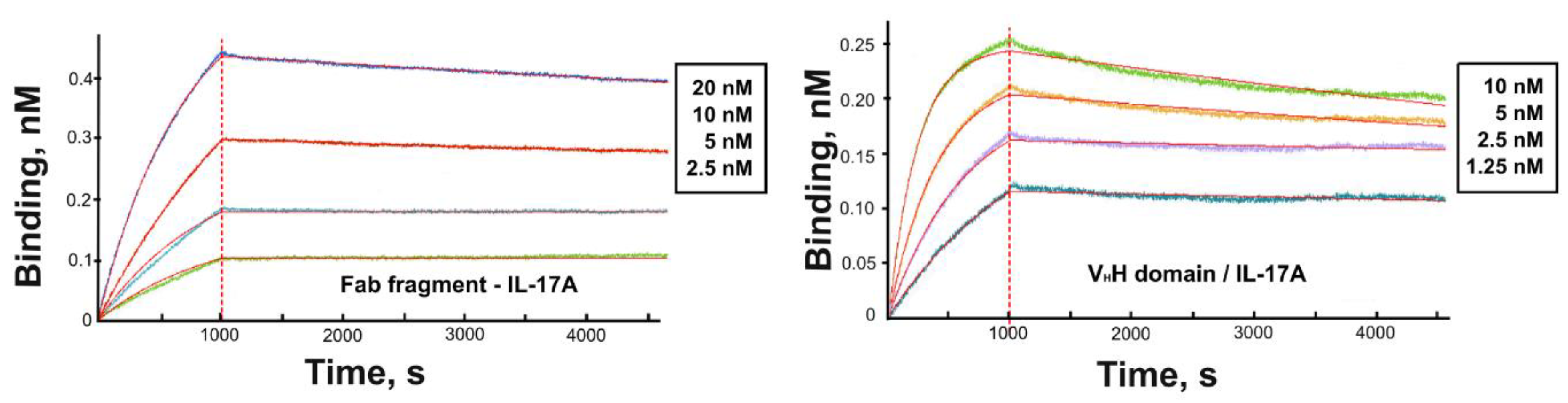
3.2. Kinetic Analysis
3.3. Modeling the VHH Domain/IL-17A Complex
4. Discussion
4.1. Structure of the Netakimab Fab Fragment
4.2. Modeling the VHH Domain/IL-17A Complex
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Cypowyj, S.; Picard, C.; Maródi, L.; Casanova, J.-L.; Puel, A. Immunity to Infection in IL-17-Deficient Mice and Humans. Eur. J. Immunol. 2012, 42, 2246–2254. [Google Scholar] [CrossRef]
- Beringer, A.; Noack, M.; Miossec, P. IL-17 in Chronic Inflammation: From Discovery to Targeting. Trends Mol. Med. 2016, 22, 230–241. [Google Scholar] [CrossRef]
- Paek, S.Y.; Frieder, J.; Kivelevitch, D.; Menter, M.A. IL-17 Inhibitors for Psoriasis. Semin. Cutan. Med. Surg. 2018, 37, 148–157. [Google Scholar] [CrossRef]
- Wolfson, W. Ablynx Makes Nanobodies from Llama Bodies. Chem. Biol. 2006, 13, 1243–1244. [Google Scholar] [CrossRef] [PubMed]
- Muyldermans, S. Single domain camel antibodies: Current status. J. Biotechnol. 2001, 74, 277–302. [Google Scholar] [CrossRef]
- Schmitz, K.R.; Bagchi, A.; Roovers, R.C.; van Bergen en Henegouwen, P.M.P.; Ferguson, K.M. Structural Evaluation of EGFR Inhibition Mechanisms for nanobodies/VHH Domains. Structure 2013, 21, 1214–1224. [Google Scholar] [CrossRef]
- Cortez-Retamozo, V. Efficient Cancer Therapy with a Nanobody-Based Conjugate. Cancer Res. 2004, 64, 2853–2857. [Google Scholar] [CrossRef]
- Cortez-Retamozo, V.; Lauwereys, M.; Hassanzadeh, G.; Gobert, M.; Conrath, K.; Muyldermans, S.; De Baetselier, P.; Revets, H. Efficient Tumor Targeting by Single-Domain Antibody Fragments of Camels. Int. J. Cancer 2002, 98, 456–462. [Google Scholar] [CrossRef]
- Harmsen, M.M.; Van Solt, C.B.; Fijten, H.P.D.; Van Setten, M.C. Prolonged in Vivo Residence Times of Llama Single-Domain Antibody Fragments in Pigs by Binding to Porcine Immunoglobulins. Vaccine 2005, 23, 4926–4934. [Google Scholar] [CrossRef] [PubMed]
- Richard, G.; Meyers, A.J.; McLean, M.D.; Arbabi-Ghahroudi, M.; MacKenzie, R.; Hall, J.C. In Vivo Neutralization of α-Cobratoxin with High-Affinity Llama Single-Domain Antibodies (VHHs) and a VHH-Fc Antibody. PLoS ONE 2013, 8, e69495. [Google Scholar] [CrossRef] [PubMed]
- Kontermann, R.E. Strategies to Extend Plasma Half-Lives of Recombinant Antibodies. BioDrugs 2009, 23, 93–109. [Google Scholar] [CrossRef] [PubMed]
- Hoefman, S.; Ottevaere, I.; Baumeister, J.; Sargentini-Maier, M. Pre-Clinical Intravenous Serum Pharmacokinetics of Albumin Binding and Non-Half-Life Extended Nanobodies®. Antibodies 2015, 4, 141–156. [Google Scholar] [CrossRef]
- Ulitin, A.; Evdokimov, S.; Soloviev, V.; Chernyh, Y.; Goncharova, O.; Korzhavin, D.; Chernovskaya, T.; Nemankin, T.; Ivanov, R.; Morozov, D.; et al. High Affinity and Aggregatively Stable Antibodies on the Basis of Variable Domains vl and a Derivative vhh. U.S. Patent 2016048188A1, 31 March 2016. [Google Scholar]
- International Nonproprietary Names for Pharmaceutical Substances (INN). INN: List 80; WHO Drug Information; WHO: Geneva, Switzerland, 2018; Volume 32, pp. 425–508. [Google Scholar]
- Samtsov, A.V.; Khairutdinov, V.R.; Bakulev, A.L.; Kubanov, A.A.; Karamova, A.E.; Artem’eva, A.V.; Korotaeva, T.V. Efficacy and Safety of BCD-085, a Novel Interleukin-17 Inhibitor. Results of Phase II Clinical Trial in Patients with Moderate-to-Severe Plaque Psoriasis. Vestn. Dermatol. Venerol. 2017, 5, 52–63. [Google Scholar] [CrossRef]
- Nurizzo, D.; Mairs, T.; Guijarro, M.; Rey, V.; Meyer, J.; Fajardo, P.; Chavanne, J.; Biasci, J.C.; McSweeney, S.; Mitchell, E. The ID23-1 Structural Biology Beamline at the ESRF. J. Synchrotron Radiat. 2006, 13 Pt 3, 227–238. [Google Scholar] [CrossRef]
- Gabadinho, J.; Beteva, A.; Guijarro, M.; Rey-Bakaikoa, V.; Spruce, D.; Bowler, M.W.; Brockhauser, S.; Flot, D.; Gordon, E.J.; Hall, D.R.; et al. MxCuBE: A Synchrotron Beamline Control Environment Customized for Macromolecular Crystallography Experiments. J. Synchrotron Radiat. 2010, 17, 700–707. [Google Scholar] [CrossRef]
- Bourenkov, G.P.; Popov, A.N. Optimization of Data Collection Taking Radiation Damage into Account. Acta Crystallogr. D Biol. Crystallogr. 2010, 66 Pt 4, 409–419. [Google Scholar] [CrossRef]
- Kabsch, W. XDS. Acta Crystallogr. D Biol. Crystallogr. 2010, 66 Pt 2, 125–132. [Google Scholar] [CrossRef]
- McCoy, A.J.; Grosse-Kunstleve, R.W.; Adams, P.D.; Winn, M.D.; Storoni, L.C.; Read, R.J. Phaser Crystallographic Software. J. Appl. Crystallogr. 2007, 40 Pt 4, 658–674. [Google Scholar] [CrossRef]
- Murshudov, G.N.; Skubák, P.; Lebedev, A.A.; Pannu, N.S.; Steiner, R.A.; Nicholls, R.A.; Winn, M.D.; Long, F.; Vagin, A.A. REFMAC5 for the Refinement of Macromolecular Crystal Structures. Acta Crystallogr. D Biol. Crystallogr. 2011, 67 Pt 4, 355–367. [Google Scholar] [CrossRef]
- Emsley, P.; Lohkamp, B.; Scott, W.G.; Cowtan, K. Features and Development of Coot. Acta Crystallogr. D Biol. Crystallogr. 2010, 66 Pt 4, 486–501. [Google Scholar] [CrossRef]
- The PyMOL Molecular Graphics System. Available online: https://pymol.org/2/ (accessed on 1 March 2019).
- Yan, Y.; Zhang, D.; Zhou, P.; Li, B.; Huang, S. HDOCK: A web server for protein—Protein and protein—DNA/RNA docking based on a hybrid strategy. Nucleic Acids Res. 2017, 45, 365–373. [Google Scholar] [CrossRef] [PubMed]
- Hess, B.; Kutzner, C.; van der Spoel, D.; Lindahl, E. GROMACS 4: Algorithms for Highly Efficient, Load-Balanced, and Scalable Molecular Simulation. J. Chem. Theory Comput. 2008, 4, 435–447. [Google Scholar] [CrossRef] [PubMed]
- MacKerell, A.D.; Bashford, D.; Bellott, M.; Dunbrack, R.L.; Evanseck, J.D.; Field, M.J.; Fischer, S.; Gao, J.; Guo, H.; Ha, S.; et al. All-Atom Empirical Potential for Molecular Modeling and Dynamics Studies of Proteins. J. Phys. Chem. B 1998, 102, 3586–3616. [Google Scholar] [CrossRef]
- Mackerell, A.D., Jr.; Feig, M.; Brooks, C.L., 3rd. Extending the Treatment of Backbone Energetics in Protein Force Fields: Limitations of Gas-Phase Quantum Mechanics in Reproducing Protein Conformational Distributions in Molecular Dynamics Simulations. J. Comput. Chem. 2004, 25, 1400–1415. [Google Scholar] [CrossRef] [PubMed]
- Hess, B.; Bekker, H.; Berendsen, H.; Johannes, G. LINCS: A linear constraint solver for molecular simulations. J. Comput. Chem. 1997, 18, 1463–1472. [Google Scholar] [CrossRef]
- Darden, T.; York, D.; Pedersen, L. Particle Mesh Ewald: An N log(N) Method for Ewald Sums in Large Systems. J. Chem. Phys. 1993, 98, 10089–10092. [Google Scholar] [CrossRef]
- Essmann, U.; Perera, L.; Berkowitz, M.L.; Darden, T.; Lee, H.; Pedersen, L.G. A Smooth Particle Mesh Ewald Method. J. Chem. Phys. 1995, 103, 8577–8593. [Google Scholar] [CrossRef]
- Berendsen, H.J.C.; Postma, J.P.M.; van Gunsteren, W.F.; DiNola, A.; Haak, J.R. Molecular Dynamics with Coupling to an External Bath. J. Chem. Phys. 1984, 81, 3684–3690. [Google Scholar] [CrossRef]
- PDBePISA (Proteins, Interfaces, Structures and Assemblies). Available online: http://www.ebi.ac.uk/pdbe/prot_int/pistart.html (accessed on 1 March 2019).
- Krissinel, E.; Henrick, K. Inference of Macromolecular Assemblies from Crystalline State. J. Mol. Biol. 2007, 372, 774–797. [Google Scholar] [CrossRef]
- Liu, S.; Song, X.; Chrunyk, B.A.; Shanker, S.; Hoth, L.R.; Marr, E.S.; Griffor, M.C. Crystal Structures of Interleukin 17A and Its Complex with IL-17 Receptor A. Nat. Commun. 2013, 4, 1888. [Google Scholar] [CrossRef]
- Hassanzadeh-Ghassabeh, G.; Devooght, N.; Pauw, P.; Vincke, C.; Muyldermans, S. Nanobodies and their potential applications. Nanomedicine 2013, 8, 1013–1026. [Google Scholar] [CrossRef] [PubMed]
- North, B.; Lehmann, A.; Dunbrack, R.L., Jr. A New Clustering of Antibody CDR Loop Conformations. J. Mol. Biol. 2011, 406, 228–256. [Google Scholar] [CrossRef]
- Martin, A.C.; Thornton, J.M. Structural Families in Loops of Homologous Proteins: Automatic Classification, Modelling and Application to Antibodies. J. Mol. Biol. 1996, 263, 800–815. [Google Scholar] [CrossRef] [PubMed]
- Stanfield, R.L.; Zemla, A.; Wilson, I.A.; Rupp, B. Antibody Elbow Angles Are Influenced by Their Light Chain Class. J. Mol. Biol. 2006, 357, 1566–1574. [Google Scholar] [CrossRef]
- Bailey, L.J.; Sheehy, K.M.; Dominik, P.K.; Liang, W.G.; Rui, H.; Clark, M.; Jaskolowski, M.; Kim, Y.; Deneka, D.; Tang, W.-J.; et al. Locking the Elbow: Improved Antibody Fab Fragments as Chaperones for Structure Determination. J. Mol. Biol. 2018, 430, 337–347. [Google Scholar] [CrossRef] [PubMed]
- Gerhardt, S.; Abbott, W.M.; Hargreaves, D.; Pauptit, R.A.; Davies, R.A.; Needham, M.R.C.; Langham, C.; Barker, W.; Aziz, A.; Snow, M.J.; et al. Structure of IL-17A in Complex with a Potent, Fully Human Neutralizing Antibody. J. Mol. Biol. 2009, 394, 905–921. [Google Scholar] [CrossRef]
- Ekimova, V.; Ulitin, A.; Evdokimov, S.; Sofronova, E.; Nemankin, T.; Solovyev, V.; Ustugov, I.; Nedorubov, A.; Chernykh, Y.; Goncharova, O.; et al. High Affinity Anti-Il-17a Monoclonal Antibody. In Proceedings of the Conference: PEGS, Boston, MA, USA, 4–8 May 2015. [Google Scholar]
- Padova, D.; Gram, H.; Hofstetter, H.; Jeschke, M.; Rondeau, J.; Van Den Berg, W. IL-17 Antagonistic Antibodies. U.S. Patent 7,807,155 B2, 5 October 2010. [Google Scholar]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Data Collection | |
| Wavelength (Å) | 0.97242 |
| Space group | P212121 (No. 19) |
| Unit-cell parameters (Å, ˚) | a = 90.56, b = 111.92, c = 165.43, α = γ =β = 90 |
| Resolution limits (Å) | 50.0–1.9 (2.01–1.9) |
| Completeness (%) | 99.7 (98.7) |
| Measured reflections | 815,153 (124,199) |
| Unique reflections | 132,510 (20,959) |
| Rmerge (%) | 5.3 (99.8) |
| Mean I/σ (I) | 19.2 (1.7) |
| Redundancy | 6.2 (5.9) |
| CC(1/2) | 100.0 (88.6) |
| Refinement | |
| Resolution (Å) | 47.6–1.9 (1.95–1.9) |
| No. of reflections | 125,855 (8961) |
| R factor (%) | 16.8 (31.8) |
| Free R factor (%) | 22.8 (37.6) |
| Refinement | |
| Average overall B factor (Å2) | 48.8 |
| Ramachandran plot | |
| Most favored (%) | 97.3 |
| Allowed (%) | 2.7 |
| R.m.s. deviations | |
| Bond lengths (Å) | 0.010 |
| Bond angles (º) | 1.291 |
| PDB code | 6QKD |
| Protein | ka (M−1 s−1 ) | kd (s−1) | KD (nM) |
|---|---|---|---|
| Fab fragment | 6.3 × 104 | 2.0 × 10−5 | 0.3 |
| VHH domain | 5.0 × 105 | 4.3 × 10−5 | 0.09 |
| Region Name | Residues (Kabat Database) | Cluster (Length) | Median PDB Entry |
|---|---|---|---|
| HFR1 | 1-22 | ||
| CDR-H1 | 23-35 | H1-13-3 (13 aa) | 1U0Q |
| HFR2 | 36-49 | ||
| CDR-H2 | 50-58 | H2-10-2 (10 aa) | 1SEQ |
| HFR3 | 59-92 | ||
| CDR-H3 | 93-102 | 18 aa | 1MVF |
| HFR4 | 103-113 | ||
| LFR1 | 1-23 | ||
| CDR-L1 | 24-34 | L1-12-2 (12 aa) | 2FX7 |
| LFR2 | 35-48 | ||
| CDR-L2 | 49-56 | L2-8-5 (8 aa) | 1IGM |
| LFR3 | 57-88 | ||
| CDR-L3 | 89-97 | L3-9cis7-1 (9 aa) | 1IGM |
| LFR4 | 98-109 |
| Anti-IL-17A antibody | KD (nM) | IC50 (pM) | Reference |
|---|---|---|---|
| netakimab Fab fragment | 0.3 | 66 | this article and [41] |
| VHH domain | 0.09 | 30 | this article and [13] |
| Secukinumab | 0.12 | 201 | [42] |
| CAT 2200 Fab fragment | 2.1 | 1560 | [40] |
| VHH Domain | Distance (Å) | IL-17A |
|---|---|---|
| Hydrogen bonds | ||
| Gly H42 O | 3.27 | Gln A94 NE2 |
| Lys H43 NZ | 2.92 | Glu A95 OE2 |
| Glu H85 OE1 | 2.66 | Lys B38 NZ |
| Arg H97 NH2 | 3.09 | Pro B59 O |
| Asp H99 OD1 | 2.58 | Arg B101 NH2 |
| Asp H99 OD2 | 3.19 | Asn B108 ND2 |
| Thr H100A OG1 | 3.55 | Asn A17 ND2 |
| Gly H100F O | 2.90 | Arg B101 NH1 |
| Asp H100G OD1 | 2.73 | Arg B101 NH2 |
| Ser H102 OG | 2.69 | Glu B57 OE2 |
| Ser H102 OG | 3.01 | Arg B55 NH1 |
| Nonpolar (distance corresponds to the closest atom pair) | ||
| Thr H100A CG2 | 3.70 | Ile B28 CD |
| Thr H100A CG2 | 4.63 | Leu B26 CG |
| Tyr H100C CB | 3.56 | Ile B28 CD |
| Tyr H100D CE1 | 3.53 | Tyr B62 CD2 |
| Tyr H100D CZ | 3.88 | Phe B110 CZ |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kostareva, O.; Kolyadenko, I.; Ulitin, A.; Ekimova, V.; Evdokimov, S.; Garber, M.; Tishchenko, S.; Gabdulkhakov, A. Fab Fragment of VHH-Based Antibody Netakimab: Crystal Structure and Modeling Interaction with Cytokine IL-17A. Crystals 2019, 9, 177. https://doi.org/10.3390/cryst9030177
Kostareva O, Kolyadenko I, Ulitin A, Ekimova V, Evdokimov S, Garber M, Tishchenko S, Gabdulkhakov A. Fab Fragment of VHH-Based Antibody Netakimab: Crystal Structure and Modeling Interaction with Cytokine IL-17A. Crystals. 2019; 9(3):177. https://doi.org/10.3390/cryst9030177
Chicago/Turabian StyleKostareva, Olga, Ilya Kolyadenko, Andrey Ulitin, Victoria Ekimova, Stanislav Evdokimov, Maria Garber, Svetlana Tishchenko, and Azat Gabdulkhakov. 2019. "Fab Fragment of VHH-Based Antibody Netakimab: Crystal Structure and Modeling Interaction with Cytokine IL-17A" Crystals 9, no. 3: 177. https://doi.org/10.3390/cryst9030177
APA StyleKostareva, O., Kolyadenko, I., Ulitin, A., Ekimova, V., Evdokimov, S., Garber, M., Tishchenko, S., & Gabdulkhakov, A. (2019). Fab Fragment of VHH-Based Antibody Netakimab: Crystal Structure and Modeling Interaction with Cytokine IL-17A. Crystals, 9(3), 177. https://doi.org/10.3390/cryst9030177