Abstract
How many strong C−I⋯N halogen bonds can one 1,3,5-trifluoro-2,4,6-triiodobenzene molecule form in a crystal structure? To answer this question, we investigated in detail the noncovalent interactions between 1,3,5-trifluoro-2,4,6-triiodobenzene and a series of 1,10-phenanthroline derivatives by employing a combined theoretical and experimental method. The results of the quantum chemical calculations and crystallographic experiments clearly show that there is a structural competition between a C−I⋯N halogen bond and π⋯π stacking interaction. For example, when there are much stronger π⋯π stacking interactions between two 1,10-phenanthroline derivative molecules or between two 1,3,5-trifluoro-2,4,6-triiodobenzene molecules in the crystal structures, then one 1,3,5-trifluoro-2,4,6-triiodobenzene molecule forms only one C−I⋯N halogen bond with one 1,10-phenanthroline derivative molecule. Another example is when π⋯π stacking interactions in the crystal structures are not much stronger, one 1,3,5-trifluoro-2,4,6-triiodobenzene molecule can form two C−I⋯N halogen bonds with two 1,10-phenanthroline derivative molecules.
1. Introduction
The pioneering work of Resnati, Metrangolo, and coworkers has led to a surge of interest in one of the most important noncovalent interactions—the halogen bond [1,2]. The key role of the halogen bond in molecular materials assembly, drug design, and other related fields has been unveiled in the last thirty years [1,2,3,4,5,6,7,8,9,10,11,12,13,14,15,16,17,18,19,20]. The strength of the halogen-bond donor increases in the order, F < Cl < Br < I. On the other hand, the strength of the halogen bonds can be enhanced by introducing electron-withdrawing substituents to the halogen-bond donors, such as fluorine atom, nitro groups, cyano groups, etc. [6]. These findings have been confirmed by both experiments and theoretical calculations [6]. Therefore, perfluorohaloarenes are particularly good halogen-bond donors. As one of the most commonly used halogen-bond donors, 1,3,5-trifluoro-2,4,6-triiodobenzene (TFTIB), has been utilized in the construction of various supramolecular architectures with intriguing topology and interesting properties [21]. The TFTIB molecule has three C−I bonds. It is natural to assume that TFTIB is a tritopic halogen-bonding donor and it can form three strong C−I⋯N halogen bonds with the Lewis bases containing electron-rich nitrogen atoms. To gain insight into the factors controlling the formation of multiple halogen bonds with a single TFTIB molecule, van der Boom and coworkers studied the cocrystals formed by TFTIB with a series of pyridyl derivatives [22,23]. They found that the cocrystals that were obtained consistently contained two C−I⋯N halogen bonds instead of the anticipated three C−I⋯N halogen bonds [22,23]. In a later study, Bruce et al. argued that a single TFTIB molecule might be able to form three halogen bonds based on the observations that three C−I⋯N halogen bonds were utilized in the complex with 4-(N,N-dimethylamino)pyridine [24]. In 2014, Aakeröy and coworkers reported that a cocrystal formed by TFTIB with 1,1’-bibenzyl-2,2’-biimidazole, in which a single TFTIB molecule formed three C−I⋯N halogen bonds with three 1,1’-bibenzyl-2,2’-biimidazole molecules [25]. In a recent study, Hidalgo et al. reported a series of five crystal structures of 1:1 halogen-bonded complexes between TFTIB and 4-[5-(4-alkoxyphenyl)-1,3,4-oxadiazole-2-yl]pyridine. In each of the five crystal structures, one TFTIB molecule formed three C−I⋯N halogen bonds with neutral halogen atom acceptors [26]. Unfortunately, the latter two studies did not discuss the factors controlling the formation of multiple halogen bonds with a single TFTIB molecule. It is worth noting that other studies showed that if much stronger ionic halogen bonds were employed, then all three iodine atoms of TFTIB could be halogen bonded, provided that the voids generated in the network were filled by a suitable cation [27,28,29].
Increasing the number of iodine atoms in a single aromatic molecule does not always result in the formation of multiple halogen bonds, as mentioned above. Therefore, what are the factors determining whether a single aromatic molecule is a monotopic, ditopic, or tritopic halogen-bond donor? Employing a combined experimental and theoretical approach, van der Boom and coworkers concluded that the reluctance of the formation of a supramolecular assembly having a third C−I⋯N halogen bond does not depend on the size of the bispyridine donor systems or the weakening of the C−I⋯N noncovalent interactions. More pyridine moieties coordinated to TFTIB might be a contributing factor in the consistent formation of two rather than three C−I⋯N halogen bonds [22,23]. However, the experimental and theoretical study of the halogen-bonded complex between TFTIB and 4-(N,N-dimethylamino)pyridine showed that the weakening of the C−I⋯N halogen bonds, as more halogen-bond acceptors coordinate to TFTIB, was not an obstacle to the formation of multiple halogen bonds in polyiodo systems [24]. Our previous theoretical calculations showed that, with the increasing number of iodine atoms, the π⋯π stacking interaction between two C6FxI(6−x) (x = 0, 1, 2, 3, 4 or 5) molecules became much stronger, and the π⋯π stacking interaction between two TFTIB molecules was stronger than the halogen bond between TFTIB and pyridine [30,31]. The subsequent crystallographic study confirmed the above theoretical predictions, and showed that a strong C−I⋯N halogen bond successfully competed with the π⋯π interaction between two 1,3-diiodotetrafluorobenzene molecules, while the π⋯π interaction between two TFTIB molecules competed successfully with the strong C−I⋯N halogen bond [32].
From the discussion above, it seems that the structural competition between the π⋯π interaction and C−I⋯N halogen bond plays a dominant role in determining the formation of two or three C−I⋯N halogen bonds. We still need more related studies to provide multiple, converging findings. In the present study, we investigated the structural competition between the π⋯π interaction and C−I⋯N halogen bond in the crystal structures of the cocrystals formed by TFTIB with 2-chloro-1,10-phenanthroline (ClPHEN), 2-bromo-1,10-phenanthroline (BrPHEN), 2-methyl-1,10-phenanthroline (CH3PHEN), and 2,9-dimethyl-1,10-phenanthroline (DCH3PHEN), respectively, by employing a combined theoretical and experimental method.
2. Materials and Methods
2.1. Quantum Chemical Calculation
The complexes formed by TFTIB with pyridine and 4-(N,N-dimethylamino)pyridine were selected as models to study the cooperativity or anticooperativity of the halogen bonds. The structures of all these model complexes were fully optimized at the ωB97X-D/def2-TZVPP level of theory [33,34,35]. The binding energies were also calculated at this theory level. For the halogen-bonded or π-stacked complexes in the crystal structures, their structures were not optimized and only single-point binding energies were calculated at the ωB97X-D/def2-TZVPP theory level. The binding energies were calculated with the supermolecule method and corrected for basis set superposition error using the Boys and Bernardi function counterpoise method [36]. The reliability of the ωB97X-D/def2-TZVPP calculations for the study of the noncovalent interactions can be found in our previous paper [31]. An “ultrafine” integration grid was used for all the ωB97X-D/def2-TZVPP calculations to avoid possible integration grid errors. The bonding characteristic of the C−I⋯N halogen bonds was analyzed using the natural bond orbital (NBO) theory of Weinhold and co-workers [37]. NBO analyses were carried out at the ωB97X-D/def2-TZVPP level of theory using ωB97X-D/def2-TZVPP geometry. All the calculations were performed with the GAUSSIAN 09 program package [38].
2.2. Crystal Preparation
The halogen atom donor TFTIB and the halogen atom acceptors ClPHEN, BrPHEN, CH3PHEN, and DCH3PHEN were purchased from Aldrich Chemical Co., and used without further purification. The solvent for crystallization was trichloromethane and was used as received. The halogen atom donor (0.50 mmol) and halogen atom acceptor (0.50 mmol) were dissolved in 15 mL trichloromethane at room temperature. After filtration, the solution was slowly evaporated at room temperature. The single crystals suitable for X-ray diffraction studies were obtained in about two days.
2.3. Measurement
Single crystal X-ray diffraction data were collected on a Rigaku AFC10 diffractometer (Rigaku Corporation, Tokyo, Japan) equipped with Rigaku Supranava X-ray generator (graphite-monochromatic Mo-Kα radiation, λ = 0.71073 Å) at room temperature. All structures were solved and refined by a combination of direct methods and difference Fourier syntheses, using the SHELX-2014 and Olex2.0 programs [39,40]. Anisotropic thermal parameters were assigned to all non-hydrogen atoms. The hydrogen atoms were set in calculated positions and refined as riding atoms with a common isotropic thermal parameter. Crystallographic data have been deposited at the Cambridge Crystallographic Data Centre (deposition numbers CCDC 1891219, 1891220, 1891221, and 1891222). Copies of the data can be obtained free of charge via http://www.ccdc.cam.ac.uk/conts/retrieving.html.
3. Results and Discussion
3.1. Anticooperativity of the Halogen Bonds
The interconnected halogen bonds may not only enhance, but also reduce the strengths of each other. The strengthening effect is often called cooperativity and the weakening effect is often called anticooperativity. When the interconnected halogen bonds run in the same direction, all the halogen bonds become stronger. On the contrary, the different directions of the interconnected halogen bonds will lead to local or whole anticooperativity. Evidently, the anticooperativity of the C−I⋯N halogen bonds occurs as two or three pyridine molecules coordinate to one TFTIB molecule. The density functional theory calculations have revealed a weakening of the C−I⋯N halogen bonds as more pyridine moieties coordinate to the TFTIB [22]. Is the anticooperativity of the C−I⋯N halogen bonds a contributing factor to the formation of two rather than three C−I⋯N halogen bonds? To answer this question, we still need more data.
Figure 1 illustrates the C−I bond lengths, C−I⋯N halogen bond lengths (I⋯N distances), and C−I⋯N halogen bond strengths for the monomer TFTIB and molecular clusters containing TFTIB calculated at the ωB97X-D/def2-TZVPP level of theory. As more pyridine or 4-(N,N-dimethylamino)pyridine moieties coordinate to the TFTIB, the C−I bond lengths become smaller, the C−I⋯N halogen bond lengths become larger, and the C−I⋯N halogen bond strengths become smaller. It is reasonable that the C−I bond length is proportional to the C−I⋯N halogen bond strength, and the C−I⋯N halogen bond length is inversely proportional to the C−I⋯N halogen bond strength. The NBO analysis will allow us to quantitatively evaluate the charge transfer involving the formation of the halogen bond. The results of the NBO analyses show that the charge-transfer stabilization energies for n(N)→σ*(C−I) decrease with the increase in the number of pyridine or 4-(N,N-dimethylamino)pyridine moieties, which is in accordance with the trend of general reduction in that the C−I⋯N halogen bond strengthens as more pyridines or 4-(N,N-dimethylamino)pyridines are coordinated to the TFTIB. All these data indicate the anticooperativity of the C−I⋯N halogen bonds. A comparison of the C−I⋯N halogen bond strengths can give more information. As shown in Figure 1, the C−I⋯N halogen bond strength is lower by 11% and 20%, respectively, on addition of two and three pyridine moieties, and the C−I⋯N halogen bond strength is lower by 16% and 30%, respectively, on addition of two and three 4-(N,N-dimethylamino)pyridine moieties. This means that the magnitude of the anticooperativity of the C−I⋯N halogen bonds is much larger on addition of successive 4-(N,N-dimethylamino)pyridines than on addition of successive pyridines. However, the experimental fact is that a cocrystal formed by TFTIB with 4-(N,N-dimethylamino)pyridine contains three C−I⋯N halogen bonds, whereas the cocrystals formed by TFTIB with a series of bipyridyl derivatives consistently contain two C−I⋯N halogen bonds [22,24]. Hence, it can be concluded that the anticooperativity of the C−I⋯N halogen bonds is not a contributing factor to the formation of two rather than three C−I⋯N halogen bonds.
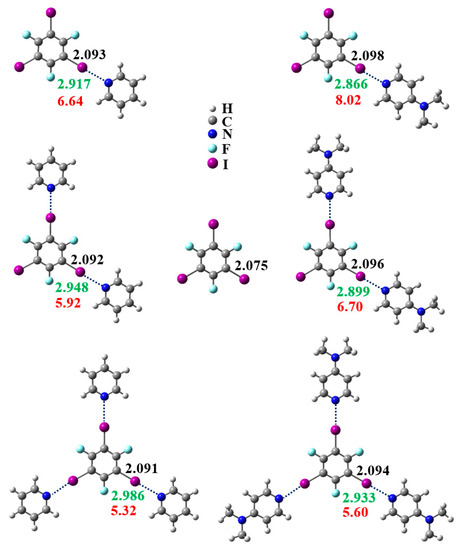
Figure 1.
The C−I bond lengths (black numbers, in Å), I⋯N distances (green numbers, in Å), and C−I⋯N halogen bond strengths (red numbers, in kcal/mol) for the monomer 1,3,5-trifluoro-2,4,6-triiodobenzene (TFTIB) and molecular clusters containing TFTIB calculated at the ωB97X-D/def2-TZVPP level of theory.
3.2. Structural Competition between C−I⋯N Halogen Bond and π⋯π Stacking Interaction in the Crystal Structures
Our density functional theory calculations have clearly demonstrated that the anticooperativity of the C−I⋯N halogen bonds was not a contributing factor in determining the number of C−I⋯N halogen bonds that one TFTIB molecule formed. Is the structural competition between a C−I⋯N halogen bond and π⋯π stacking interaction in the crystal structures the determining factor? To answer this question, in the present study we selected TFTIB as the halogen atom donor and ClPHEN, BrPHEN, CH3PHEN, and DCH3PHEN as halogen atom acceptors to synthesize a series of halogen bonded cocrystals. It was found that TFTIB crystallized readily with ClPHEN, BrPHEN, CH3PHEN, and DCH3PHEN in a trichloromethane solvent, yielding cocrystals I, II, III and IV, respectively. The crystallographic data and structure refinement parameters are summarized in Table 1.

Table 1.
Crystallographic data and structure refinement parameters for I, II, III and IV.
TFTIB with ClPHEN formed 1:1 cocrystal I. The supramolecular structure of I (Figure 2) was characterized by asymmetrical bifurcated halogen bonds, π⋯π stacking interaction between two TFTIB molecules, and π⋯π stacking interaction between two ClPHEN molecules. The molecule ClPHEN is asymmetrical. It is natural that the three-center halogen bond formed by ClPHEN is asymmetrical too, and the two-center halogen bond close to the C−Cl bond is a little longer than the two-center halogen bond away from the C−Cl bond due to the steric hindrance. In the three-center halogen bond, one I⋯N distance was 2.974 Å and the other I⋯N distance was 3.363 Å, both of which were smaller than the sum of the I and N van der Waals radii (3.530 Å) [41]. The C−I⋯F halogen bonds, C−I⋯Cl halogen bonds, and C−H⋯F hydrogen bonds were also observed in the crystal structure of I, but these interactions were too weak to be considered in this study. Although, these weak noncovalent interactions also played an important role in the construction of the three-dimensional framework of I. According to our calculations at the ωB97X-D/def2-TZVPP theory level, the strengths of these weak noncovalent interactions were all smaller than 2.00 kcal/mol. It was anticipated that one TFTIB molecule would form three asymmetrical bifurcated halogen bonds with three ClPHEN molecules in the crystal structure of I. In fact, only one asymmetrical bifurcated halogen bond was observed. At the ωB97X-D/def2-TZVPP level of theory, the binding energy of the asymmetrical bifurcated halogen bond was 7.87 kcal/mol, the binding energy of the π-stacked TFTIB dimer was 10.55 kcal/mol, and the binding energy of the π-stacked ClPHEN dimer was 16.11 kcal/mol. The π⋯π stacking interaction between two TFTIB molecules was as much as 1.34 times stronger than the asymmetrical bifurcated halogen bond, and the π⋯π stacking interaction between two ClPHEN molecules was over two times stronger than the asymmetrical bifurcated halogen bond. Evidently, the formation of the π-stacked structures had priority over the formation of the halogen bonded structures. Both experiment and calculation show that the π⋯π stacking interactions between the TFTIB molecules and the π⋯π stacking interactions between the ClPHEN molecules can successfully compete with the strong C−I⋯N halogen bonds. It is the structural competition between the π⋯π stacking interaction and strong C−I⋯N halogen bond that determines how many halogen bonds one TFTIB molecule can form.
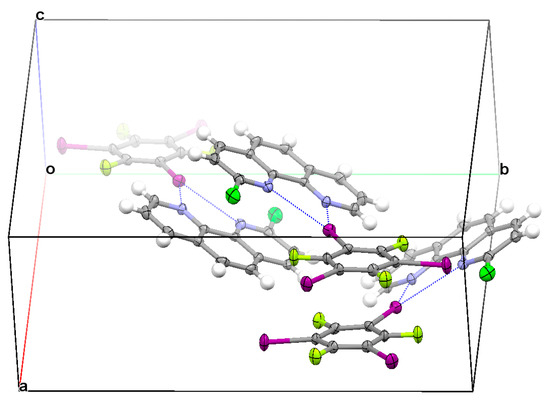
Figure 2.
Part of the crystal structure of I showing the formation of an asymmetrical bifurcated halogen bond, π⋯π stacking interaction between two TFTIB molecules, and π⋯π stacking interaction between two ClPHEN molecules
As shown in Table 1 and Figure 3, the crystal structure of II was almost the same as the crystal structure of I, differing only in that the substituent of 1,10-phenanthroline was changed from chloro to bromo. Similarly, the asymmetrical bifurcated halogen, π⋯π stacking interaction between two TFTIB molecules, and π⋯π stacking interaction between two BrPHEN molecules were also found in the crystal structure of II (Figure 3). In the three-center halogen bond, one I⋯N distance close to the C−Br bond was 3.377 Å and the other I⋯N distance away from the C−Br bond was 2.973 Å, each of them was similar to the corresponding I⋯N distance in the crystal structure of I. At the ωB97X-D/def2-TZVPP level of theory, the binding energy of the asymmetrical bifurcated halogen bond was 8.15 kcal/mol, the binding energy of the π-stacked TFTIB dimer was 10.52 kcal/mol, and the binding energy of the π-stacked BrPHEN dimer was 16.95 kcal/mol. The π⋯π stacking interaction between two TFTIB molecules was as much as 1.29 times stronger than the asymmetrical bifurcated halogen bond, and the π⋯π stacking interaction between two BrPHEN molecules was over two times stronger than the asymmetrical bifurcated halogen bond. Again, it was the stronger π⋯π stacking interactions between the TFTIB molecules and stronger π⋯π stacking interactions between the BrPHEN molecules that led one TFTIB molecule to form only one asymmetrical bifurcated halogen bond.
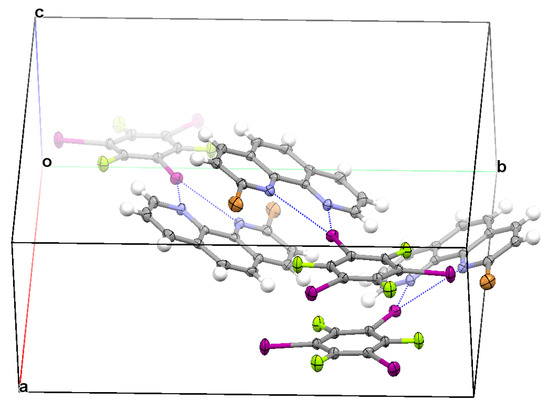
Figure 3.
Part of the crystal structure of II showing the formation of asymmetrical bifurcated halogen bond, π⋯π stacking interaction between two TFTIB molecules, and π⋯π stacking interaction between two BrPHEN molecules.
When the substituent of 1,10-phenanthroline was changed from chloro or bromo to methyl, cocrystal III was formed. Cocrystal III was also monoclinic. In the crystal structure of III, one TFTIB molecule also formed only one asymmetrical bifurcated halogen bond (Figure 4). The methyl group in CH3PHEN was electron-donating, so the asymmetrical bifurcated halogen bond in the crystal structure of III should have been a little stronger than that in the crystal structure of I or II. This can be seen from the shorter I⋯N distances in the asymmetrical bifurcated halogen bond in the crystal structure of III. In the three-center halogen bond, one I⋯N distance close to the methyl group was 3.352 Å and the other I⋯N distance away from the methyl group was 2.920 Å. Correspondingly, the binding energy of the asymmetrical bifurcated halogen bond was 9.23 kcal/mol at the ωB97X-D/def2-TZVPP level of theory. At the same time, the binding energy of the π-stacked TFTIB dimer was 9.25 kcal/mol and the binding energy of the π-stacked CH3PHEN dimer was 11.96 kcal/mol. Both experiment and calculation show that the π⋯π stacking interactions successfully competed with the strong C−I⋯N halogen bonds in the crystal structure of III.
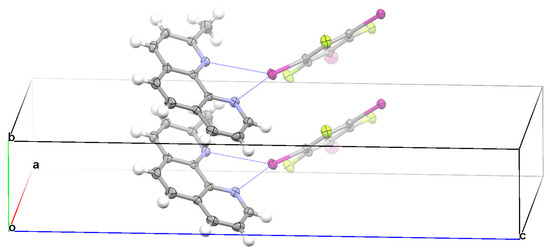
Figure 4.
Part of the crystal structure of III showing the formation of an asymmetrical bifurcated halogen bond, π⋯π stacking interaction between two TFTIB molecules, and π⋯π stacking interaction between two CH3PHEN molecules.
Crystallization of TFTIB with DCH3PHEN in the trichloromethane solvent yielded cocrystal IV. Cocrystal IV had the symmetry of triclinic space group P-1. It can be clearly seen from Figure 5 that the asymmetrical bifurcated halogen bond, monocoordinate C−I⋯N halogen bond, and π⋯π stacking interaction between two TFTIB molecules were formed in the crystal structure of IV. At the ωB97X-D/def2-TZVPP level of theory, the binding energy of the asymmetrical bifurcated halogen bond was 7.48 kcal/mol, the binding energy of the monocoordinate C−I⋯N halogen bond was 7.50 kcal/mol, and the binding energy of the π-stacked TFTIB dimer was 8.52 kcal/mol. The π⋯π stacking interaction between two TFTIB molecules was slightly stronger than both the asymmetrical bifurcated halogen bond and the monocoordinate C−I⋯N halogen bond. As shown in Figure 5, the π⋯π stacking interaction between two TFTIB molecules is different from the ones in Figure 2, Figure 3 and Figure 4. Here, the I atom of one TFTIB molecule is on the top of the electron-deficient six-membered ring of the other TFTIB molecule. In this case, the π⋯π stacking interaction between two TFTIB molecules can also be interpreted as two pairs of attractive lp(I)-π-hole interactions. Different from the case in the crystal structure of I, II or III, there was no π⋯π stacking interaction between two DCH3PHEN molecules in the crystal structure of IV, and one TFTIB molecule formed two halogen bonds with two different DCH3PHEN molecules. Evidently, the lack of the π⋯π stacking interaction between two DCH3PHEN molecules was related directly to the increase of the number of C−I⋯N halogen bonds that one TFTIB molecule formed. This again proves that it is the structural competition between the π⋯π stacking interaction and strong C−I⋯N halogen bond that determines how many halogen bonds one TFTIB molecule can form.
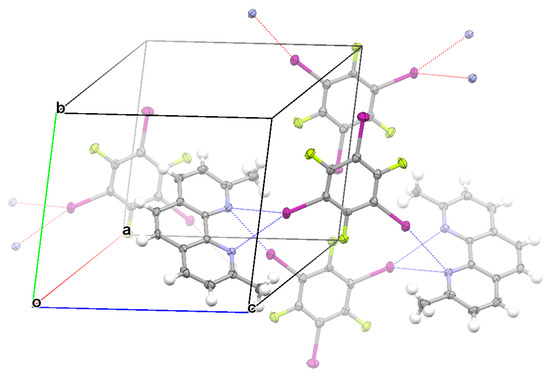
Figure 5.
Part of the crystal structure of IV showing the formation of an asymmetrical bifurcated halogen bond, monocoordinate halogen bond, and π⋯π stacking interaction between two TFTIB molecules.
4. Conclusions
In order to answer the question of how many halogen bonds one TFTIB molecule can form in the crystal structures, in the present study, we have investigated the noncovalent interactions in the complexes formed by TFTIB with ClPHEN, BrPHEN, CH3PHEN, and DCH3PHEN, respectively, by employing a combined theoretical and experimental method. The density functional theory calculations show that the anticooperativity of the C−I⋯N halogen bonds was not a contributing factor to the formation of two rather than three C−I⋯N halogen bonds. Both crystallographic experiments and density functional theory calculations clearly show that it was the structural competition between the π⋯π stacking interaction and the strong C−I⋯N halogen bond that determined how many halogen bonds one TFTIB molecule formed. Here, it must be stressed that other noncovalent interactions can also compete with the strong C−I⋯N halogen bond in the formation of supramolecular architectures. Considering that the π⋯π stacking interaction is one of the most important and widely studied intermolecular interactions in crystal engineering, we only focused on the structural competition between the π⋯π stacking interaction and the strong C−I⋯N halogen bond in this study.
The TFTIB molecule is a commonly used halogen bond donor in crystal growth and design. We believe that the results reported in this study provide very useful information for further application of the halogen bond in the fields of supramolecular chemistry, crystal engineering, and optoelectronic functional materials.
Author Contributions
Y.Z. synthesized the cocrystals, performed the X-ray single diffraction studies and carried out the quantum chemical calculations. J.-G.W. resolved the crystal structures. W.W. designed the experiments and conceived the project. Y.Z. and W.W. made equal contributions in writing of manuscript.
Acknowledgments
This work was supported by the National Science Foundation of China (21773104) and the Program for Science & Technology Innovation Talents in Universities of Henan Province (13HASTIT015). Computer time was provided by National Supercomputing Center in Shenzhen.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Amico, V.; Meille, S.V.; Corradi, E.; Messina, M.T.; Resnati, G. Perfluorocarbon-Hydrocarbon Self-Assembling. 1D Infinite Chain Formation Driven by Nitrogen⋯Iodine Interactions. J. Am. Chem. Soc. 1998, 120, 8261–8262. [Google Scholar] [CrossRef]
- Farina, A.; Meille, S.V.; Messina, M.T.; Metrangolo, P.; Resnati, G.; Vecchio, G. Resolution of Racemic 1,2-Dibromohexafluoropropane through Halogen-Bonded Supramolecular Helices. Angew. Chem. Int. Ed. 1999, 38, 2433–2436. [Google Scholar] [CrossRef]
- Wang, W.; Wong, N.B.; Zheng, W.; Tian, A. Theoretical Study on the Blueshifting Halogen Bond. J. Phys. Chem. A 2004, 108, 1799–1805. [Google Scholar] [CrossRef]
- Clark, T.; Hennemann, M.; Murray, J.S.; Politzer, P. Halogen Bonding: The σ-Hole. J. Mol. Model. 2007, 13, 291–296. [Google Scholar] [CrossRef] [PubMed]
- Riley, K.E.; Hobza, P. Investigations into the Nature of Halogen Bonding Including Symmetry Adapted Perturbation Theory Analyses. J. Chem. Theory Comput. 2008, 4, 232–242. [Google Scholar] [CrossRef] [PubMed]
- Metrangolo, P.; Meyer, F.; Pilati, T.; Resnati, G.; Terraneo, G. Halogen Bonding in Supramolecular Chemistry. Angew. Chem. Int. Ed. 2008, 47, 6114–6127. [Google Scholar] [CrossRef] [PubMed]
- Politzer, P.; Murray, J.S.; Clark, T. Halogen Bonding: An Electrostatically-Driven Highly Directional Noncovalent Interaction. Phys. Chem. Chem. Phys. 2010, 12, 7748–7757. [Google Scholar] [CrossRef] [PubMed]
- Legon, A.C. The halogen bond: an interim perspective. Phys. Chem. Chem. Phys. 2010, 12, 7736–7747. [Google Scholar] [CrossRef] [PubMed]
- Bertani, R.; Sgarbossa, P.; Venzo, A.; Lelj, F.; Amati, M.; Resnati, G.; Pilati, T.; Metrangolo, P.; Terraneo, G. Halogen Bonding in Metal–Organic–Supramolecular Networks. Coord. Chem. Rev. 2010, 254, 677–695. [Google Scholar] [CrossRef]
- Cavallo, G.; Metrangolo, P.; Pilati, T.; Resnati, G.; Sansotera, M.; Terraneo, G. Halogen Bonding: A General Route in Anion Recognition and Coordination. Chem. Soc. Rev. 2010, 39, 3772–3783. [Google Scholar] [CrossRef]
- Wang, W. Halogen Bond Involving Hypervalent Halogen: CSD Search and Theoretical Study. J. Phys. Chem. A 2011, 115, 9294–9299. [Google Scholar] [CrossRef] [PubMed]
- Pennington, W.T.; Resnati, G.; Taylor, M.S. Halogen Bonding: From Self-Assembly to Materials and Biomolecules. CrystEngComm 2013, 15, 3057. [Google Scholar] [CrossRef]
- Desiraju, G.R.; Ho, P.S.; Kloo, L.; Legon, A.C.; Marquardt, R.; Metrangolo, P.; Politzer, P.; Resnati, G.; Rissanen, K. Definition of the Halogen Bond. Pure Appl. Chem. 2013, 85, 1711–1713. [Google Scholar] [CrossRef]
- Cavallo, G.; Metrangolo, P.; Pilati, T.; Resnati, G.; Terraneo, G. Naming Interactions from the Electrophilic Site. Cryst. Growth Des. 2014, 14, 2697–2702. [Google Scholar] [CrossRef]
- Giese, M.; Albrecht, M.; Rissanen, K. Anion−π Interactions with Fluoroarenes. Chem. Rev. 2015, 115, 8867–8895. [Google Scholar] [CrossRef] [PubMed]
- Gilday, L.C.; Robinson, S.W.; Barendt, T.A.; Langton, M.J.; Mullaney, B.R.; Beer, P.D. Halogen Bonding in Supramolecular Chemistry. Chem. Rev. 2015, 115, 7118–7195. [Google Scholar] [CrossRef] [PubMed]
- Cavallo, G.; Metrangolo, P.; Milani, R.; Pilati, T.; Priimagi, A.; Resnati, G.; Terraneo, G. The Halogen Bond. Chem. Rev. 2016, 116, 2478–2601. [Google Scholar] [CrossRef]
- Wang, H.; Wang, W.; Jin, W.J. σ-Hole Bond vs π-Hole Bond: A Comparison Based on Halogen Bond. Chem. Rev. 2016, 116, 5072–5104. [Google Scholar] [CrossRef]
- Kolář, M.H.; Hobza, P. Computer Modeling of Halogen Bonds and Other σ-Hole Interactions. Chem. Rev. 2016, 116, 5155–5187. [Google Scholar] [CrossRef]
- Resnati, G.; Pennington, W.T. The Halogen Bond: A New Avenue in Recognition and Self-Assembly. New J. Chem. 2018, 42, 10461–10462. [Google Scholar] [CrossRef]
- Ding, X.H.; Ou, C.J.; Wang, S.; Xie, L.H.; Lin, J.Y.; Wang, J.P.; Huang, W. Co-Crystallization of 1,3,5-Trifluoro-2,4,6-triiodobenzene (1,3,5-TFTIB) with a Variety of Lewis Bases through Halogen-Bonding Interactions. CrystEngComm 2017, 19, 5504–5521. [Google Scholar] [CrossRef]
- Lucassen, A.C.B.; Karton, A.; Leitus, G.; Shimon, L.J.W.; Martin, J.M.L.; van der Boom, M.E. Co-Crystallization of Sym-Triiodo-Trifluorobenzene with Bipyridyl Donors: Consistent Formation of Two Instead of Anticipated Three N⋯I Halogen Bonds. Cryst. Growth Des. 2007, 7, 386–392. [Google Scholar] [CrossRef]
- Vartanian, M.; Lucassen, A.C.B.; Shimon, L.J.W.; van der Boom, M.E. Cocrystallization of a Tripyridyl Donor with Perfluorinated Iodobenzene Derivatives: Formation of Different N⋯I Halogen Bonds Determining Network vs Plain Packing Crystals. Cryst. Growth Des. 2008, 8, 786–790. [Google Scholar] [CrossRef]
- Roper, L.C.; Präsang, C.; Kozhevnikov, V.N.; Whitwood, A.C.; Karadakov, P.B.; Bruce, D.W. Experimental and Theoretical Study of Halogen-Bonded Complexes of DMAP with Di- and Triiodofluorobenzenes. A Complex with a Very Short N⋯I Halogen Bond. Cryst. Growth Des. 2010, 10, 3710–3720. [Google Scholar] [CrossRef]
- Aakeröy, C.B.; Wijethunga, T.K.; Desper, J. Practical Crystal Engineering Using Halogen Bonding: A Hierarchy Based on Calculated Molecular Electrostatic Potential Surfaces. J. Mol. Struct. 2014, 1072, 20–27. [Google Scholar] [CrossRef]
- Hidalgo, P.I.; Leal, S.; Jiménez, C.A.; Vöhringer-Martinez, E.; Herrera, B.; Pasán, J.; Ruiz-Pérez, C.; Bruce, D.W. Extending the Halogen-Bonded Supramolecular Synthon Concept to 1,3,4-Oxadiazole Derivatives. CrystEngComm 2016, 18, 42–47. [Google Scholar] [CrossRef]
- Triguero, S.; Llusar, R.; Polo, V.; Formigué, M. Halogen Bonding Interactions of sym-Triiodotrifluorobenzene with Halide Anions: A Combined Structural and Theoretical Study. Cryst. Growth Des. 2008, 8, 2241–2247. [Google Scholar] [CrossRef]
- Metrangolo, P.; Meyer, F.; Pilati, T.; Resnati, G.; Terraneo, G. Mutual Induced Coordination in Halogen-Bonded Anionic Assemblies with (6,3) Cation-Templated Topologies. Chem. Commun. 2008, 1635–1637. [Google Scholar] [CrossRef]
- Cauliez, P.; Polo, V.; Roisnel, T.; Llusar, R.; Formigué, M. The Thiocyanate Anion as a Polydentate Halogen Bond Acceptor. CrystEngComm 2010, 12, 558–566. [Google Scholar] [CrossRef]
- Wang, W.; Zhang, Y.; Wang, Y.B. The π⋯π Stacking Interactions between Homogeneous Dimers of C6FxI(6-x) (x =0, 1, 2, 3, 4 and 5): A Comparative Study with the Halogen Bond. J. Phys. Chem. A 2012, 116, 12486–12491. [Google Scholar] [CrossRef]
- Wang, W.; Zhang, Y.; Wang, Y.B. Unexpected Strong Stacking Interactions between the Homogeneous Dimers of C6FxI(6-x) (x =0, 1, 2, 3, 4 and 5). Comput. Theor. Chem. 2013, 1023, 88–94. [Google Scholar] [CrossRef]
- Ji, B.; Wang, W.; Deng, D.; Zhang, Y.; Cao, L.; Zhou, L.; Ruan, C.; Li, T. Structural Competition between π⋯π Interactions and Halogen Bonds: A Crystallographic Study. CrystEngComm 2013, 15, 769–774. [Google Scholar] [CrossRef]
- Chai, J.-D.; Head-Gordon, M. Systematic Optimization of Long-Range Corrected Hybrid Density Functionals. J. Chem. Phys. 2008, 128, 084106. [Google Scholar] [CrossRef] [PubMed]
- Chai, J.-D.; Head-Gordon, M. Long-Range Corrected Hybrid Density Functionals with Damped Atom–Atom Dispersion Corrections. Phys. Chem. Chem. Phys. 2008, 10, 6615–6620. [Google Scholar] [CrossRef] [PubMed]
- Weigend, F.; Ahlrichs, R. Balanced Basis Sets of Split Valence, Triple Zeta Valence and Quadruple Zeta Valence Quality for H to Rn: Design and Assessment of Accuracy. Phys. Chem. Chem. Phys. 2005, 7, 3297–3305. [Google Scholar] [CrossRef] [PubMed]
- Boys, S.F.; Bernardi, F. The Calculation of Small Molecular Interactions by the Difference of Separate Total Energies. Some Procedures with Reduced Errors. Mol. Phys. 1970, 19, 553–566. [Google Scholar] [CrossRef]
- Reed, A.E.; Curitss, L.A.; Weinhold, F. Intermolecular Interactions from a Natural Bond Orbital, Donor-Acceptor Viewpoint. Chem. Rev. 1998, 88, 899–926. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09, Revision C.01; Gaussian, Inc.: Wallingford, CT, USA, 2010. [Google Scholar]
- Sheldrick, G.M. SHELXT–Integrated Space-Group and Crystal-Structure Determination. Acta Crystallogr. 2015, A71, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Dolomanov, O.V.; Bourhis, L.J.; Gildea, R.J.; Howard, J.A.K.; Puschmann, H. OLEX2: A Complete Structure Solution, Refinement and Analysis Program. J. Appl. Cryst. 2009, 42, 339–341. [Google Scholar] [CrossRef]
- Bondi, A. van der Waals Volumes and Radii. J. Phys. Chem. 1964, 68, 441–451. [Google Scholar] [CrossRef]





© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).