Noncovalent Interactions between 1,3,5-Trifluoro-2,4,6-triiodobenzene and a Series of 1,10-Phenanthroline Derivatives: A Combined Theoretical and Experimental Study


Abstract
1. Introduction
2. Materials and Methods
2.1. Quantum Chemical Calculation
2.2. Crystal Preparation
2.3. Measurement
3. Results and Discussion
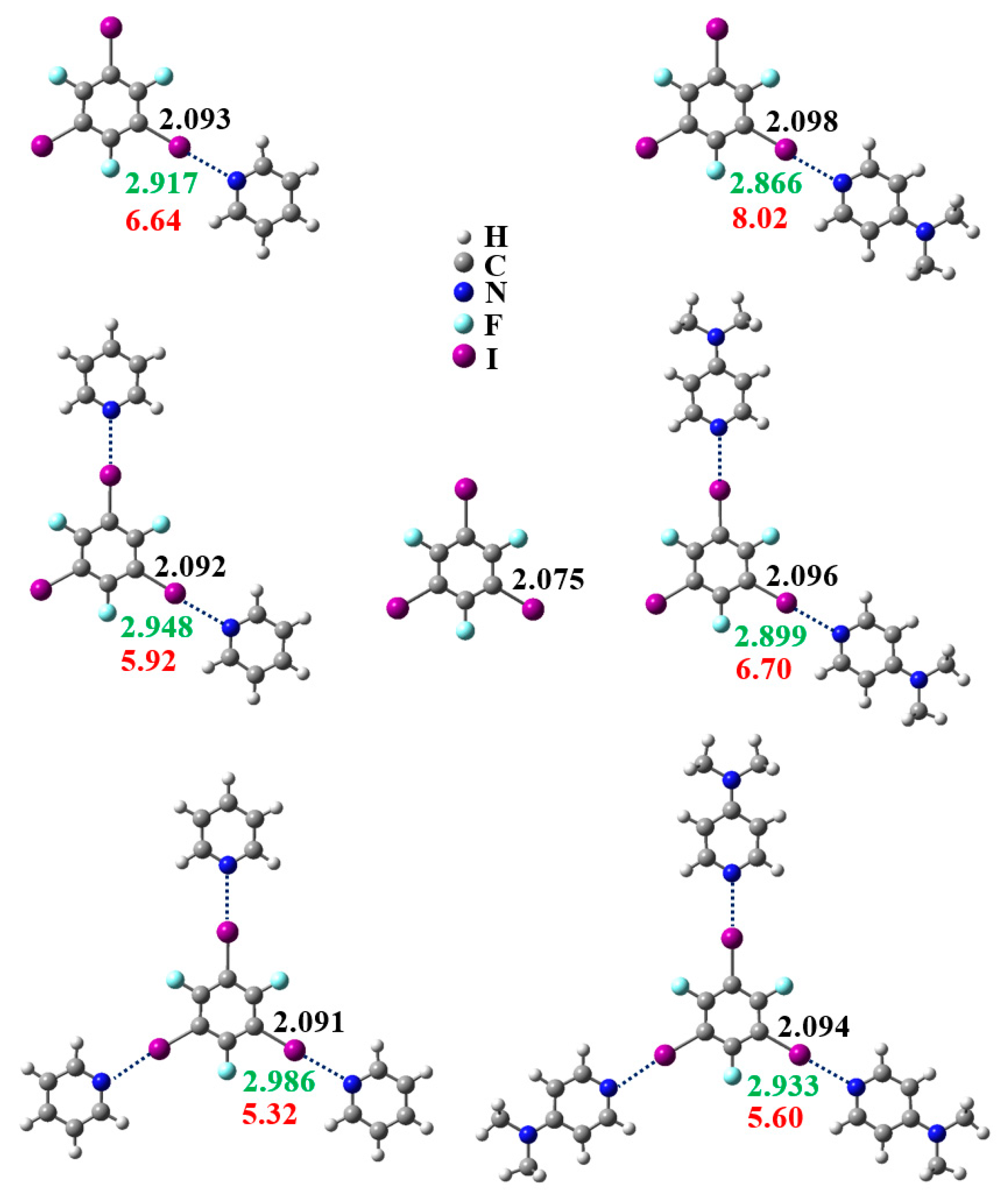
3.1. Anticooperativity of the Halogen Bonds
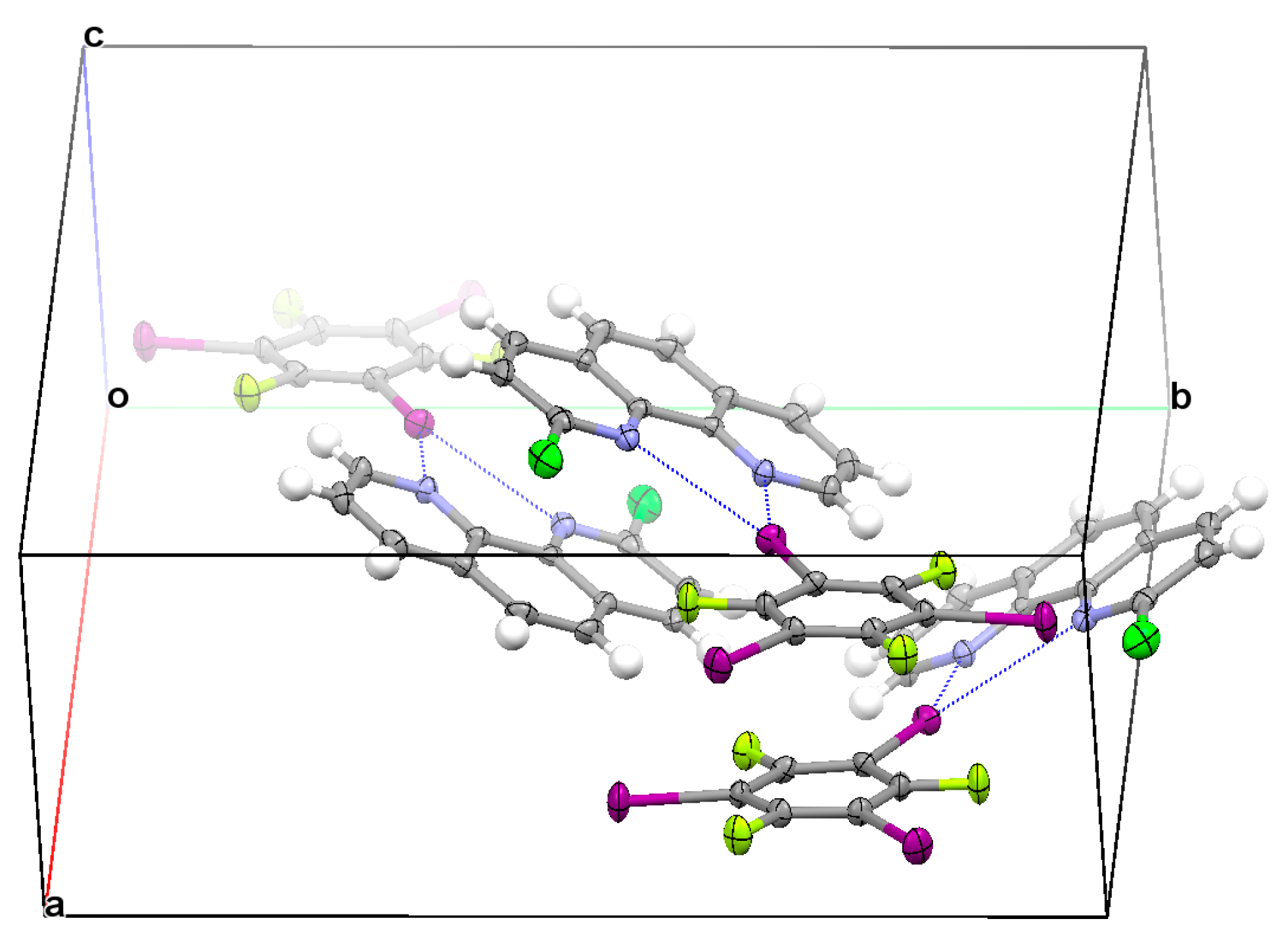
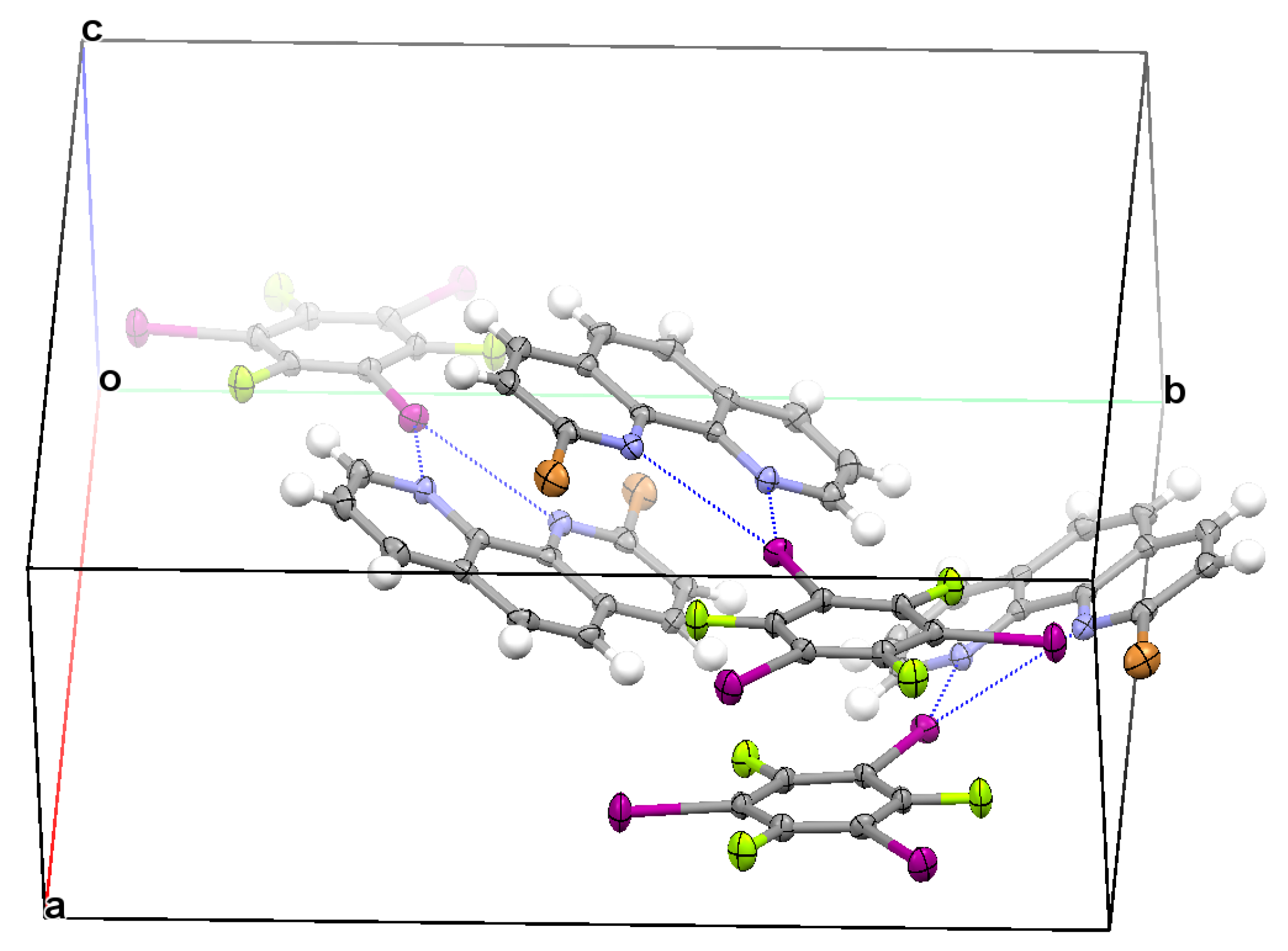
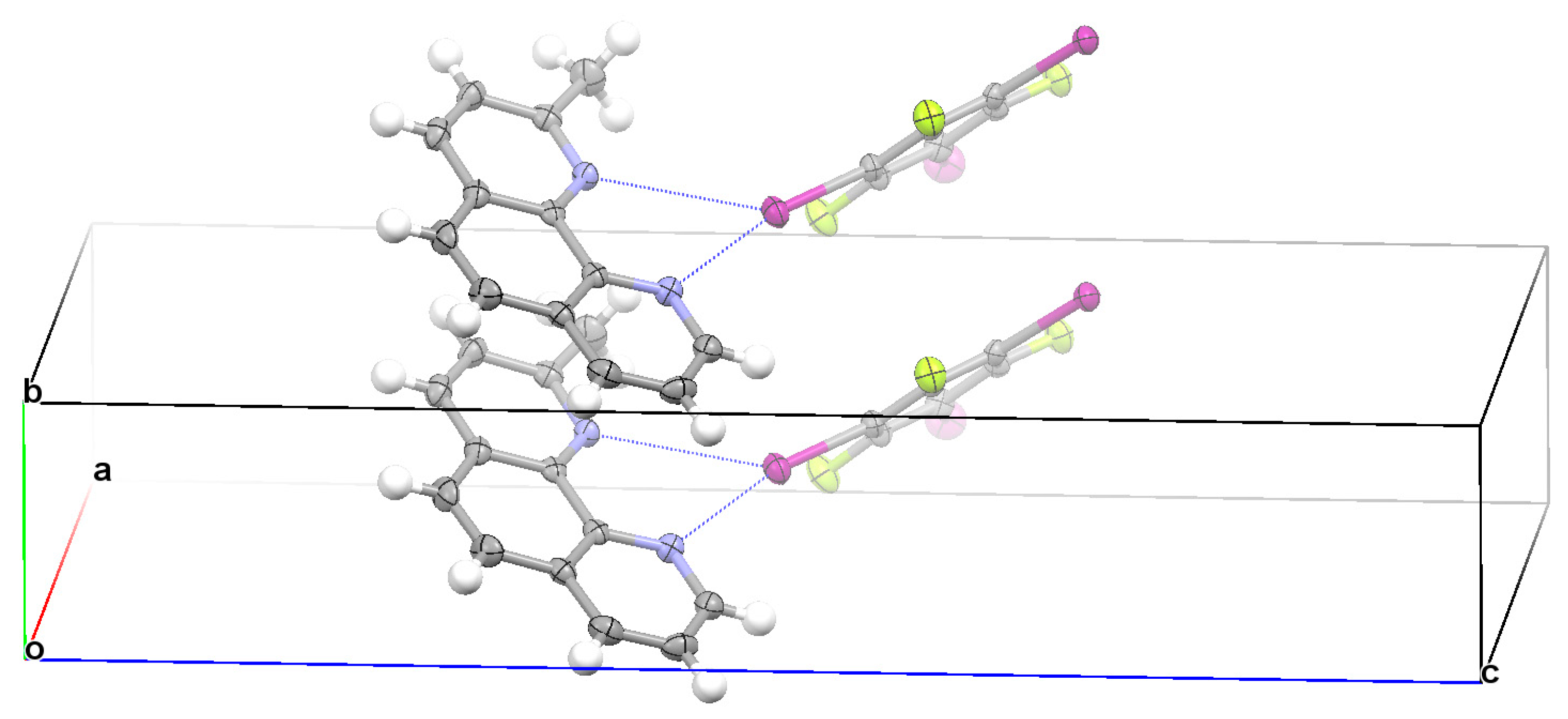
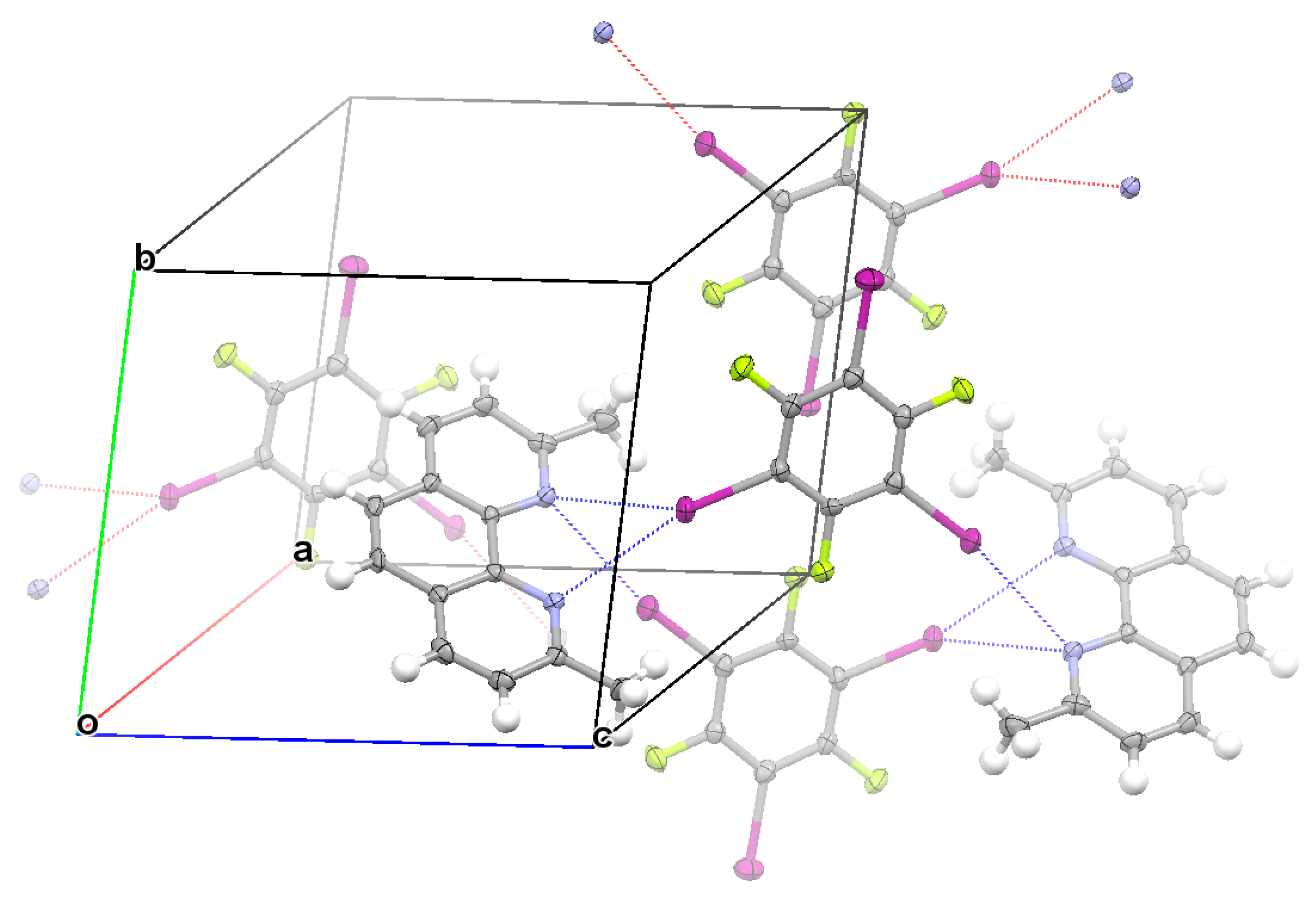
3.2. Structural Competition between C−I⋯N Halogen Bond and π⋯π Stacking Interaction in the Crystal Structures
4. Conclusions
Author Contributions
Acknowledgments
Conflicts of Interest
References
- Amico, V.; Meille, S.V.; Corradi, E.; Messina, M.T.; Resnati, G. Perfluorocarbon-Hydrocarbon Self-Assembling. 1D Infinite Chain Formation Driven by Nitrogen⋯Iodine Interactions. J. Am. Chem. Soc. 1998, 120, 8261–8262. [Google Scholar] [CrossRef]
- Farina, A.; Meille, S.V.; Messina, M.T.; Metrangolo, P.; Resnati, G.; Vecchio, G. Resolution of Racemic 1,2-Dibromohexafluoropropane through Halogen-Bonded Supramolecular Helices. Angew. Chem. Int. Ed. 1999, 38, 2433–2436. [Google Scholar] [CrossRef]
- Wang, W.; Wong, N.B.; Zheng, W.; Tian, A. Theoretical Study on the Blueshifting Halogen Bond. J. Phys. Chem. A 2004, 108, 1799–1805. [Google Scholar] [CrossRef]
- Clark, T.; Hennemann, M.; Murray, J.S.; Politzer, P. Halogen Bonding: The σ-Hole. J. Mol. Model. 2007, 13, 291–296. [Google Scholar] [CrossRef] [PubMed]
- Riley, K.E.; Hobza, P. Investigations into the Nature of Halogen Bonding Including Symmetry Adapted Perturbation Theory Analyses. J. Chem. Theory Comput. 2008, 4, 232–242. [Google Scholar] [CrossRef] [PubMed]
- Metrangolo, P.; Meyer, F.; Pilati, T.; Resnati, G.; Terraneo, G. Halogen Bonding in Supramolecular Chemistry. Angew. Chem. Int. Ed. 2008, 47, 6114–6127. [Google Scholar] [CrossRef] [PubMed]
- Politzer, P.; Murray, J.S.; Clark, T. Halogen Bonding: An Electrostatically-Driven Highly Directional Noncovalent Interaction. Phys. Chem. Chem. Phys. 2010, 12, 7748–7757. [Google Scholar] [CrossRef] [PubMed]
- Legon, A.C. The halogen bond: an interim perspective. Phys. Chem. Chem. Phys. 2010, 12, 7736–7747. [Google Scholar] [CrossRef] [PubMed]
- Bertani, R.; Sgarbossa, P.; Venzo, A.; Lelj, F.; Amati, M.; Resnati, G.; Pilati, T.; Metrangolo, P.; Terraneo, G. Halogen Bonding in Metal–Organic–Supramolecular Networks. Coord. Chem. Rev. 2010, 254, 677–695. [Google Scholar] [CrossRef]
- Cavallo, G.; Metrangolo, P.; Pilati, T.; Resnati, G.; Sansotera, M.; Terraneo, G. Halogen Bonding: A General Route in Anion Recognition and Coordination. Chem. Soc. Rev. 2010, 39, 3772–3783. [Google Scholar] [CrossRef]
- Wang, W. Halogen Bond Involving Hypervalent Halogen: CSD Search and Theoretical Study. J. Phys. Chem. A 2011, 115, 9294–9299. [Google Scholar] [CrossRef] [PubMed]
- Pennington, W.T.; Resnati, G.; Taylor, M.S. Halogen Bonding: From Self-Assembly to Materials and Biomolecules. CrystEngComm 2013, 15, 3057. [Google Scholar] [CrossRef]
- Desiraju, G.R.; Ho, P.S.; Kloo, L.; Legon, A.C.; Marquardt, R.; Metrangolo, P.; Politzer, P.; Resnati, G.; Rissanen, K. Definition of the Halogen Bond. Pure Appl. Chem. 2013, 85, 1711–1713. [Google Scholar] [CrossRef]
- Cavallo, G.; Metrangolo, P.; Pilati, T.; Resnati, G.; Terraneo, G. Naming Interactions from the Electrophilic Site. Cryst. Growth Des. 2014, 14, 2697–2702. [Google Scholar] [CrossRef]
- Giese, M.; Albrecht, M.; Rissanen, K. Anion−π Interactions with Fluoroarenes. Chem. Rev. 2015, 115, 8867–8895. [Google Scholar] [CrossRef] [PubMed]
- Gilday, L.C.; Robinson, S.W.; Barendt, T.A.; Langton, M.J.; Mullaney, B.R.; Beer, P.D. Halogen Bonding in Supramolecular Chemistry. Chem. Rev. 2015, 115, 7118–7195. [Google Scholar] [CrossRef] [PubMed]
- Cavallo, G.; Metrangolo, P.; Milani, R.; Pilati, T.; Priimagi, A.; Resnati, G.; Terraneo, G. The Halogen Bond. Chem. Rev. 2016, 116, 2478–2601. [Google Scholar] [CrossRef]
- Wang, H.; Wang, W.; Jin, W.J. σ-Hole Bond vs π-Hole Bond: A Comparison Based on Halogen Bond. Chem. Rev. 2016, 116, 5072–5104. [Google Scholar] [CrossRef]
- Kolář, M.H.; Hobza, P. Computer Modeling of Halogen Bonds and Other σ-Hole Interactions. Chem. Rev. 2016, 116, 5155–5187. [Google Scholar] [CrossRef]
- Resnati, G.; Pennington, W.T. The Halogen Bond: A New Avenue in Recognition and Self-Assembly. New J. Chem. 2018, 42, 10461–10462. [Google Scholar] [CrossRef]
- Ding, X.H.; Ou, C.J.; Wang, S.; Xie, L.H.; Lin, J.Y.; Wang, J.P.; Huang, W. Co-Crystallization of 1,3,5-Trifluoro-2,4,6-triiodobenzene (1,3,5-TFTIB) with a Variety of Lewis Bases through Halogen-Bonding Interactions. CrystEngComm 2017, 19, 5504–5521. [Google Scholar] [CrossRef]
- Lucassen, A.C.B.; Karton, A.; Leitus, G.; Shimon, L.J.W.; Martin, J.M.L.; van der Boom, M.E. Co-Crystallization of Sym-Triiodo-Trifluorobenzene with Bipyridyl Donors: Consistent Formation of Two Instead of Anticipated Three N⋯I Halogen Bonds. Cryst. Growth Des. 2007, 7, 386–392. [Google Scholar] [CrossRef]
- Vartanian, M.; Lucassen, A.C.B.; Shimon, L.J.W.; van der Boom, M.E. Cocrystallization of a Tripyridyl Donor with Perfluorinated Iodobenzene Derivatives: Formation of Different N⋯I Halogen Bonds Determining Network vs Plain Packing Crystals. Cryst. Growth Des. 2008, 8, 786–790. [Google Scholar] [CrossRef]
- Roper, L.C.; Präsang, C.; Kozhevnikov, V.N.; Whitwood, A.C.; Karadakov, P.B.; Bruce, D.W. Experimental and Theoretical Study of Halogen-Bonded Complexes of DMAP with Di- and Triiodofluorobenzenes. A Complex with a Very Short N⋯I Halogen Bond. Cryst. Growth Des. 2010, 10, 3710–3720. [Google Scholar] [CrossRef]
- Aakeröy, C.B.; Wijethunga, T.K.; Desper, J. Practical Crystal Engineering Using Halogen Bonding: A Hierarchy Based on Calculated Molecular Electrostatic Potential Surfaces. J. Mol. Struct. 2014, 1072, 20–27. [Google Scholar] [CrossRef]
- Hidalgo, P.I.; Leal, S.; Jiménez, C.A.; Vöhringer-Martinez, E.; Herrera, B.; Pasán, J.; Ruiz-Pérez, C.; Bruce, D.W. Extending the Halogen-Bonded Supramolecular Synthon Concept to 1,3,4-Oxadiazole Derivatives. CrystEngComm 2016, 18, 42–47. [Google Scholar] [CrossRef]
- Triguero, S.; Llusar, R.; Polo, V.; Formigué, M. Halogen Bonding Interactions of sym-Triiodotrifluorobenzene with Halide Anions: A Combined Structural and Theoretical Study. Cryst. Growth Des. 2008, 8, 2241–2247. [Google Scholar] [CrossRef]
- Metrangolo, P.; Meyer, F.; Pilati, T.; Resnati, G.; Terraneo, G. Mutual Induced Coordination in Halogen-Bonded Anionic Assemblies with (6,3) Cation-Templated Topologies. Chem. Commun. 2008, 1635–1637. [Google Scholar] [CrossRef]
- Cauliez, P.; Polo, V.; Roisnel, T.; Llusar, R.; Formigué, M. The Thiocyanate Anion as a Polydentate Halogen Bond Acceptor. CrystEngComm 2010, 12, 558–566. [Google Scholar] [CrossRef]
- Wang, W.; Zhang, Y.; Wang, Y.B. The π⋯π Stacking Interactions between Homogeneous Dimers of C6FxI(6-x) (x =0, 1, 2, 3, 4 and 5): A Comparative Study with the Halogen Bond. J. Phys. Chem. A 2012, 116, 12486–12491. [Google Scholar] [CrossRef]
- Wang, W.; Zhang, Y.; Wang, Y.B. Unexpected Strong Stacking Interactions between the Homogeneous Dimers of C6FxI(6-x) (x =0, 1, 2, 3, 4 and 5). Comput. Theor. Chem. 2013, 1023, 88–94. [Google Scholar] [CrossRef]
- Ji, B.; Wang, W.; Deng, D.; Zhang, Y.; Cao, L.; Zhou, L.; Ruan, C.; Li, T. Structural Competition between π⋯π Interactions and Halogen Bonds: A Crystallographic Study. CrystEngComm 2013, 15, 769–774. [Google Scholar] [CrossRef]
- Chai, J.-D.; Head-Gordon, M. Systematic Optimization of Long-Range Corrected Hybrid Density Functionals. J. Chem. Phys. 2008, 128, 084106. [Google Scholar] [CrossRef] [PubMed]
- Chai, J.-D.; Head-Gordon, M. Long-Range Corrected Hybrid Density Functionals with Damped Atom–Atom Dispersion Corrections. Phys. Chem. Chem. Phys. 2008, 10, 6615–6620. [Google Scholar] [CrossRef] [PubMed]
- Weigend, F.; Ahlrichs, R. Balanced Basis Sets of Split Valence, Triple Zeta Valence and Quadruple Zeta Valence Quality for H to Rn: Design and Assessment of Accuracy. Phys. Chem. Chem. Phys. 2005, 7, 3297–3305. [Google Scholar] [CrossRef] [PubMed]
- Boys, S.F.; Bernardi, F. The Calculation of Small Molecular Interactions by the Difference of Separate Total Energies. Some Procedures with Reduced Errors. Mol. Phys. 1970, 19, 553–566. [Google Scholar] [CrossRef]
- Reed, A.E.; Curitss, L.A.; Weinhold, F. Intermolecular Interactions from a Natural Bond Orbital, Donor-Acceptor Viewpoint. Chem. Rev. 1998, 88, 899–926. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09, Revision C.01; Gaussian, Inc.: Wallingford, CT, USA, 2010. [Google Scholar]
- Sheldrick, G.M. SHELXT–Integrated Space-Group and Crystal-Structure Determination. Acta Crystallogr. 2015, A71, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Dolomanov, O.V.; Bourhis, L.J.; Gildea, R.J.; Howard, J.A.K.; Puschmann, H. OLEX2: A Complete Structure Solution, Refinement and Analysis Program. J. Appl. Cryst. 2009, 42, 339–341. [Google Scholar] [CrossRef]
- Bondi, A. van der Waals Volumes and Radii. J. Phys. Chem. 1964, 68, 441–451. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cocrystal | I | II | III | IV |
|---|---|---|---|---|
| CCDC No. | 1891221 | 1891219 | 1891220 | 1891222 |
| Formula | C18H7ClF3I3N2 | C18H7BrF3I3N2 | C19H10F3I3N2 | C20H12F3I3N2 |
| Formula weight | 724.41 | 768.87 | 703.99 | 718.02 |
| Crystal size/mm3 | 0.24 × 0.15 × 0.017 | 0.30 × 0.27 × 0.26 | 0.27 × 0.26 × 0.19 | 0.31 × 0.28 × 0.24 |
| Crystal system | monoclinic | monoclinic | monoclinic | triclinic |
| Space group | P21/c | P21/c | P21/n | P-1 |
| a/Å | 14.2733(5) | 14.2515(4) | 17.9454(9) | 9.6529(7) |
| b/Å | 18.2696(6) | 18.2923(6) | 4.4461(3) | 9.6760(5) |
| c/Å | 7.6371(4) | 7.6939(3) | 25.0595(14) | 11.8940(7) |
| α/° | 90 | 90 | 90 | 81.258(5) |
| β/° | 91.662(3) | 90.725(3) | 93.433(5) | 88.575(5) |
| γ/° | 90 | 90 | 90 | 72.537(6) |
| Volume/Å3 | 1990.69(13) | 2005.57(12) | 1995.81(19) | 1047.15(12) |
| Z | 4 | 4 | 4 | 2 |
| ρcalc/g cm–3 | 2.417 | 2.546 | 2.343 | 2.277 |
| T/K | 287.12(10) | 293(2) | 289.78(10) | 293(2) |
| 2θ Range for data collection/° | 6.52–50.992 | 6.92–51.00 | 6.92–51.00 | 6.612–50.994 |
| Reflections collected | 10704 | 11373 | 10763 | 12066 |
| No. unique data [R(int)] | 3622 [0.0282] | 3729 [0.0273] | 3689 [0.0384] | 3892 [0.0379] |
| Final R (I > 2σ(I)) | 0.0319 | 0.0302 | 0.0392 | 0.0508 |
| Final wR2 (all data) | 0.0611 | 0.0567 | 0.0877 | 0.1550 |
| Goodness-of-fit | 1.065 | 1.039 | 1.113 | 1.072 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, Y.; Wang, J.-G.; Wang, W. Noncovalent Interactions between 1,3,5-Trifluoro-2,4,6-triiodobenzene and a Series of 1,10-Phenanthroline Derivatives: A Combined Theoretical and Experimental Study. Crystals 2019, 9, 140. https://doi.org/10.3390/cryst9030140
Zhang Y, Wang J-G, Wang W. Noncovalent Interactions between 1,3,5-Trifluoro-2,4,6-triiodobenzene and a Series of 1,10-Phenanthroline Derivatives: A Combined Theoretical and Experimental Study. Crystals. 2019; 9(3):140. https://doi.org/10.3390/cryst9030140
Chicago/Turabian StyleZhang, Yu, Jian-Ge Wang, and Weizhou Wang. 2019. "Noncovalent Interactions between 1,3,5-Trifluoro-2,4,6-triiodobenzene and a Series of 1,10-Phenanthroline Derivatives: A Combined Theoretical and Experimental Study" Crystals 9, no. 3: 140. https://doi.org/10.3390/cryst9030140
APA StyleZhang, Y., Wang, J.-G., & Wang, W. (2019). Noncovalent Interactions between 1,3,5-Trifluoro-2,4,6-triiodobenzene and a Series of 1,10-Phenanthroline Derivatives: A Combined Theoretical and Experimental Study. Crystals, 9(3), 140. https://doi.org/10.3390/cryst9030140