Symmetric Fluoroborate and its Boron Modification: Crystal and Electronic Structures



Abstract
1. Introduction
2. Materials and Methods
2.1. Synthesis
2.2. X-ray Single-Crystal Diffraction
2.3. Theoretical Calculations
2.4. Hishfeld Surface Analysis; Pairwise Energies and their Energy Frameworks
3. Results and Discussions

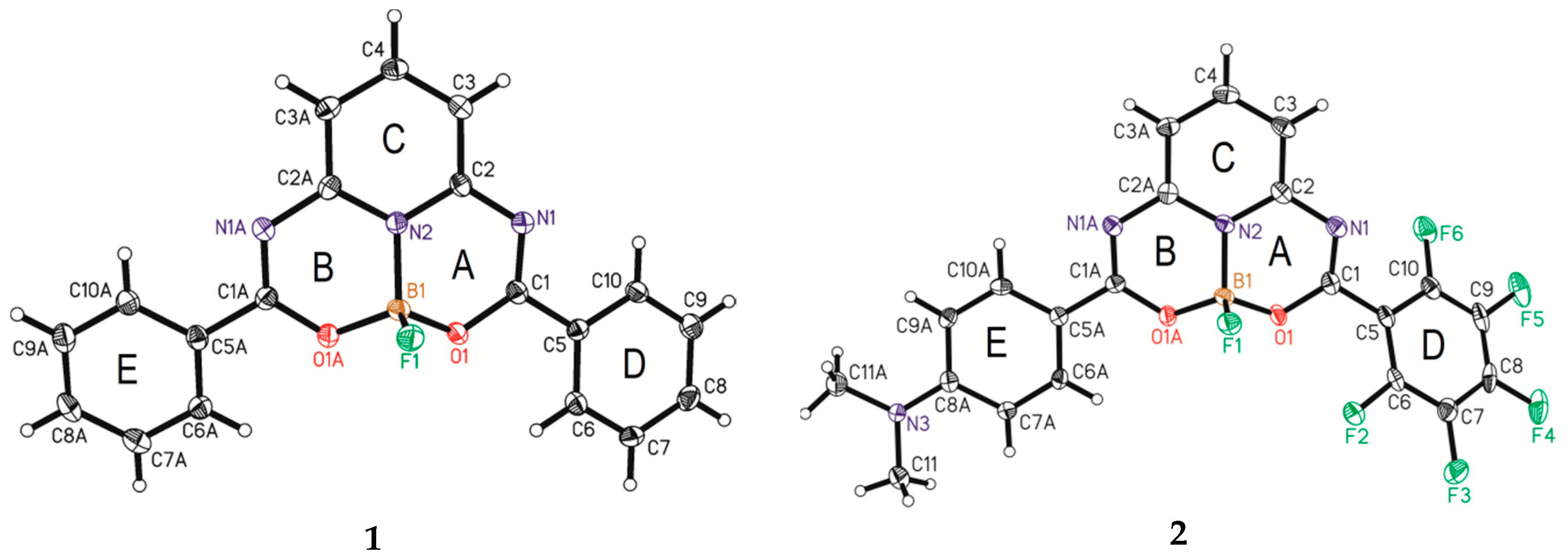
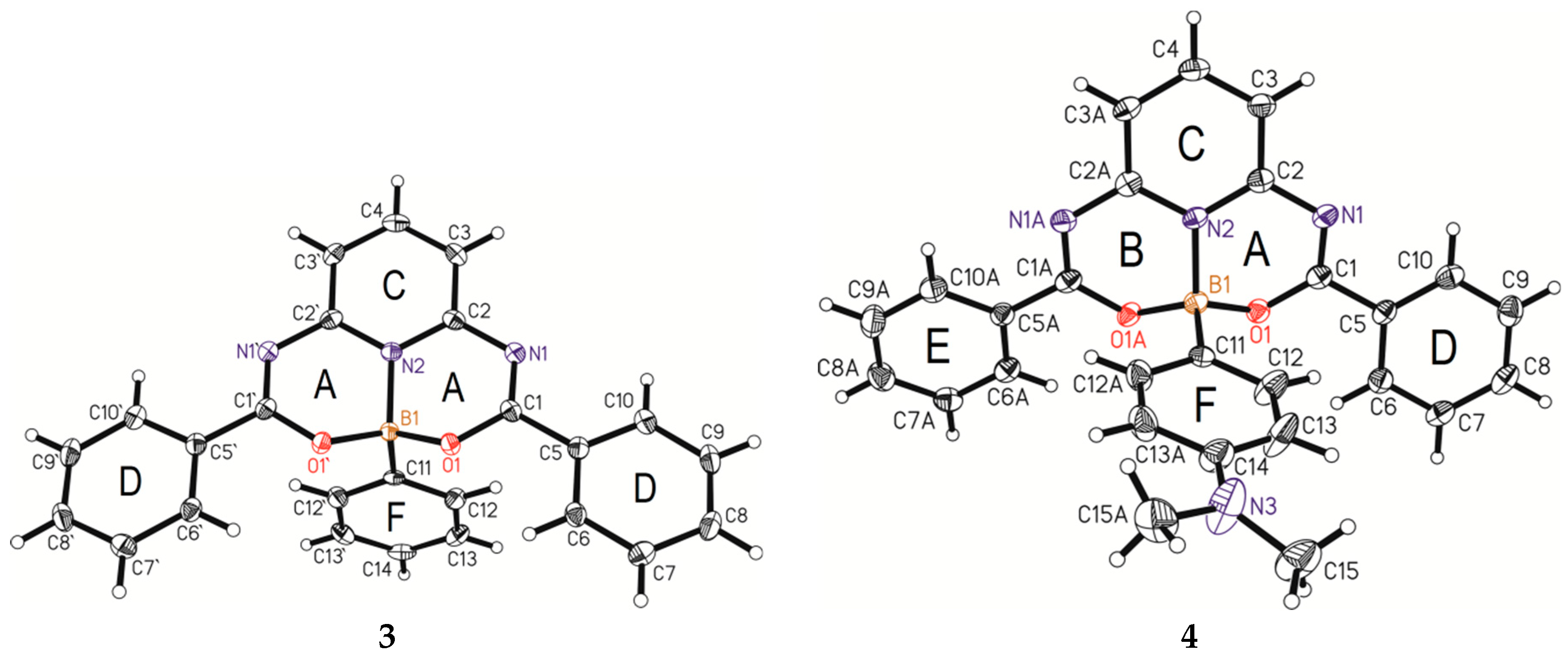
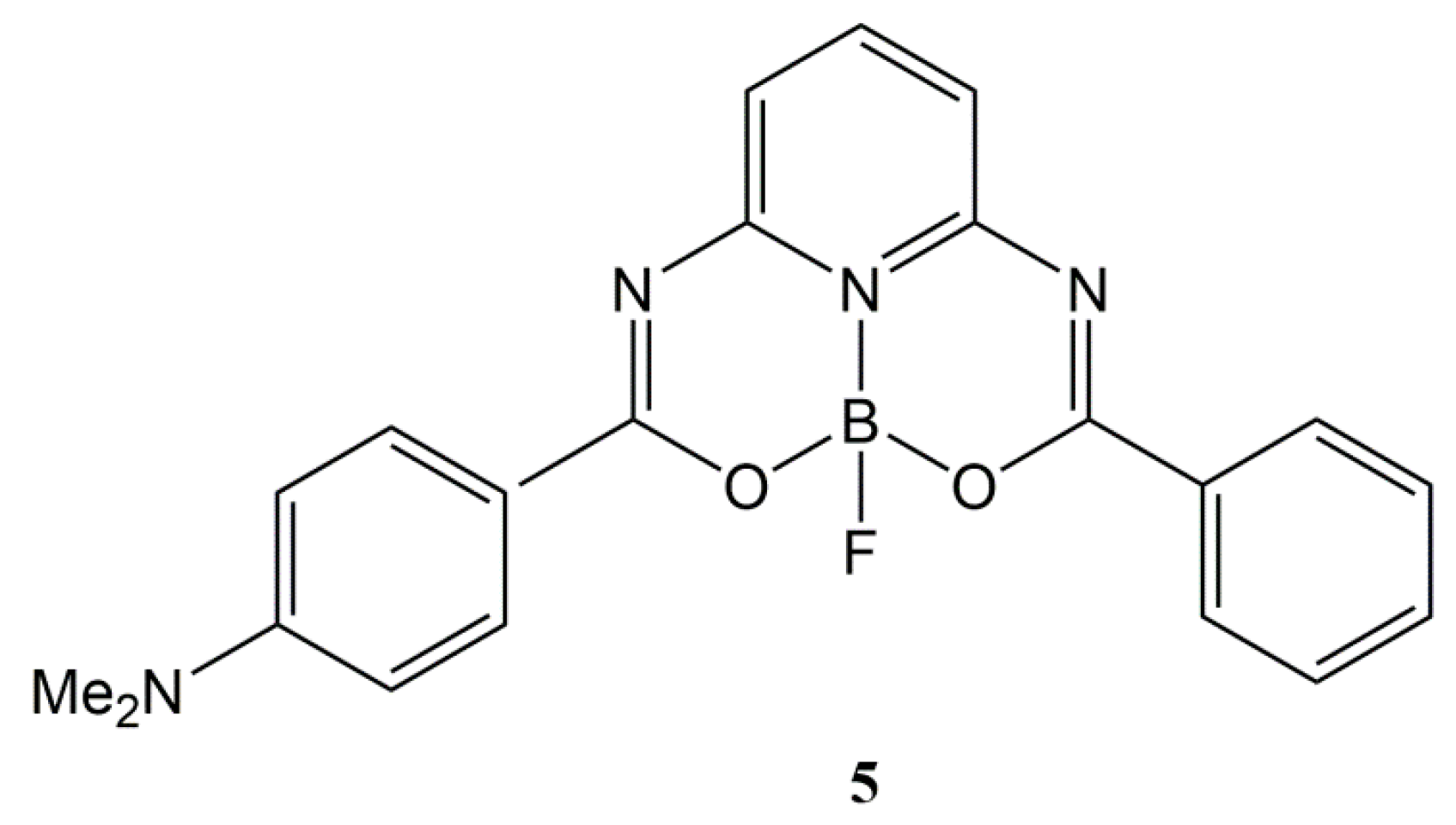
3.1. X-ray Single-Crystal Analysis
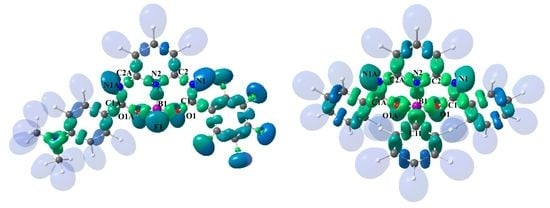
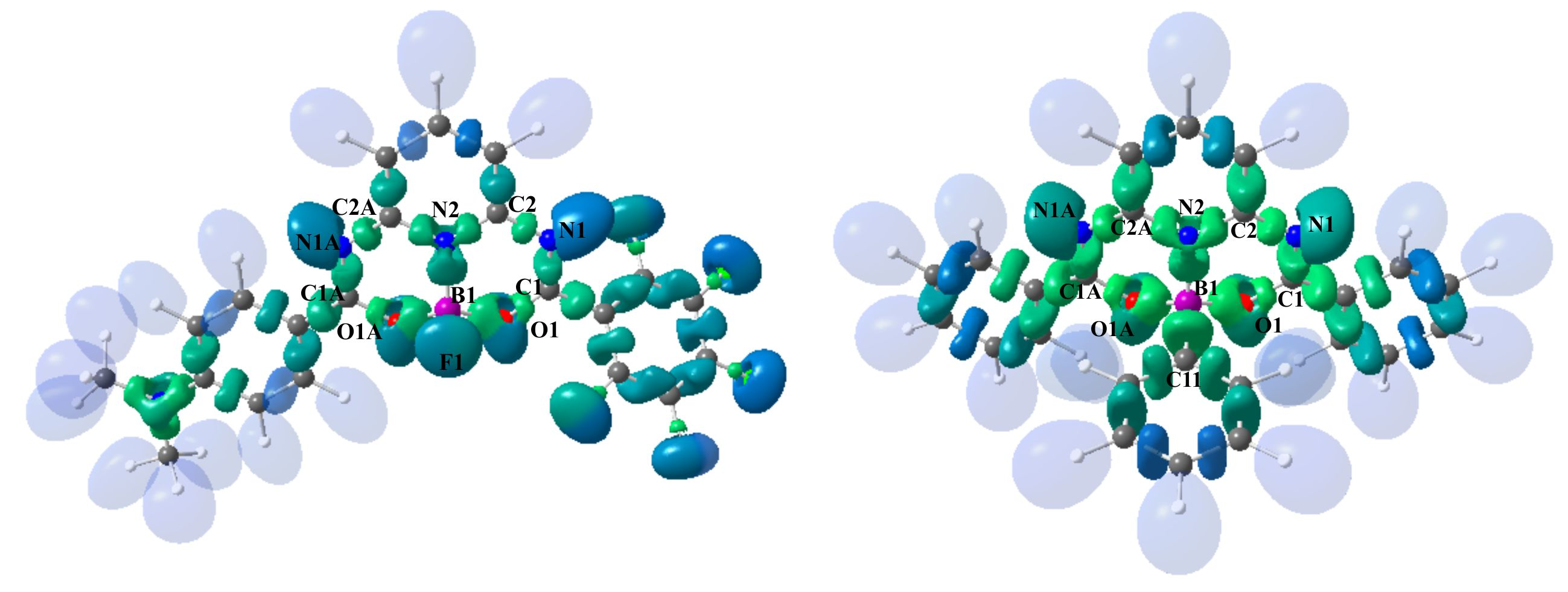
3.2. Theoretical Electron-Density Analysis
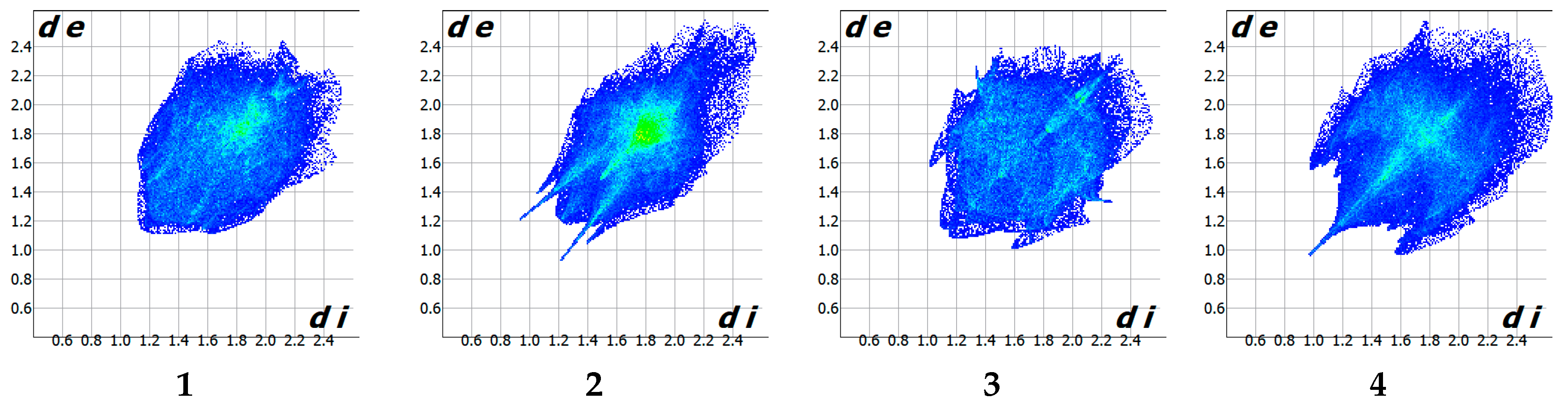
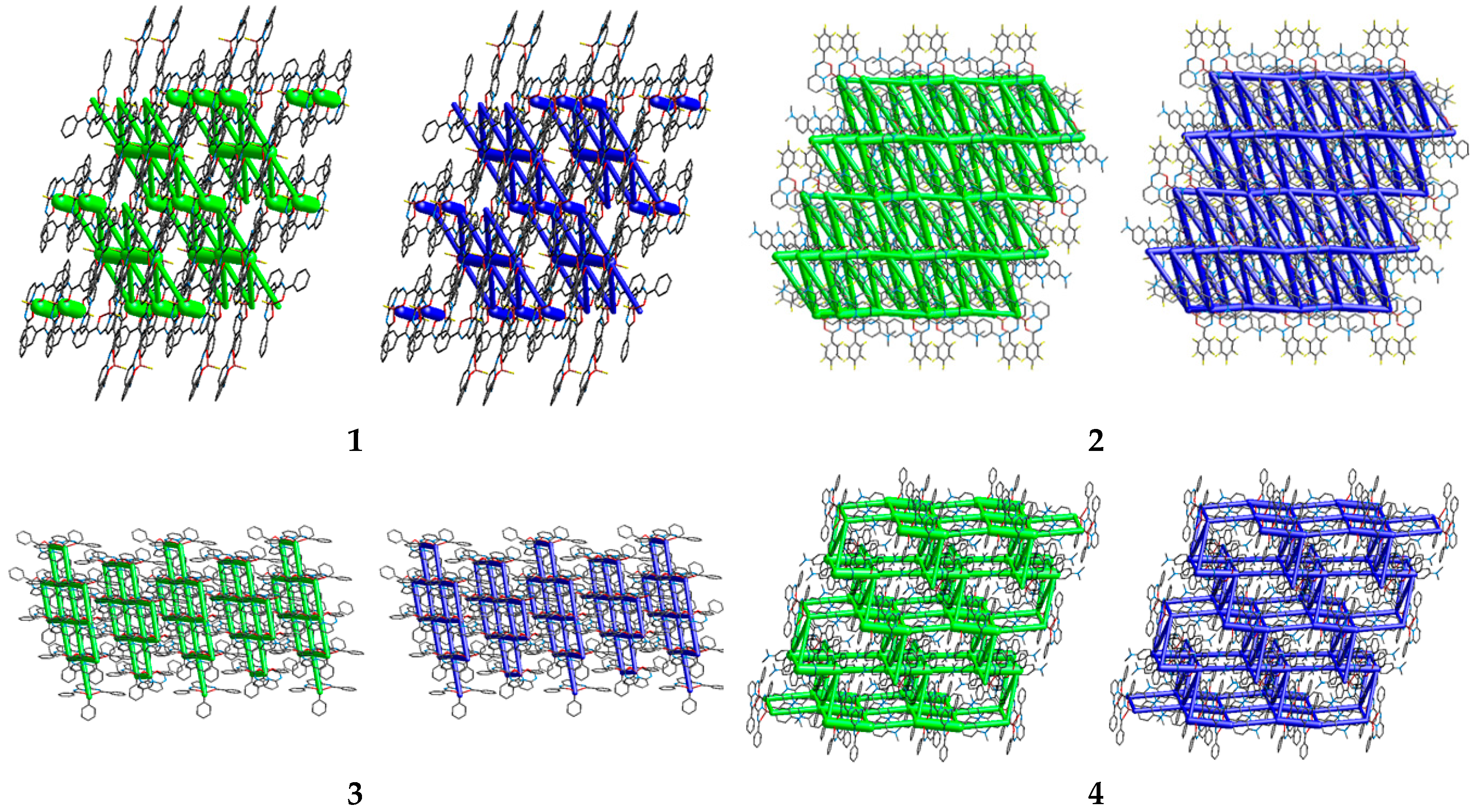
3.3. Intemolecular Interactions and Crystal Packing Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Loudet, A.; Burgess, K. BODIPY Dyes and Their Derivatives: Syntheses and Spectroscopic Properties. Chem. Rev. 2007, 107, 4891–4932. [Google Scholar] [CrossRef] [PubMed]
- Ulrich, G.; Ziessel, R.; Harriman, A. The Chemistry of Fluorescent Bodipy Dyes: Versatility Unsurpassed. Angew. Chem. Int. Ed. 2008, 47, 1184–1201. [Google Scholar] [CrossRef] [PubMed]
- Rouxel, C.; Mongin, O.; Hameau, A.; Ouali, A.; Blanchard-Desce, M.; Majoral, J.P.; Caminade, A.M. BF2 complexes of 1,3-diketones on the surface of phosphorus dendrimers: Synthesis and study of the photoluminescence properties. Can. J. Chem. 2017, 95, 948–953. [Google Scholar] [CrossRef]
- Ono, K.; Yoshikawa, K.; Tsuji, Y.; Yamaguchi, H.; Uozumi, R.; Tomura, M.; Tagaa, K.; Saitoa, K. Synthesis and photoluminescence properties of BF2 complexes with 1,3-diketone ligands. Tetrahedron 2007, 63, 9354–9358. [Google Scholar] [CrossRef]
- Mayoral, M.J.; Ovejero, P.; Campo, J.A.; Heras, J.V.; Oliveira, E.; Pedras, B.; Lodeiro, C.; Cano, M. Exploring photophysical properties of new boron and palladium(II) complexes with b-diketone pyridine type ligands: From liquid crystals to metal fluorescence probes. J. Mater. Chem. 2011, 21, 1255–1263. [Google Scholar] [CrossRef]
- Maeda, H.; Fujiia, Y.; Mihashia, Y. Diol-substituted boron complexes of dipyrrolyl diketones as anion receptors and covalently linked ‘pivotal’ dimers. Chem. Commun. 2008, 4285–4287. [Google Scholar] [CrossRef] [PubMed]
- Grabarz, A.M.; Jędrzejewska, B.; Zakrzewska, A.; Zaleśny, R.; Laurent, A.D.; Jacquemin, D.; Ośmiałowski, B. Photophysical Properties of Phenacylphenantridine Difluoroboranyls: Effect of Substituent and Double Benzannulation. J. Org. Chem. 2017, 82, 1529–1537. [Google Scholar] [CrossRef]
- Ośmiałowski, B.; Zakrzewska, A.; Jędrzejewska, B.; Grabarz, A.; Zaleśny, R.; Bartkowiak, W.; Kolehmainen, E. Influence of Substituent and Benzoannulation on Photophysical Properties of 1-Benzoylmethyleneisoquinoline Difluoroborates. J. Org. Chem. 2015, 80, 2072–2080. [Google Scholar]
- Bai, G.; Yu, C.; Cheng, C.; Hao, E.; Wei, Y.; Mu, X.; Jiao, L. Syntheses and photophysical properties of BF2 complexes of curcumin analogues. Org. Biomol. Chem. 2014, 12, 1618–1626. [Google Scholar] [CrossRef]
- Nosova, E.V.; Moshkina, T.N.; Lipunova, G.N.; Baklanova, I.V.; Slepukhin, P.A.; Charushin, V.N. Synthesis, structure and photoluminescent properties of BF2 and BPh2 complexes with N,O-benzazine ligands. J. Fluor. Chem. 2015, 175, 145–151. [Google Scholar] [CrossRef]
- Crandall, L.A.; Dawadi, M.B.; Burrell, T.; Odoom, A.; Ziegler, C.J. Structure and electronics in dimeric boron expanded azine and salphen complexes. Photochem. Photobiol. Sci. 2017, 16, 627–632. [Google Scholar] [CrossRef] [PubMed]
- Dhanunjayarao, K.; Mukundam, V.; Ramesh, M.; Venkatasubbaiah, K. Synthesis and Optical Properties of Salicylaldimine-Based Diboron Complexes. Eur. J. Inorg. Chem. 2014, 2014, 539–545. [Google Scholar] [CrossRef]
- Frath, D.; Massue, J.; Ulrich, G.; Ziessel, R. Luminescent Materials: Locking p-Conjugated and Heterocyclic Ligands with Boron (III). Angew. Chem. Int. Ed. 2014, 53, 2290–2310. [Google Scholar] [CrossRef] [PubMed]
- Zakrzewska, A.; Zaleśny, R.; Kolehmainen, E.; Ośmiałowski, B.; Jędrzejewska, B.; Pietrzak, M. Substituent effects on the photophysical properties of fluorescent 2-benzoylmethylenequinoline difluoroboranes: A combined experimental and quantum chemical study. Dye. Pigment. 2013, 99, 957–965. [Google Scholar] [CrossRef]
- Grabarz, A.M.; Laurent, A.D.; Jedrzejewska, B.; Zakrzewska, A.; Jacquemin, D.; Osmialowski, B. The influence of the π-conjugated spacer on photophysical properties of difluoroboranyls derived from amides carrying donor group. J. Org. Chem. 2016, 81, 2280–2292. [Google Scholar] [CrossRef]
- Courtis, A.M.; Santos, S.A.; Guan, Y.; Hendricks, J.A.; Ghosh, B.; Szantai-Kis, D.M.; Reis, S.A.; Shah, J.V.; Mazitschek, R. Monoalkoxy BODIPYs A Fluorophore Class for Bioimaging. Bioconjugate Chem. 2014, 25, 1043–1051. [Google Scholar] [CrossRef]
- Goze, C.; Ulrich, G.; Mallon, L.J.; Allen, B.D.; Harriman, A.; Ziessel, R. Synthesis and Photophysical Properties of Borondipyrromethene Dyes Bearing Aryl Substituents at the Boron Center. J. Am. Chem. Soc. 2006, 128, 10231–10239. [Google Scholar] [CrossRef]
- Li, L.; Nguyen, B.; Burgess, K. Functionalization of the 4, 4-difluoro-4-bora-3a, 4a-diaza-s-indacene (BODIPY) core. Bioorg. Med. Chem. Lett. 2008, 18, 3112–3116. [Google Scholar] [CrossRef]
- Goze, C.; Ulrich, G.; Ziessel, R. Tetrahedral Boron Chemistry for the Preparation of Highly Efficient “Cascatelle” devices. J. Org. Chem. 2007, 72, 313–322. [Google Scholar] [CrossRef]
- Brizet, B.; Bernhard, C.; Volkova, Y.; Rousselin, Y.; Harvey, P.D.; Goze, C.; Denat, F. Boron functionalization of BODIPY by various alcohols and phenols. Org. Biomol. Chem. 2013, 11, 7729–7737. [Google Scholar] [CrossRef]
- Maeda, C.; Nagahata, K.; Takaishi, K.; Ema, T. Synthesis of chiral carbazole-based BODIPYs showing circularly polarized luminescence. Chem. Commun. 2019, 55, 3136–3139. [Google Scholar] [CrossRef] [PubMed]
- More, A.B.; Mula, S.; Thakare, S.; Sekar, N.; Ray, A.K.; Chattopadhyay, S. Masking and Demasking Strategies for the BF2-BODIPYs as a Tool for BODIPY Fluorophores. J. Org. Chem. 2014, 79, 10981–10987. [Google Scholar] [CrossRef] [PubMed]
- Crawford, S.M.; Thompson, A. Conversion of 4, 4-Difluoro-4-bora-3a, 4a-diaza-s-indacenes (F-BODIPYs) to Dipyrrins with a Microwave-Promoted Deprotection Strategy. Org. Lett. 2010, 12, 1424–1427. [Google Scholar] [CrossRef] [PubMed]
- Smithen, D.A.; Baker, A.E.G.; Offman, M.; Crawford, S.M.; Cameron, T.S.; Thompson, A. Use of F-BODIPYs as a Protection Strategy for Dipyrrins: Optimization of BF2 Removal. J. Org. Chem. 2012, 77, 3439–3453. [Google Scholar] [CrossRef]
- Kawatani, M.; Kamiya, M.; Takahashi, H.; Urano, Y. Factors affecting the uncaging efficiency of 500 nm light-activatable BODIPY caging group. Bioorg. Med. Chem. Lett. 2018, 28, 1–5. [Google Scholar] [CrossRef]
- Kim, H.; Burghart, A.; Welch, M.B.; Reibenspies, J.; Burgess, K. Synthesis and spectroscopic properties of a new 4-bora-3a,4a-diaza-s-indacene (BODIPY) dye. Chem. Commun. 1999, 1889–1890. [Google Scholar] [CrossRef]
- Okujima, T.; Shida, Y.; Ohara, K.; Tomimori, Y.; Nishioka, M.; Mori, S.; Nakae, T.; Uno, H. Synthesis of NIR-emitting O-chelated BODIPYs fused with benzene and acenaphthylene. J. Porphyr. Phthalocyanines 2014, 18, 752–761. [Google Scholar] [CrossRef]
- Lundrigan, T.; Crawford, S.M.; Cameron, T.S.; Thompson, A. Cl-BODIPYs: A BODIPY class enabling facile B-substitution. Chem. Commun. 2012, 48, 1003–1005. [Google Scholar] [CrossRef]
- Ulrich, G.; Goze, C.; Guardigli, M.; Roda, A.; Ziessel, R. Pyrromethene Dialkynyl Borane Complexes for “Cascatelle” Energy Transfer and Protein Labeling. Angew. Chem. Int. Ed. 2005, 44, 3694–3698. [Google Scholar] [CrossRef]
- Davies, L.H.; Stewart, B.; Harrington, R.W.; Clegg, W.; Higham, L.J. Air-Stable, Highly Fluorescent Primary Phosphanes. Angew. Chem. Int. Ed. 2012, 51, 4921–4924. [Google Scholar] [CrossRef]
- Haefele, A.; Ulrich, G.; Retailleau, P.; Ziessel, R. Synthesis of multi-branched dipyrromethene dyes with soluble diethynylphenyl links. Tetrahedron Lett. 2008, 49, 3716–3721. [Google Scholar] [CrossRef]
- Goze, C.; Ulrich, G.; Ziessel, R. Unusual Fluorescent Monomeric and Dimeric Dialkynyl Dipyrromethene-Borane Complexes. Org. Lett. 2006, 8, 4445–4448. [Google Scholar] [CrossRef] [PubMed]
- Zhang, G.; Wang, M.; Fronczek, F.R.; Smith, K.M.; Vicente, M.G.H. Lewis-Acid-Catalyzed BODIPY Boron Functionalization Using Trimethylsilyl Nucleophiles. Inorg. Chem. 2018, 57, 14493–14496. [Google Scholar] [CrossRef] [PubMed]
- Ray, C.; Díaz-Casado, L.; Avellanal-Zaballa, E.; Banuelos, J.; Cerdán, L.; García-Moreno, I.; Moreno, F.; Maroto, B.L.; López-Arbeloa, I.; Moya, S. N-BODIPYs Come into Play: Smart Dyes for Photonic Materials. Chem. Eur. J. 2017, 23, 9383–9390. [Google Scholar] [CrossRef]
- Chen, N.; Zhang, W.; Chen, S.; Wu, Q.; Yu, C.; Wei, Y.; Xu, Y.; Hao, E.; Jiao, L. Sterically Protected N2O-Type Benzopyrromethene Boron Complexes from Boronic Acids with Intense Red/Near-Infrared Fluorescence. Org. Lett. 2017, 19, 2026–2029. [Google Scholar] [CrossRef]
- Claessens, C.G.; González-Rodríguez, D.; Torres, T. Subphthalocyanines: Singular Nonplanar Aromatic Compounds—Synthesis, Reactivity, and Physical Properties. Chem. Rev. 2002, 102, 835–853. [Google Scholar] [CrossRef]
- Zhang, S.; Wang, Y.; Meng, F.; Cheng, Y.; Dai, C.; Zhu, C. Circularly polarized luminescence of AIE-active chiral O-BODIPYs induced via intramolecular energy transfer. Chem. Commun. 2015, 51, 9014–9017. [Google Scholar] [CrossRef]
- Groves, B.R.; Crawford, S.M.; Lundrigan, T.; Matta, C.F.; Sowlati-Hashjinb, S.; Thompson, A. Synthesis and characterisation of the unsubstituted dipyrrin and 4, 4-dichloro-4-bora-3a, 4a-diaza-s-indacene: Improved synthesis and functionalisation of the simplest BODIPY framework. Chem. Commun. 2013, 49, 816–818. [Google Scholar] [CrossRef]
- Bednarska, J.; Zaleśny, R.; Wielgus, M.; Jędrzejewska, B.; Puttreddy, R.; Rissanen, K.; Bartkowiak, W.; Agren, H.; Ośmiałowski, B. Two-photon absorption of BF2-carrying compounds: Insights from theory and experiment. Phys. Chem. Chem. Phys. 2017, 19, 5705–5708. [Google Scholar] [CrossRef]
- Kim, H.M.; Cho, B.R. Small-Molecule Two-Photon Probes for Bioimaging Applications. Chem. Rev. 2015, 115, 5014–5055. [Google Scholar] [CrossRef]
- Li, Y.-X.; Zhang, H.; Yu, M.-N.; Wang, S.-S.; Liu, Y.-R.; Lin, D.-Q.; Xie, L.-H.; Lin, Z.-Q.; Huang, W. Supramolecular steric hindrance effect on morphologies and photophysical behaviors of spirocyclic aromatic hydrocarbon nanocrystals. Nanoscale 2019, 11, 5158–5162. [Google Scholar] [CrossRef] [PubMed]
- Xie, L.-H.; Huang, W. Supramolecular Steric Hindrance at Bulky Organic/Polymer Semiconductors and Devices. In Non-Covalent Interactions in the Synthesis and Design of New Compounds, 1st ed.; Maharramov, A.M., Mahmudov, K.T., Kopylovich, M.N., Pombeiro, A.J.L., Eds.; John Wiley & Sons Inc.: Hoboken, NJ, USA, 2016; pp. 443–455. [Google Scholar] [CrossRef]
- Glotzbach, C.; Kauscher, U.; Voskuhl, J.; Kehr, N.S.; Stuart, M.C.A.; Frohlich, R.; Galla, H.J.; Ravoo, B.J.; Nagura, K.; Saito, S.; et al. Fluorescent Modular Boron Systems Based on NNN- and ONO- Tridentate Ligands: Self-Assembly and Cell Imaging. J. Org. Chem. 2013, 78, 4410–4418. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, K.; Osuka, A. Subporpholactone, Subporpholactam, Imidazolosubporphyrin, and Iridium Complexes of Imidazolosubporphyrin: Formation of Iridium Carbene Complexes. Angew. Chem. Int. Ed. 2017, 57, 338–342. [Google Scholar] [CrossRef] [PubMed]
- Inokuma, Y.; Kwon, J.H.; Ahn, T.K.; Yoo, M.-C.; Kim, D.; Osuka, A. Tribenzosubporphines: Synthesis and Characterization. Angew. Chem. Int. Ed. 2006, 45, 961–964. [Google Scholar] [CrossRef] [PubMed]
- Inokuma, Y.; Yoon, Z.S.; Kim, D.; Osuka, A. Meso-Aryl-Substituted Subporphyrins: Synthesis, Structures, and Large Substituent Effects on Their Electronic Properties. J. Am. Chem. Soc. 2007, 129, 4747–4761. [Google Scholar] [CrossRef]
- Zhang, H.; Huo, C.; Zhang, J.; Zhang, P.; Tian, W.; Wang, Y. Efficient single-layer electroluminescent device based on a bipolar emitting boron-containing material. Chem. Commun. 2006, 281–283. [Google Scholar] [CrossRef]
- Chouaib Tahtaoui, C.; Thomas, C.; Rohmer, F.; Klotz, P.; Duportail, G.; Mély, Y.; Bonnet, D.; Hibert, M. Convenient Method to Access New 4, 4-Dialkoxyand 4, 4-Diaryloxy-diaza-s-indacene Dyes: Synthesis and Spectroscopic Evaluation. J. Org. Chem. 2007, 72, 269–272. [Google Scholar] [CrossRef]
- Sánchez-Carnerero, E.M.; Gartzia-Rivero, L.; Moreno, F.; Maroto, B.L.; Agarrabeitia, A.R.; Ortiz, M.J.; Banuelos, J.; López-Arbeloa, I.; Moya, S. Spiranic BODIPYs: A ground-breaking design to improve the energy transfer in molecular Cassettes. Chem. Commun. 2014, 50, 12765–12767. [Google Scholar] [CrossRef]
- Bader, R.F.W. Atoms in molecules. In A Quantum Theory; Oxford University Press: Oxford, UK, 1990. [Google Scholar]
- Kohout, M. A measure of electron localizability. Int. J. Quantum Chem. 2004, 97, 651–658. [Google Scholar] [CrossRef]
- Kohout, M.; Wagner, F.R.; Grin, Y. Electron localizability indicator for correlated wavefunctions III: Singlet and triplet pairs. Theor. Chem. Acc. 2008, 119, 413–420. [Google Scholar] [CrossRef]
- Spackman, M.A.; Jayatilaka, D. Hirshfeld surface analysis. CrystEngComm 2009, 11, 19–32. [Google Scholar] [CrossRef]
- Turner, M.J.; Grabowsky, S.; Jayatilaka, D.; Spackman, M.A. Accurate and efficient model energies for exploring intermolecular interactions in molecular crystals. J. Phys. Chem. Lett. 2014, 5, 4249–4255. [Google Scholar] [CrossRef]
- Turner, M.J.; Thomas, S.P.; Shi, M.W.; Jayatilaka, D.; Spackman, M.A. Energy frameworks: Insights into interaction anisotropy and the mechanical properties of molecular crystals. Chem. Commun. 2015, 51, 3735–3738. [Google Scholar] [CrossRef] [PubMed]
- CrysAlis CCD, Version 1.171.33.57; Oxford Diffraction Ltd.: Abingdon, UK, 2005.
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Cryst. Sect. C 2015, 71, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Sheldrick, G.M. A short history of SHELX. Acta Cryst. Sect. A 2008, 64, 112–122. [Google Scholar] [CrossRef] [PubMed]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09; Rev. D.01; Gaussian Inc.: Wallingford, CT, USA, 2013. [Google Scholar]
- Keith, T.A. AIMAll, Version 16.05.18; TK Gristmill Software: Overland Park, KS, USA, 2014.
- Kohout, M. Dgrid-4.6. Available online: http://www.cpfs.mpg.de/~kohout/dgrid.html (accessed on 15 May 2015).
- Turner, M.J.; McKinnon, J.J.; Wolff, S.K.; Grimwood, D.J.; Spackman, P.R.; Jayatilaka, D.; Spackman, M.A. CrystalExplorer 17; University of Western Australia: Crawley, Australia, 2017. [Google Scholar]
- Mackenzie, C.F.; Spackman, P.R.; Jayatilaka, D.; Spackman, M.A. CrystalExplorer model energies and energy frameworks: Extension to metal coordination compounds, organic salts, solvates and open-shell systems. IUCr J. 2017, 4, 575–587. [Google Scholar] [CrossRef]
- Barros, L.W.T.; Cardoso, T.A.S.; Bihlmeier, A.; Wagner, D.; Kölmel, D.K.; Hörner, A.; Bräse, S.; Cruz, C.H.B.; Padilha, L.A. Two-photon absorption in a series of 2, 6-disubstituted BODIPY dyes. Phys. Chem. Chem. Phys. 2017, 19, 21683–21690. [Google Scholar] [CrossRef]
- Descalzo, A.B.; Xu, H.-J.; Xue, Z.-L.; Hoffmann, K.; Shen, Z.; Weller, M.G.; You, X.-Z.; Rurack, K. Phenanthrene-Fused Boron-Dipyrromethenes as Bright Long-Wavelength Fliorophores. Org. Lett. 2008, 10, 1581–1584. [Google Scholar] [CrossRef]
- Cremer, D.; Pople, J.A. General definition of ring puckering coordinates. J. Am. Chem. Soc. 1975, 97, 1354–1358. [Google Scholar] [CrossRef]
- Duax, L.; Norton, D.A. Atlas of Steroid Structure; IFI/Plenum: New York, NY, USA, 1975; Volume 1, pp. 16–22. [Google Scholar]
- Raub, S.; Jansen, G. A quantitative measure of bond polarity from the electron localization function and the theory of atoms in molecules. Theor. Chem. Acc. 2001, 106, 223–232. [Google Scholar] [CrossRef]
- Vidal, I.; Melchor, S.; Dobado, J.A. On the nature of metal-carbon bonding: AIM and ELF analyses of MCHn (n = 1–3) compounds containing early transition metals. J. Phys. Chem. A 2005, 109, 7500–7508. [Google Scholar] [CrossRef] [PubMed]
- Chęcińska, L.; Mebs, S.; Ośmiałowski, B.; Zakrzewska, A.; Ejsmont, K.; Kohout, M. Tuning the Electronic Properties of the Dative NB Bond with Associated OB Interaction: Electron Localizability Indicator from X-Ray Wavefunction Refinement. Chem. Phys. Chem. 2016, 17, 1–13. [Google Scholar] [CrossRef]
- Bader, R.F.W.; Slee, T.S.; Cremer, D.; Kraka, E. Description of conjugation and hyperconjugation in terms of electron distributions. J. Am. Chem. Soc. 1983, 105, 5061–5068. [Google Scholar] [CrossRef]
- Bader, R.F.W.; Stephens, M.E. Spatial localization of the electronic pair and number distributions in molecules. J. Am. Chem. Soc. 1975, 97, 7391–7399. [Google Scholar] [CrossRef]
- Fradera, X.; Austen, M.A.; Bader, R.F.W. The Lewis model and beyond. J. Phys. Chem. A 1999, 103, 304–314. [Google Scholar] [CrossRef]
- Espinosa, E.; Alkorta, I.; Elguero, J.; Molins, E. From weak to strong interactions: A comprehensive analysis of the topological and energetic properties of the electron density distribution involving X–H…F–Y systems. J. Chem. Phys. 2002, 117, 5529–5542. [Google Scholar] [CrossRef]
- Yuan, Z.; Entwistle, C.D.; Collings, J.C.; Albesa-Jové, D.; Batsanov, A.S.; Howard, J.A.K.; Taylor, N.J.; Kaiser, H.M.; Kaufmann, D.E.; Poon, S.Y.; et al. Synthesis, Crystal Structures, Linear and Nonlinear Optical Properties, and Theoretical Studies of (p-R-Phenyl)-, (p-R-Phenylethynyl)-, and (E)-[2-(p-R-Phenyl-ethenyl]dimesitylboranes and Related Compounds. Chem. Eur. J. 2006, 12, 2758–2771. [Google Scholar] [CrossRef]
- Hübschle, C.B.; Luger, P. Moliso-A program for colour-mapped iso-surfaces. J. Appl. Cryst. 2006, 39, 901–904. [Google Scholar] [CrossRef]
- Spackman, M.A.; McKinnon, J.J. Fingerprinting intermolecular interactions in molecular crystals. CrystEngComm 2002, 4, 378–392. [Google Scholar] [CrossRef]
- Chęcińska, L.; Jóźwiak, A.; Ciechańska, M.; Paulmann, C.; Holstein, J.J.; Dittrich, B.; Małecka, M. Quantifying intermolecular interactions for isoindole derivatives: Substituent effect vs, crystal packing. Z. Krist. 2018, 233, 675–687. [Google Scholar] [CrossRef]
- Thomas, S.P.; Spackman, P.R.; Jayatilaka, D.; Spackman, M.A. Accurate Lattice Energies for Molecular Crystals from Experimental Crystal Structures. J. Chem. Theory Comput. 2018, 14, 1614–1623. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Comp./Bond | d | ρbcp | ∇2ρbcp | G/ρbcp | H/ρbcp | ε | δ | V001ELI | ELIpop | ΔELI | RJI |
|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | |||||||||||
| N1–B1 | 1.549 | 1.07 | 4.3 | 1.23 | −0.96 | 0.00 | 0.29 | 7.51 | 2.98 | 0.003 | 94.2 |
| O1/1A–B1 | 1.461 | 1.12 | 11.3 | 1.57 | −0.96 | 0.04 | 0.29 | 2.17 | 1.95 | 0.029 | 92.9 |
| F1–B1 | 1.384 | 1.20 | 19.7 | 1.93 | −0.77 | 0.01 | 0.31 | 1.11 | 1.18 | 0.007 | 89.6 |
| N2–C2/2A | 1.362 | 2.17 | −24.5 | 0.57 | −1.36 | 0.15 | 1.12 | 3.30 | 2.32 | 0.018 | 71.5 |
| N1/1A–C1/1A | 1.303 | 2.53 | −30.5 | 0.59 | −1.44 | 0.20 | 1.35 | 5.07 | 2.66 | 0.061 | 62.2 |
| N2/2A–C2/2A | 1.368 | 2.21 | −23.7 | 0.43 | −1.18 | 0.09 | 1.15 | 2.76 | 2.14 | 0.046 | 60.8 |
| O1/1A–C1/1A | 1.324 | 2.17 | −17.0 | 1.03 | −1.58 | 0.04 | 0.97 | 1.43 | 1.59 | 0.024 | 75.5 |
| 2 | |||||||||||
| N2–B1 | 1.550 | 1.07 | 4.2 | 1.23 | −0.95 | 0.01 | 0.29 | 7.56 | 2.99 | 0.006 | 94.2 |
| O1A–B1 | 1.454 | 1.15 | 11.5 | 1.58 | −0.88 | 0.04 | 0.30 | 2.17 | 1.94 | 0.029 | 92.8 |
| O1–B1 | 1.473 | 1.09 | 10.8 | 1.55 | −0.86 | 0.04 | 0.29 | 2.18 | 1.95 | 0.025 | 93.1 |
| F1–B1 | 1.382 | 1.21 | 19.9 | 1.93 | −0.78 | 0.01 | 0.31 | 1.11 | 1.19 | 0.007 | 89.6 |
| N2–C2/2A | 1.364 | 2.17 | −24.3 | 0.57 | −1.35 | 0.14 | 1.11 | 3.25 | 2.33 | 0.019 | 71.3 |
| N1A–C1A | 1.311 | 2.49 | −29.8 | 0.56 | −1.40 | 0.19 | 1.32 | 4.63 | 2.59 | 0.058 | 61.8 |
| N1A–C2A | 1.359 | 2.25 | −24.4 | 0.44 | −1.20 | 0.10 | 1.18 | 2.89 | 2.18 | 0.047 | 60.4 |
| N1–C1 | 1.292 | 2.58 | −31.1 | 0.63 | −1.48 | 0.25 | 1.40 | 6.04 | 2.81 | 0.060 | 60.8 |
| N1–C2 | 1.378 | 2.16 | −22.8 | 0.42 | −1.16 | 0.08 | 1.12 | 2.63 | 2.09 | 0.044 | 61.3 |
| O1A–C1A | 1.328 | 2.14 | −17.0 | 1.02 | −1.57 | 0.03 | 0.96 | 1.41 | 1.58 | 0.024 | 75.7 |
| O1–C1 | 1.318 | 2.21 | −17.8 | 1.03 | −1.59 | 0.07 | 0.98 | 1.47 | 1.61 | 0.024 | 74.6 |
| 3 | |||||||||||
| N2–B1 | 1.572 | 1.00 | 4.8 | 1.25 | −0.91 | 0.10 | 0.31 | 7.12 | 2.90 | 0.011 | 94.8 |
| O1/1A–B1 | 1.480 | 1.05 | 11.7 | 1.60 | −0.82 | 0.13 | 0.31 | 2.17 | 1.94 | 0.037 | 93.7 |
| C11–B1 | 1.615 | 1.21 | −9.3 | 0.55 | −1.08 | 0.02 | 0.46 | 5.69 | 2.28 | 0.007 | 82.9 |
| N2–C2/2A | 1.362 | 2.18 | −24.5 | 0.57 | −1.36 | 0.15 | 1.12 | 3.49 | 2.38 | 0.017 | 71.9 |
| N1/1A–C1/1A | 1.304 | 2.52 | −30.4 | 0.58 | −1.43 | 0.20 | 1.34 | 4.66 | 2.65 | 0.058 | 62.0 |
| N2/2A–C2/2A | 1.369 | 2.20 | −23.5 | 0.43 | −1.17 | 0.09 | 1.15 | 2.78 | 2.13 | 0.050 | 60.7 |
| O–C | 1.318 | 2.20 | −16.7 | 1.06 | −1.60 | 0.04 | 0.98 | 1.46 | 1.61 | 0.025 | 75.5 |
| 4 | |||||||||||
| N2–B1 | 1.558 | 1.02 | 5.3 | 1.28 | −0.92 | 0.11 | 0.31 | 6.73 | 2.92 | 0.006 | 94.8 |
| O1/1A–B1 | 1.488 | 1.03 | 11.3 | 1.59 | −0.81 | 0.14 | 0.31 | 2.11 | 1.92 | 0.037 | 94.0 |
| C11–B1 | 1.602 | 1.24 | −9.4 | 0.56 | −1.09 | 0.04 | 0.46 | 5.47 | 2.31 | 0.010 | 82.9 |
| N2–C2/2A | 1.361 | 2.18 | −24.5 | 0.57 | −1.36 | 0.15 | 1.12 | 3.32 | 2.36 | 0.017 | 71.7 |
| N1/1A–C1/1A | 1.306 | 2.51 | −30.3 | 0.58 | −1.42 | 0.20 | 1.34 | 4.89 | 2.64 | 0.060 | 62.0 |
| N2/2A–C2/2A | 1.368 | 2.21 | −23.6 | 0.42 | −1.17 | 0.09 | 1.15 | 2.78 | 2.13 | 0.048 | 60.4 |
| O–C | 1.315 | 2.21 | −16.9 | 1.07 | −1.60 | 0.04 | 0.98 | 1.51 | 1.63 | 0.024 | 75.4 |
| 5 | |||||||||||
| N2–B1 | 1.548 | 1.08 | 4.3 | 1.23 | −0.96 | 0.00 | 0.29 | 7.64 | 3.01 | 0.002 | 94.2 |
| O1A–B1 | 1.458 | 1.14 | 11.4 | 1.58 | −0.88 | 0.04 | 0.29 | 2.16 | 1.94 | 0.029 | 92.8 |
| O1–B1 | 1.464 | 1.12 | 11.2 | 1.57 | −0.87 | 0.04 | 0.29 | 2.17 | 1.95 | 0.029 | 93.0 |
| F1–B1 | 1.386 | 1.19 | 19.6 | 1.92 | −0.77 | 0.01 | 0.31 | 1.11 | 1.18 | 0.007 | 89.7 |
| N2–C2/2A | 1.363 | 2.17 | −24.4 | 0.57 | −1.35 | 0.15 | 1.11 | 3.28 | 2.33 | 0.018 | 71.6 |
| N1A–C1A | 1.310 | 2.49 | −30.0 | 0.57 | −1.41 | 0.19 | 1.33 | 4.72 | 2.60 | 0.059 | 61.8 |
| N1A–C2A | 1.362 | 2.24 | −24.2 | 0.44 | −1.19 | 0.10 | 1.17 | 2.85 | 2.16 | 0.047 | 60.5 |
| N1–C1 | 1.302 | 2.53 | −30.5 | 0.60 | −1.44 | 0.21 | 1.36 | 5.13 | 2.68 | 0.061 | 62.2 |
| N1–C2 | 1.370 | 2.20 | −23.5 | 0.43 | −1.17 | 0.09 | 1.15 | 2.74 | 2.13 | 0.046 | 61.0 |
| O1A–C1A | 1.328 | 2.15 | −17.1 | 1.02 | −1.58 | 0.03 | 0.96 | 1.41 | 1.58 | 0.023 | 75.4 |
| O1–C1 | 1.323 | 2.17 | −17.1 | 1.03 | −1.58 | 0.04 | 0.97 | 1.43 | 1.59 | 0.024 | 75.4 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dziuk, B.; Ośmiałowski, B.; Zarychta, B.; Ejsmont, K.; Chęcińska, L. Symmetric Fluoroborate and its Boron Modification: Crystal and Electronic Structures. Crystals 2019, 9, 662. https://doi.org/10.3390/cryst9120662
Dziuk B, Ośmiałowski B, Zarychta B, Ejsmont K, Chęcińska L. Symmetric Fluoroborate and its Boron Modification: Crystal and Electronic Structures. Crystals. 2019; 9(12):662. https://doi.org/10.3390/cryst9120662
Chicago/Turabian StyleDziuk, Błażej, Borys Ośmiałowski, Bartosz Zarychta, Krzysztof Ejsmont, and Lilianna Chęcińska. 2019. "Symmetric Fluoroborate and its Boron Modification: Crystal and Electronic Structures" Crystals 9, no. 12: 662. https://doi.org/10.3390/cryst9120662
APA StyleDziuk, B., Ośmiałowski, B., Zarychta, B., Ejsmont, K., & Chęcińska, L. (2019). Symmetric Fluoroborate and its Boron Modification: Crystal and Electronic Structures. Crystals, 9(12), 662. https://doi.org/10.3390/cryst9120662