Intermolecular Interactions in Ionic Crystals of Nucleobase Chlorides—Combining Topological Analysis of Electron Densities with Energies of Electrostatic Interactions



Abstract
1. Introduction
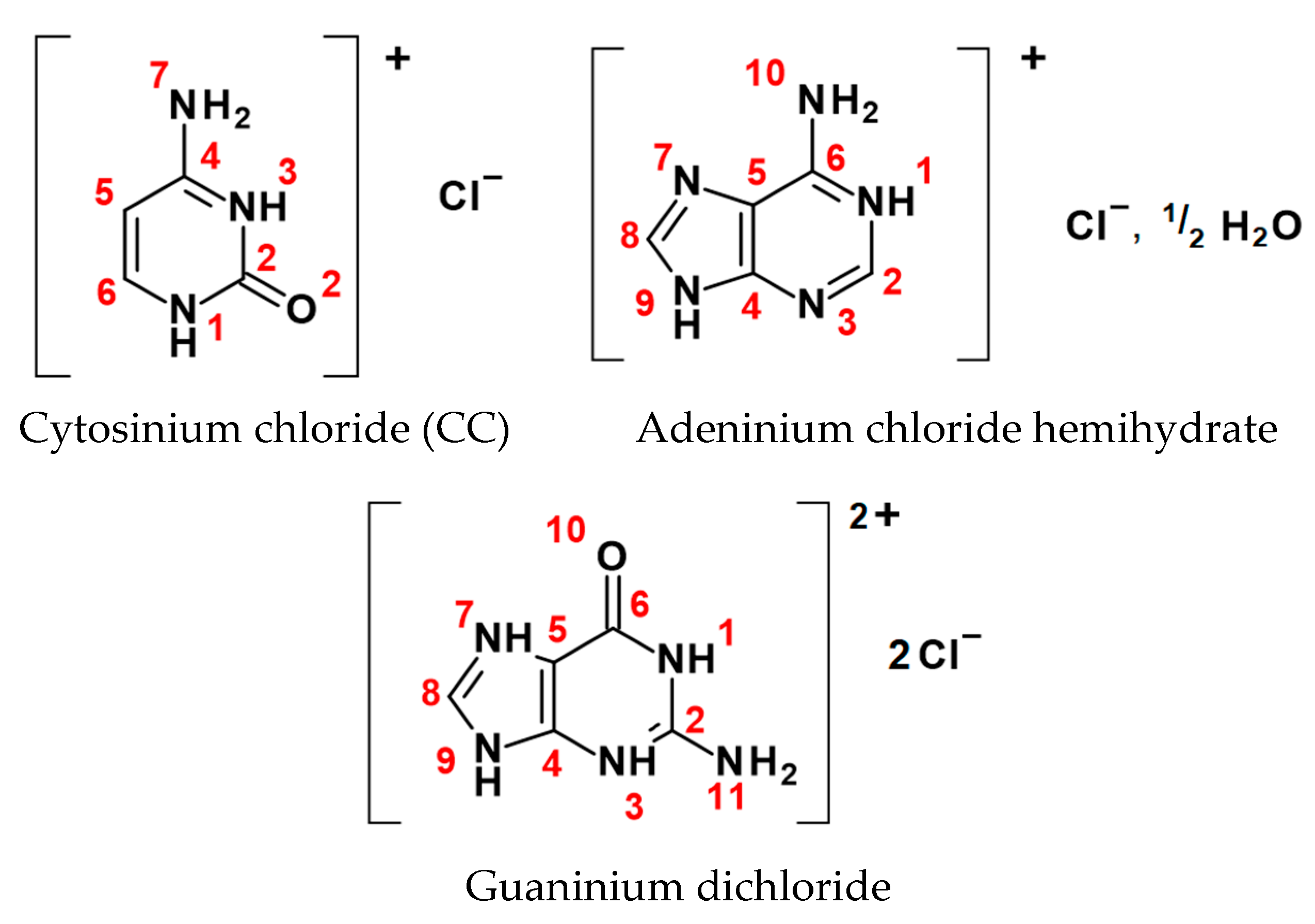
2. Materials and Methods
2.1. Crystal Charge Density Models
2.2. QTAIM Analysis
2.3. Intermolecular Interaction Energies and Electrostatic Contributions to Them
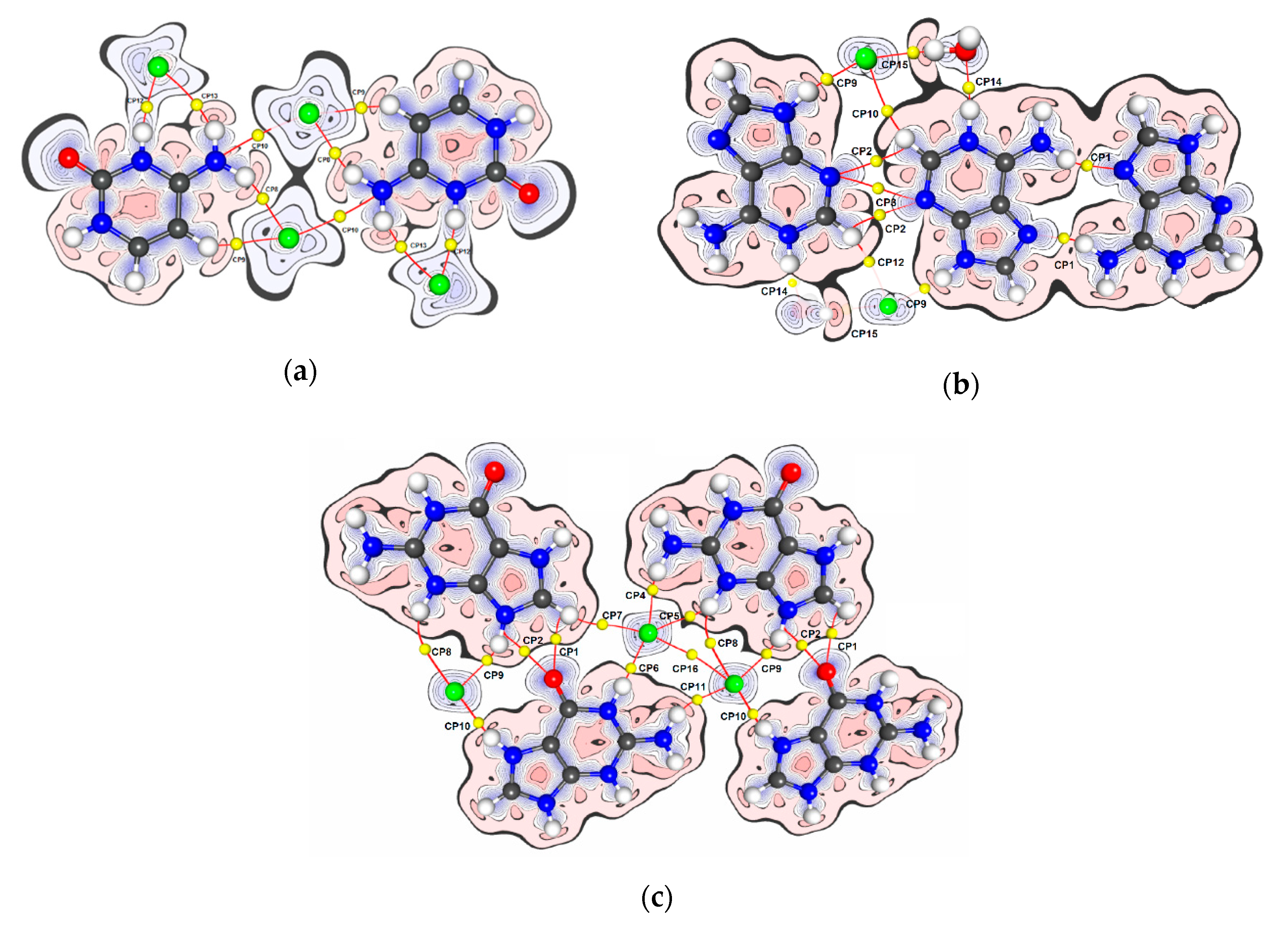
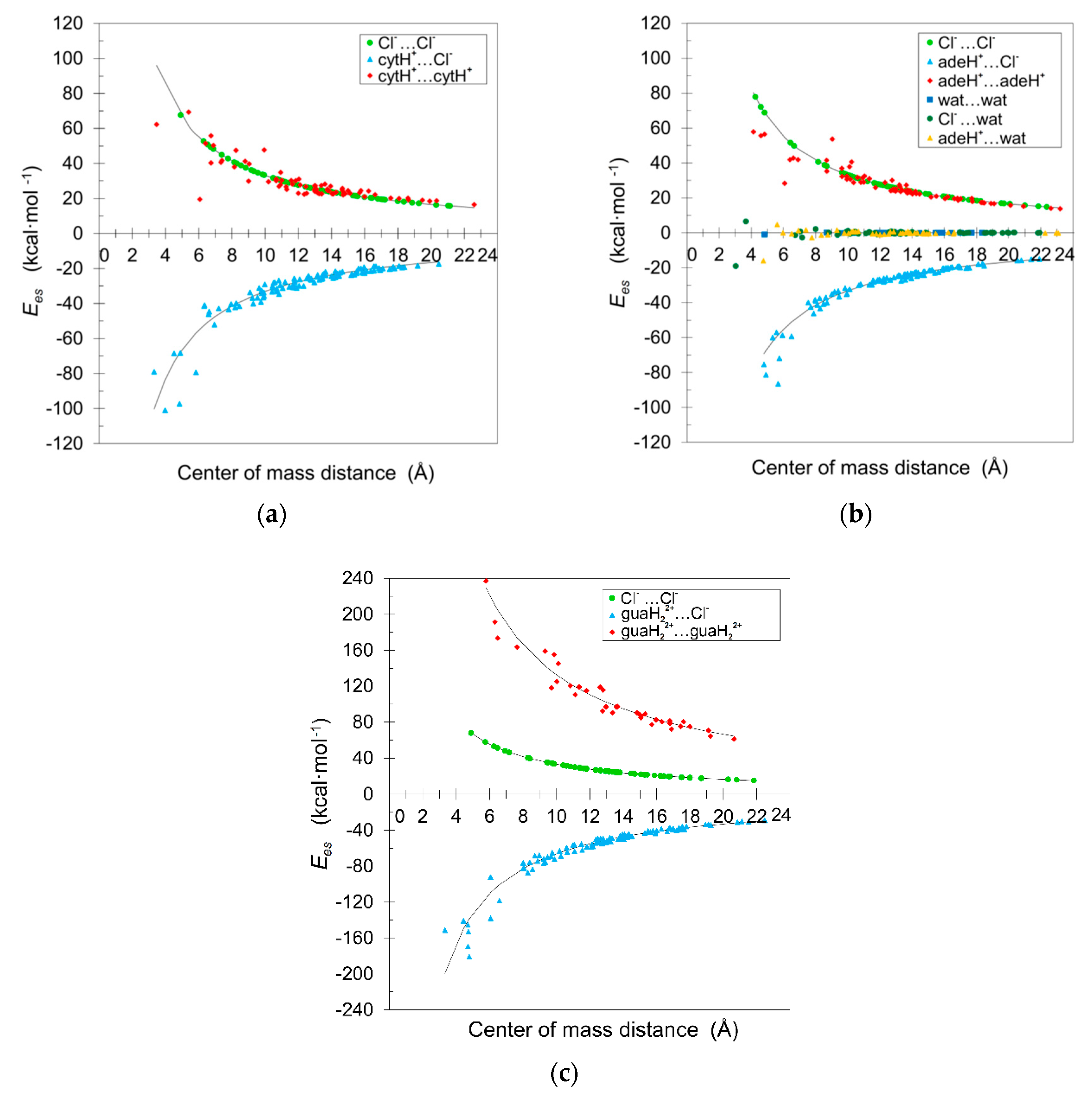
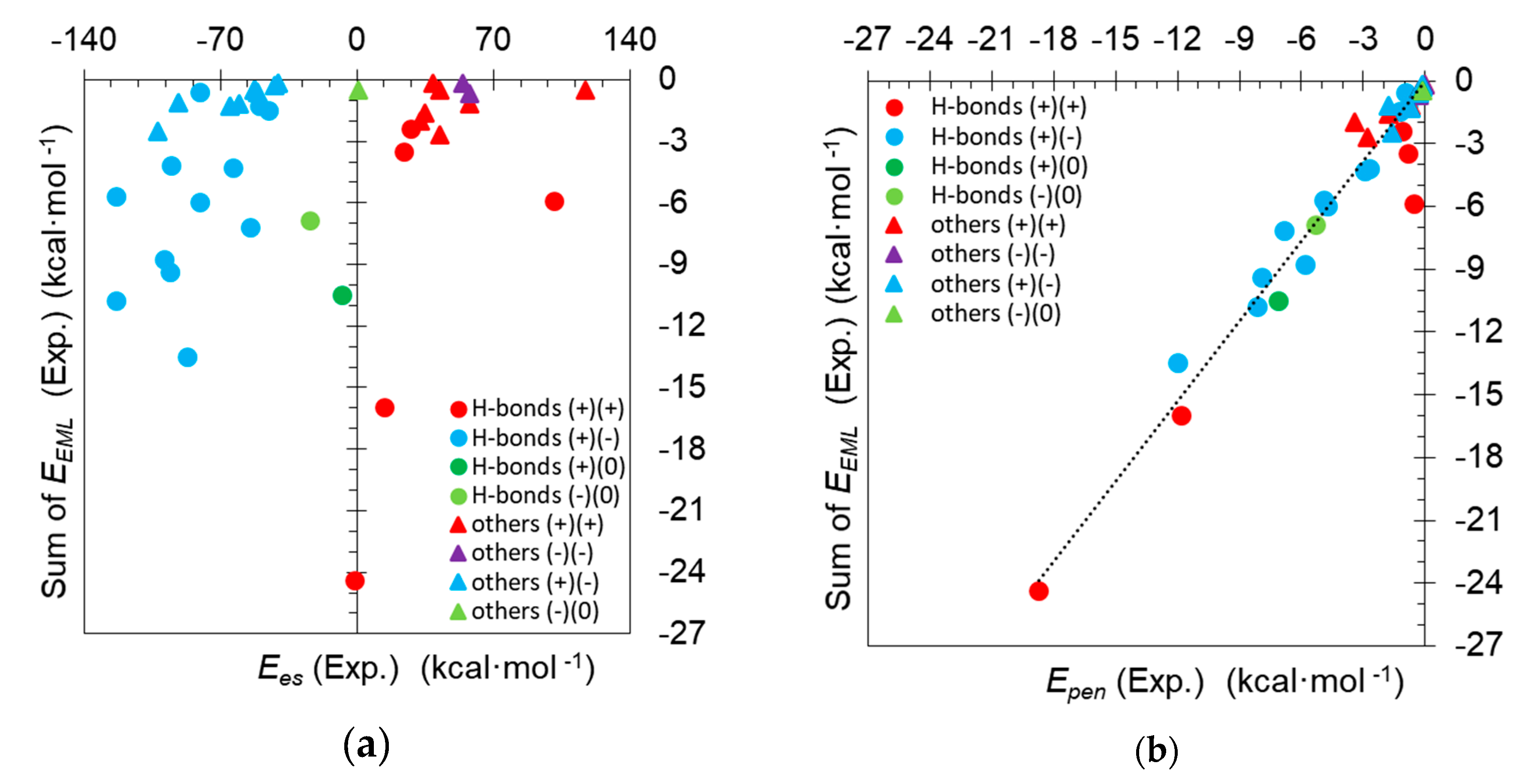
3. Results and Discussion
3.1. Topological Analysis of Electron Density
3.2. Interaction Energies for Dimers
3.3. A Missing Link between Topology and Energy Analyses
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Desiraju, G.R. Crystal engineering: From molecule to crystal. J. Am. Chem. Soc. 2013, 135, 9952–9967. [Google Scholar] [CrossRef] [PubMed]
- Desiraju, G.R. Supramolecular synthons in crystal engineering—A new organic synthesis. Angew. Chem. Int. Ed. Engl. 1995, 34, 23112327. [Google Scholar] [CrossRef]
- Dunitz, J.D.; Gavezzotti, A. Supramolecular synthons: Validation and ranking of intermolecular interaction energies. Cryst. Growth Des. 2012, 12, 5873–5877. [Google Scholar] [CrossRef]
- Dunitz, J.D.; Gavezzotti, A. How molecules stick together in organic crystals: Weak intermolecular interactions. Chem. Soc. Rev. 2009, 38, 2622–2633. [Google Scholar] [CrossRef] [PubMed]
- Dunitz, J.D. Intermolecular atom–atom bonds in crystals? IUCrJ 2015, 2, 157–158. [Google Scholar] [CrossRef] [PubMed]
- Lecomte, C.; Espinosa, E.; Matta, C.F. On atom–atom short contact bonding interactions in crystals. IUCrJ 2015, 2, 161–163. [Google Scholar] [CrossRef] [PubMed]
- Moggach, S.A.; Marshall, W.G.; Rogers, D.M.; Parsons, S. How focussing on hydrogen bonding interactions in amino acids can miss the bigger picture: A high-pressure neutron powder diffraction study of ε-glycine. Cryst. Eng. Comm. 2015, 17, 5315–5328. [Google Scholar] [CrossRef]
- Thakuer, T.S.; Dubey, R.; Desiraju, G.R. Intermolecular atom–atom bonds in crystals–a chemical perspective. IUCrJ 2015, 2, 159–160. [Google Scholar] [CrossRef]
- Gavezzotti, A. The lines-of-force landscape of interactions between molecules in crystals; cohesive versus tolerant and collateral damage contact. Acta Crystallogr. B 2010, 66, 396–406. [Google Scholar] [CrossRef]
- Gamrad, W.; Dreier, A.; Goddard, R.; Porschke, K.-R. Cation–Cation Pairing by N-C-H⋅⋅⋅O Hydrogen Bonds. Angew. Chem. Int. Ed. Engl. 2015, 54, 4482–4487. [Google Scholar] [CrossRef]
- Panini, P.; Venugopala, K.N.; Odhav, B.; Chopra, D. Polymorphism in two biologically active dihydropyrimidinium hydrochloride derivatives: Quantitative inputs towards the energetics associated with crystal packing. Acta Crystallogr. B 2014, 70, 681–696. [Google Scholar] [CrossRef] [PubMed]
- Dunitz, J.D.; Gavezzotti, A.; Rizzato, S. “Coulombic Compression”, a Pervasive Force in Ionic Solids. A Study of Anion Stacking in Croconate Salts. Cryst. Growth Des. 2014, 14, 357–366. [Google Scholar] [CrossRef]
- Mata, I.; Molins, E.; Alkorta, I.; Espinosa, E. The Paradox of Hydrogen-Bonded Anion–Anion Aggregates in Oxoanions: A Fundamental Electrostatic Problem Explained in Terms of Electrophilic···Nucleophilic Interactions. J. Phys. Chem. A 2015, 119, 183–194. [Google Scholar] [CrossRef] [PubMed]
- Mata, I.; Alkorta, I.; Molins, E.; Espinosa, E. Tracing environment effects that influence the stability of anion–anion complexes: The case of phosphate–phosphate interactions. Chem. Phys. Lett. 2013, 555, 106–109. [Google Scholar] [CrossRef]
- Mata, I.; Alkorta, I.; Molins, E.; Espinosa, E. Electrostatics at the Origin of the Stability of Phosphate-Phosphate Complexes Locked by Hydrogen Bonds. ChemPhysChem 2012, 13, 1421–1424. [Google Scholar] [CrossRef]
- Macchi, P.; Iversen, B.B.; Sironi, A.; Chakoumakos, B.C.; Larsen, F.K. Interanionic O–H⋅⋅⋅O Interactions: The Charge Density Point of View. Angew. Chem. Int. Ed. Engl. 2000, 39, 2719–2722. [Google Scholar] [CrossRef]
- D’Oria, E.; Bragga, D.; Novoa, J.J. Protonated nucleobases are not fully ionized and may form stable base pairs in the crystalline state. Cryst. Eng. Comm. 2012, 14, 792–798. [Google Scholar]
- Contreras-Garcia, J.; Calatayud, M.; Piquemal, J.-P.; Recio, J.M. Ionic interactions: Comparative topological approach. Comput. Theor. Chem. 2012, 998, 193–201. [Google Scholar] [CrossRef]
- Alkorta, I.; Mata, I.; Molins, E.; Espinosa, E. Charged versus Neutral Hydrogen-Bonded Complexes: Is There a Difference in the Nature of the Hydrogen Bonds? Chem. Eur. J. 2016, 22, 9226–9234. [Google Scholar] [CrossRef]
- Braga, D.; Grepioni, F.; Novoa, J. Inter-anion O–H–···O– hydrogen bond like interactions: The breakdown of the strength–length analogy. J. Chem. Commun. 1998, 1959–1960. [Google Scholar] [CrossRef]
- Quiñonero, D.; Alkorta, I.; Elguero, J. Cation–cation and anion–anion complexes stabilized by halogen bonds. Phys. Chem. Chem. Phys. 2016, 18, 27939–27950. [Google Scholar] [CrossRef]
- Weinhold, F. Polyion Covalency: Exotic Species from the Unexplored World of Electrostatically Shielded Molecular Ion Chemistry. Angew. Chem. 2016, 129, 14769–14773. [Google Scholar] [CrossRef]
- Grimme, S.; Djukic, J.P. Cation–cation “attraction”: When London dispersion attraction wins over Coulomb repulsion. Inorg. Chem. 2011, 50, 2619–2628. [Google Scholar] [CrossRef] [PubMed]
- Shokri, A.; Ramezani, M.; Fattahi, A.; Kass, S.R. Electrostatically Defying Cation–Cation Clusters: Can Likes Attract in a Low-Polarity Environment? J. Phys. Chem. A 2013, 117, 9252–9258. [Google Scholar] [CrossRef] [PubMed]
- Braga, D.; Bazzi, C.; Grepioni, F.; Novoa, J. Electrostatic compression on non-covalent interactions: The case of π stacks involving ions. J. New J. Chem. 1999, 23, 577–579. [Google Scholar] [CrossRef]
- Steiner, T. Hydrogen-bond distances to halide ions in organic and organometallic crystal structures: Up-to-date database study. Acta Crystallogr. B 1998, 54, 456–463. [Google Scholar] [CrossRef]
- Braga, D.; Maini, L.; Grepioni, F. Croconic acid and alkali metal croconate salts: Some new insights into an old story. Chem. Eur. J. 2002, 8, 1804–1812. [Google Scholar] [CrossRef]
- Rychkov, D.; Arkhipov, S.; Boldyreva, E. Structure-forming units of amino acid maleates. Case study of l-valinium hydrogen maleate. Acta Crystallogr. B 2016, 72, 160–163. [Google Scholar] [CrossRef]
- Gavezzotti, A. Pillars of crystal engineering: Crystal energies and symmetry operators. Cryst. Eng. Comm. 2018, 20, 2511–2518. [Google Scholar] [CrossRef]
- Kumar, P.; Cabaj, M.K.; Pazio, A.; Dominiak, P.M. Protonated nucleobases are not fully ionized in their chloride salt crystals and form metastable base pairs further stabilized by the surrounding anions. IUCRJ 2018, 5, 449–469. [Google Scholar] [CrossRef]
- Stewart, R.F.; Bentley, J.; Goodman, B. Generalized x-ray scattering factors in diatomic molecules. J. Chem. Phys. 1975, 63, 3786–3793. [Google Scholar] [CrossRef]
- Hansen, N.K.; Coppens, P. Testing aspherical atom refinements on small-molecule data sets. Acta Crystallogr. A 1978, A34, 909–921. [Google Scholar] [CrossRef]
- Volkov, A.; Li, X.; Koritsanszky, T.; Coppens, P. Ab Initio Quality Electrostatic Atomic and Molecular Properties Including Intermolecular Energies from a Transferable Theoretical Pseudoatom Databank. J. Phys. Chem. A 2004, 108, 4283–4300. [Google Scholar] [CrossRef]
- Jarzembska, K.N.; Dominiak, P.M. New version of the theoretical databank of transferable aspherical pseudoatoms, UBDB2011–towards nucleic acid modelling. Acta Crystallogr. A 2012, 68, 139–147. [Google Scholar] [CrossRef]
- Dovesi, R.; Orlando, R.; Erba, A.; Zicovich-Wilson, C.M.; Civalleri, B.; Casassa, S.; Maschio, L.; Ferrabone, M.; De La Pierre, M.; D’Arco, P.; et al. CRYSTAL14: A program for the ab initio investigation of crystalline solids. Int. J. Quantum Chem. 2014, 114, 1287–1317. [Google Scholar] [CrossRef]
- Dovesi, R.; Saunders, V.R.; Roetti, C.; Orlando, R.; Zicovich-Wilson, C.M.; Pascale, F.; Civalleri, B.; Doll, K.; Harrison, N.M.; Bush, I.J.; et al. CRYSTAL14 User’s Manual; University of Torino: Torino, Italy, 2014. [Google Scholar]
- Becke, A.D. Density-functional exchange-energy approximation with correct asymptotic behavior. Phys. Rev. A 1988, 38, 3098–3100. [Google Scholar] [CrossRef]
- Perdew, J.P. Density-functional approximation for the correlation energy of the inhomogeneous electron gas. Phys. Rev. B 1986, 33, 8822–8824. [Google Scholar] [CrossRef]
- Lee, C.; Yang, W.; Parr, R.G. Density-functional exchange-energy approximation with correct asymptotic behaviour. Phys. Rev. B 1988, 37, 785–789. [Google Scholar] [CrossRef]
- Dunning, J.T.H. Gaussian basis sets for use in correlated molecular calculations. I. The atoms boron through neon and hydrogen. J. Chem. Phys. 1989, 90, 1007–1023. [Google Scholar] [CrossRef]
- Volkov, A.; Macchi, P.; Farrugia, L.J.; Gatti, C.; Mallinson, P.; Richter, T.; Koritsanszky, T. XD2006—A Computer Program for Multipole Refinement, Topological Analysis of Charge Densities and Evaluation of Intermolecular Energies from Experimental or Theoretical Structure Factors; University at Buffalo—The State University of New York: New York, NY, USA, 2006. [Google Scholar]
- Gatti, C.; Saunders, V.R.; Roetti, C. Crystal field effects on the topological properties of the electron density in molecular crystals: The case of urea. J. Chem. Phys. 1994, 101, 10686–10696. [Google Scholar] [CrossRef]
- Guillot, B.; Viry, L.; Guillot, R.; Lecomte, C.; Jelsch, C. Refinement of proteins at subatomic resolution with MOPRO. J. Appl. Cryst. 2001, 34, 214–223. [Google Scholar] [CrossRef]
- Jelsch, C.; Guillot, B.; Lagoutte, A.; Lecomte, C. Advances in protein and small-molecule charge-density refinement methods using MoPro. J. Appl. Cryst. 2005, 38, 38–54. [Google Scholar] [CrossRef]
- Jansen, G.; Hesselmann, A. Comment on “Using Kohn− Sham orbitals in symmetry-adapted perturbation theory to investigate intermolecular interactions”. J. Phys. Chem. A 2001, 105, 11156–11157. [Google Scholar] [CrossRef]
- Williams, H.L.; Chabalowski, C.F. Using Kohn-Sham orbitals in symmetry-adapted perturbation theory to investigate intermolecular interactions. J. Phys. Chem. A 2001, 105, 646–659. [Google Scholar] [CrossRef]
- Jeziorski, B.; Moszynski, R.; Szalewicz, K. Perturbation theory approach to intermolecular potential energy surfaces of van der Waals complexes. Chem. Rev. 1994, 94, 1887–1930. [Google Scholar] [CrossRef]
- Werner, H.-J.; Knowles, P.J.; Knizia, G.; Manby, F.R.; Schütz, M. Molpro: A general-purpose quantum chemistry program package. WIREs Comput. Mol. Sci. 2012, 2, 242–253. [Google Scholar] [CrossRef]
- Heßelmann, A.; Jansen, G. First-order intermolecular interaction energies from Kohn-Sham orbitals. Chem. Phys. Lett. 2002, 357, 464–470. [Google Scholar]
- Gross, E.K.U.; Dobson, J.F.; Petersilka, M. Density Functional Theory of Time–Dependent Phenomena; Springer: Berlin/Heidelberg, Germany, 1996; pp. 81–172. [Google Scholar]
- Kendall, R.A.; Dunning, J.T.H.; Harrison, R.J. Electron affinities of the first-row atoms revisited. Systematic basis sets and wave functions. J. Chem. Phys. 1992, 96, 6796–6806. [Google Scholar] [CrossRef]
- Gavezzotti, A. Efficient computer modeling of organic materials. The atom–atom, Coulomb–London–Pauli (AA-CLP) model for intermolecular electrostatic-polarization, dispersion and repulsion energies. New J. Chem. 2011, 35, 1360–1368. [Google Scholar] [CrossRef]
- Hariharan, P.C.; Pople, J.A. The influence of polarization functions on molecular orbital hydrogenation energies. Theor. Chim. Acta 2003, 28, 213–222. [Google Scholar] [CrossRef]
- Gavezzotti, A. Towards a realistic model for the quantitative evaluation of intermolecular potentials and for the rationalization of organic crystal structures. Part I. Philosophy. Cryst. Eng. Comm. 2003, 5, 429–438. [Google Scholar] [CrossRef]
- Gavezzotti, A. Towards a realistic model for the quantitative evaluation ofintermolecular potentials and for the rationalization of organic crystal structures. Part II. Crystal energy landscapes. Cryst. Eng. Comm. 2003, 5, 439–446. [Google Scholar] [CrossRef]
- Gavezzotti, A. Calculation of Intermolecular Interaction Energies by Direct Numerical Integration over Electron Densities. 2. An Improved Polarization Model and the Evaluation of Dispersion and Repulsion Energies. J. Phys. Chem. B 2003, 107, 2344–2353. [Google Scholar] [CrossRef]
- Gavezzotti, A.Z. Calculation of lattice energies of organic crystals: The PIXEL integration method in comparison with more traditional methods. Kristallographie 2003, 220, 499–510. [Google Scholar] [CrossRef]
- Turner, M.J.; McKinnon, J.J.; Wolff, S.K.; Grimwood, D.J.; Spackman, P.R.; Jayatilaka, D.; Spackman, M.A. CrystalExplorer17. University of Western Australia, 2017. Available online: http://hirshfeldsurface.net (accessed on 10 December 2019).
- Jarzembska, K.N.; Kubsik, M.; Kaminski, R.; Wozniak, K.; Dominiak, P.M. From a single molecule to molecular crystal architectures: Structural and energetic studies of selected uracil derivatives. Cryst. Growth Des. 2012, 12, 2508–2524. [Google Scholar] [CrossRef]
- Jarzembska, K.N.; Kamiski, R.; Wenger, E.; Lecomte, C.; Dominiak, P.M. Interplay between charge density distribution, crystal structure energetic features, and crystal morphology of 6-methyl-2-thiouracil. J. Phys. Chem. C 2013, 117, 7764–7775. [Google Scholar] [CrossRef]
- Espinosa, E.; Lecomte, C.; Molins, E. Experimental electron density overlapping in hydrogen bonds: Topology vs. energetics. Chem. Phys. Lett. 1999, 300, 745–748. [Google Scholar] [CrossRef]
- Abramov, Y.A. On the possibility of kinetic energy density evaluation from the experimental electron-density distribution. Acta Crystallogr. A 1997, 53, 264–272. [Google Scholar] [CrossRef]
- Jarzembska, K.N.; Goral, A.M.; Gajda, R.; Dominiak, P.M. Hoogsteen–Watson–Crick 9-methyladenine: 1-methylthymine complex: Charge density study in the context of crystal engineering and nucleic acid base pairing. Cryst. Growth Des. 2013, 13, 239–254. [Google Scholar] [CrossRef]
- Spackman, M.A. How reliable are intermolecular interaction energies estimated from topological analysis of experimental electron densities? Cryst. Growth Des. 2015, 15, 5624–5628. [Google Scholar] [CrossRef]
- Bojarowski, S.A.; Kumar, P.; Dominiak, P.M. Interplay of point multipole moments and charge penetration for intermolecular electrostatic interaction energies from the University at Buffalo pseudoatom databank model of electron density. Acta Cryst. B 2017, 73, 598–609. [Google Scholar] [CrossRef] [PubMed]
- Bader, R.F.W. Bond paths are not chemical bonds. J. Phys. Chem. A 2009, 113, 10391–10396. [Google Scholar] [CrossRef] [PubMed]
- Bader, R.F.W. Definition of molecular structure: By choice or by appeal to observation? J. Phys. Chem. A 2010, 114, 7431–7444. [Google Scholar] [CrossRef] [PubMed]
- Gatti, C.; Forni, A. Revealing the Intermolecular Bonds in Molecular Crystals Through Charge Density Methods. Intermolecular Interactions in Crystals; Novoa, J.J., Ed.; The Royal Society of Chemistry: London, UK, 2018. [Google Scholar]
- Pendas, A.M.; Francosco, E.; Blanco, M.A.; Gatti, C. Bond paths as privileged exchange channels. Chem. Eur. J. 2007, 13, 9362–9371. [Google Scholar] [CrossRef] [PubMed]
- Wood, P.A.; Francis, D.; Marshall, W.G.; Moggach, S.A.; Parson, S.; Pidcock, E.; Rohl, A.L. A study of the high-pressure polymorphs of L-serine using ab initio structures and PIXEL calculations. Cryst. Eng. Comm. 2008, 10, 1154–1166. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Dimer | No. BCP | Interacting Atoms | Interaction Type | Rab | d1BPL | d2BPL | ρ(r)BCP | ∇2ρ(r)BCP | ε | EEML |
|---|---|---|---|---|---|---|---|---|---|---|
| Cytosinium chloride (CC) | ||||||||||
| AA1 | CP1 | H1...O2 | HB | 1.776 | 0.640 | 1.136 | 0.307 | 1.64 | 0.07 | −12.2 |
| AA2 | CP2 | C4...C2 | π...π | 3.312 | 1.668 | 1.752 | 0.047 | 0.43 | 0.72 | −0.9 |
| AA2 | CP3 | N3...N3 | π...π | 3.312 | 1.656 | 1.656 | 0.039 | 0.52 | 14.39 | −0.9 |
| AA3 | CP4 | H6...O2 | HB | 2.327 | 0.955 | 1.392 | 0.082 | 1.11 | 0.36 | −2.4 |
| AA4 | CP5 | N7...C5 | π...π | 3.433 | 1.688 | 1.797 | 0.029 | 0.39 | 0.65 | −0.6 |
| AA5 | CP6 | N7...N1 | π...π | 3.765 | 1.893 | 1.905 | 0.011 | 0.17 | 1.54 | −0.2 |
| AA6 | CP7 | H6...C5 | CH... π | 3.007 | 1.285 | 1.802 | 0.030 | 0.28 | 0.20 | −0.5 |
| AB1 | CP8 | H7B...Cl1 | HB | 2.362 | 0.867 | 1.508 | 0.138 | 0.86 | 0.05 | −3.7 |
| AB1 | CP9 | H5...Cl1 | HB | 2.545 | 0.924 | 1.633 | 0.084 | 1.01 | 0.29 | −2.3 |
| AB2 | CP10 | N7...Cl1 | NH2...Cl- | 3.308 | 1.575 | 1.738 | 0.051 | 0.74 | 0.63 | −1.3 |
| AB3 | CP11 | C2...Cl1 | π...Cl− | 3.340 | 1.578 | 1.767 | 0.053 | 0.58 | 0.32 | −1.2 |
| AB4 | CP12 | H3...Cl1 | HB | 2.022 | 0.690 | 1.337 | 0.304 | 1.46 | 0.05 | −11.9 |
| AB4 | CP13 | H7A...Cl1 | HB | 2.619 | 0.973 | 1.699 | 0.063 | 0.80 | 0.58 | −1.6 |
| AB5 | CP14 | C6...Cl1 | π...Cl− | 3.729 | 1.932 | 2.003 | 0.024 | 0.36 | 0.68 | −0.5 |
| AB6 | CP15 | C6...Cl1 | π...Cl− | 3.691 | 1.840 | 1.946 | 0.024 | 0.29 | 0.40 | −0.5 |
| Adeninium chloride hemihydrate (ACH) | ||||||||||
| AA1 | CP1 | H10A...N7 | HB | 1.937 | 0.708 | 1.235 | 0.225 | 1.67 | 0.03 | −8.0 |
| AA2 | CP2 | H2...N3 | HB | 2.734 | 1.358 | 1.562 | 0.048 | 0.72 | 0.20 | −1.2 |
| AA2 | CP3 | N3...N3 | 3.150 | 1.585 | 1.585 | 0.040 | 0.67 | 0.53 | −1.1 | |
| AA3 | − | |||||||||
| AA4 | CP4 | N3...N7 | π...π | 3.217 | 1.609 | 1.619 | 0.043 | 0.56 | 0.81 | −1.0 |
| AA4 | CP5 | C2...N10 | π...π | 3.438 | 1.767 | 1.917 | 0.030 | 0.37 | 3.05 | −0.6 |
| AA5 | CP6 | N7…N10 | π...π | 3.340 | 1.631 | 1.980 | 0.045 | 0.52 | 1.15 | −1.0 |
| AB1 | CP7 | H10B...Cl1 | HB | 2.255 | 0.785 | 1.480 | 0.144 | 1.26 | 0.06 | −4.3 |
| AB2 | CP8 | H8...Cl1 | HB | 2.606 | 0.936 | 1.683 | 0.066 | 0.65 | 0.15 | −1.5 |
| AB3 | CP9 | H9...Cl1 | HB | 2.109 | 0.738 | 1.378 | 0.221 | 1.04 | 0.00 | −7.2 |
| AB4 | CP10 | H2...Cl1 | HB | 2.699 | 0.999 | 1.706 | 0.054 | 0.65 | 0.04 | −1.3 |
| AB5 | CP11 | N1...Cl1 | π...Cl− | 3.569 | 1.724 | 1.866 | 0.027 | 0.38 | 0.67 | −0.6 |
| AB6 | CP12 | C8...Cl1 | π...Cl− | 3.950 | 1.952 | 2.083 | 0.012 | 0.15 | 0.42 | −0.2 |
| AB7 | CP13 | N9...Cl1 | π...Cl− | 3.826 | 1.812 | 2.035 | 0.014 | 0.22 | 3.09 | −0,3 |
| AW1 | CP14 | H1...O1 | HB | 1.834 | 0.678 | 1.159 | 0.268 | 2.01 | 0.02 | −10.5 |
| BW1 | CP15 | H1A...Cl1 | HB | 2.118 | 0.704 | 1.418 | 0.217 | 0.96 | 0.02 | −6.9 |
| BW2 | CP16 | O1...Cl1 | 3.608 | 1.680 | 1.932 | 0.021 | 0.32 | 0.67 | −0.5 | |
| BB1 | CP17 | Cl1...Cl1 | Cl−...Cl− | 4.262 | 2.132 | 2.132 | 0.010 | 0.14 | 0.09 | −0.2 |
| Guaninium dichloride (GDC) | ||||||||||
| AA1 | CP1 | H8...O10 | HB | 2.246 | 1.179 | 1.304 | 0.083 | 1.69 | 0.08 | −3.1 |
| AA1 | CP2 | N9...O10 | 2.759 | 1.725 | 1.324 | 0.080 | 1.52 | 0.65 | −2.8 | |
| AA2 | CP3 | O10...N11 | π...π | 3.418 | 1.731 | 1.737 | 0.023 | 0.33 | 0.72 | −0.5 |
| AB1 | CP4 | H11B...Cl1 | HB | 2.084 | 0.676 | 1.413 | 0.193 | 1.54 | 0.07 | −6.5 |
| AB1 | CP5 | H3...Cl1 | HB | 2.274 | 0.821 | 1.467 | 0.151 | 0.98 | 0.03 | −4.3 |
| AB2 | CP6 | H1...Cl1 | HB | 2.036 | 0.664 | 1.373 | 0.231 | 2.13 | 0.02 | −8.8 |
| AB3 | CP7 | H8...Cl1 | HB | 2.826 | 1.083 | 1.758 | 0.047 | 0.27 | 0.05 | −0.6 |
| AB4 | CP8 | H3...Cl2 | HB | 2.996 | 1.246 | 1.851 | 0.030 | 0.35 | 1.27 | −0.6 |
| AB4 | CP9 | H9...Cl2 | HB | 2.204 | 0.765 | 1.445 | 0.162 | 1.37 | 0.02 | −5.1 |
| AB5 | CP10 | H7...Cl2 | HB | 2.076 | 0.712 | 1.367 | 0.262 | 1.27 | 0.01 | −9.4 |
| AB6 | CP11 | H11A...Cl2 | HB | 2.274 | 0.805 | 1.478 | 0.145 | 1.11 | 0.04 | −4.2 |
| AB7 | CP12 | C8...Cl1 | π...Cl− | 3.255 | 1.517 | 1.747 | 0.049 | 0.60 | 0.75 | −1.1 |
| AB8 | CP13 | C2...Cl2 | π...Cl− | 3.424 | 1.643 | 1.826 | 0.038 | 0.47 | 2.23 | −0.8 |
| AB8 | CP14 | C4...Cl2 | π...Cl− | 3.546 | 1.951 | 1.833 | 0.036 | 0.43 | 3.26 | −0.8 |
| AB8 | CP15 | N1...Cl2 | π...Cl− | 3.483 | 1.684 | 1.815 | 0.037 | 0.51 | 4.03 | −0.9 |
| BB1 | CP16 | Cl1...Cl2 | Cl−...Cl− | 3.692 | 1.846 | 1.848 | 0.031 | 0.40 | 0.73 | −0.7 |
| Dimer | Centre of Mass Distance (Å) | Experimental | UBDB | DFT–SAPT | |||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Cytosinium Chloride (CC) | |||||||||||
| AA1 | 6.060 | −0.8 | 17.9 | 18.2 | 42.3 | 14.0 | 24.6 | 30.7 | 54.8 | 16.6 | 20.0 |
| AA2 | 3.455 | 42.8 | 45.6 | 46.5 | 74.1 | 60.8 | 63.9 | 108.0 | 96.1 | 61.9 | 55.4 |
| AA3 | 6.739 | 27.8 | 28.9 | 29.0 | 38.0 | 40.0 | 41.0 | 40.9 | 49.3 | 39.8 | 38.1 |
| AA4 | 5.385 | 57.7 | 58.4 | 55.5 | 47.5 | 68.5 | 69.3 | 65.6 | 61.6 | 68.5 | 64.3 |
| AA5 | 6.877 | 38.9 | 38.9 | 38.8 | 37.2 | 50.3 | 50.4 | 50.5 | 48.3 | 50.0 | 46.2 |
| AA6 | 6.740 | 42.7 | 43.0 | 43.1 | 38.0 | 57.6 | 57.8 | 58.1 | 49.3 | 56.2 | 52.2 |
| AB1 | 4.837 | −80.2 | −75.5 | −75.5 | −52.9 | −95.2 | −91.1 | −90.7 | −68.6 | −96.8 | −101.1 |
| AB2 | 5.828 | −65.3 | −64.6 | −64.2 | −43.9 | −76.6 | −75.9 | −75.6 | −57.0 | −79.7 | −84.5 |
| AB3 | 3.309 | −60.1 | −58.3 | −67.4 | −77.4 | −79.1 | −76.8 | −90.8 | −100.4 | −82.8 | −85.3 |
| AB4 | 3.964 | −86.7 | −74.7 | −77.0 | −64.6 | −107.7 | −100.3 | −102.0 | −83.8 | −106.2 | −108.9 |
| AB5 | 4.890 | −52.1 | −51.9 | −51.6 | −52.4 | −70.0 | −69.7 | −69.7 | −67.9 | −69.7 | −74.9 |
| AB6 | 4.513 | −52.8 | −52.5 | −52.2 | −56.7 | −70.4 | −70.1 | −69.4 | −73.6 | −70.2 | −75.3 |
| BB1 | 3.991 | 63.8 | 63.8 | 63.8 | 64.2 | 83.1 | 83.2 | 83.2 | 83.2 | 80.9 | 75.4 |
| Adeninium chloride hemihydrate (ACH) | |||||||||||
| AA1 | 6.076 | 14.4 | 26.2 | 30.4 | 34.4 | 37.0 | 44.9 | 50.9 | 54.6 | 29.5 | 33.2 |
| AA2 | 6.909 | 24.5 | 25.3 | 25.2 | 30.3 | 40.3 | 40.9 | 42.0 | 48.1 | 41.4 | 40.1 |
| AA3 | 4.596 | 33.3 | 35.7 | 15.5 | 45.5 | 52.6 | 55.2 | 42.6 | 72.3 | 55.2 | 50.7 |
| AA4 | 4.819 | 35.1 | 36.9 | 29.6 | 43.4 | 55.8 | 57.6 | 54.0 | 68.9 | 55.7 | 51.4 |
| AA5 | 4.134 | 32.8 | 36.2 | 71.3 | 50.6 | 57.6 | 60.8 | 83.0 | 80.3 | 56.3 | 51.0 |
| AB1 | 5.666 | −63.1 | −60.2 | −58.0 | −38.7 | −88.1 | −84.9 | −82.8 | −58.6 | −86.2 | −91.2 |
| AB2 | 5.927 | −45.3 | −44.1 | −43.1 | −37.0 | −58.7 | −57.1 | −56.3 | −56.0 | −61.0 | −65.9 |
| AB3 | 4.917 | −54.3 | −47.5 | −47.5 | −44.6 | −82.5 | −77.4 | −75.5 | −67.5 | −81.1 | −84.7 |
| AB4 | 5.749 | −49.8 | −49.0 | −48.5 | −38.1 | −69.0 | −67.6 | −67.7 | −57.8 | −72.7 | −78.8 |
| AB5 | 4.787 | −50.7 | −50.4 | −48.9 | −45.8 | −76.5 | −76.1 | −75.0 | −69.4 | −76.7 | −82.4 |
| AB6 | 5.561 | −40.4 | −40.3 | −41.1 | −39.4 | −57.5 | −57.4 | −57.9 | −59.7 | −57.7 | −62.3 |
| AB7 | 5.328 | −42.2 | −42.1 | −43.0 | −41.2 | −60.5 | −60.3 | −61.0 | −62.3 | −59.4 | −63.7 |
| AW1 | 4.752 | −7.7 | −0.6 | −1.2 | 4.2 | −16.9 | −12.2 | −10.7 | 0.0 | −14.8 | −11.0 |
| BW1 | 3.031 | −23.8 | −18.5 | −18.6 | −6.9 | −19.1 | −15.3 | −15.2 | 0.0 | −18.5 | −14.3 |
| BW2 | 3.659 | 0.6 | 0.7 | 0.7 | −5.7 | 8.1 | 8.2 | 8.2 | 0.0 | 5.7 | 2.4 |
| BB1 | 4.262 | 54.2 | 54.2 | 54.2 | 53.9 | 78.0 | 77.9 | 77.9 | 77.9 | 76.6 | 70.4 |
| Guaninium Dichloride (GDC) | |||||||||||
| AA1 | 7.637 | 101.2 | 101.7 | 102.6 | 115.0 | 159.0 | 160.8 | 161.8 | 173.9 | 162.1 | 154.0 |
| AA2 | 6.309 | 117.3 | 117.6 | 111.6 | 139.3 | 187.3 | 187.7 | 180.6 | 210.5 | 192.5 | 180.7 |
| AA3 | 5.781 | 165.4 | 165.5 | 164.3 | 152.0 | 245.0 | 245.2 | 242.0 | 229.8 | 239.2 | 223.2 |
| AB1 | 4.777 | −123.1 | −115.0 | −115.4 | −88.2 | −184.3 | −176.0 | −176.2 | −139.0 | −181.4 | −191.8 |
| AB2 | 4.731 | −98.5 | −92.7 | −92.7 | −89.0 | −153.3 | −146.9 | −147.1 | −140.4 | −153.6 | −164.0 |
| AB3 | 6.584 | −80.3 | −79.4 | −78.6 | −64.0 | −119.0 | −118.1 | −117.6 | −100.9 | −119.5 | −128.9 |
| AB4 | 4.702 | −123.0 | −118.1 | −116.0 | −97.3 | −177.9 | −171.1 | −170.4 | −141.2 | −169.9 | −188.4 |
| AB5 | 4.710 | −95.4 | −87.5 | −87.2 | −97.1 | −144.6 | −139.5 | −138.5 | −141.0 | −151.5 | −162.8 |
| AB6 | 6.062 | −95.3 | −92.6 | −91.1 | −75.5 | −136.3 | −133.7 | −130.8 | −109.6 | −138.5 | −148.6 |
| AB7 | 4.435 | −91.5 | −90.6 | −88.3 | −94.9 | −144.9 | −143.2 | −138.0 | −149.7 | −144.2 | −151.8 |
| AB8 | 3.306 | −102.1 | −100.5 | −111.9 | −137.5 | −151.5 | −149.1 | −160.4 | −199.7 | −156.0 | −162.5 |
| BB1 | 3.692 | 57.9 | 59.4 | 59.4 | 59.4 | 89.6 | 89.6 | 89.9 | 89.9 | 85.6 | 80.1 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kumar, P.; Cabaj, M.K.; Dominiak, P.M. Intermolecular Interactions in Ionic Crystals of Nucleobase Chlorides—Combining Topological Analysis of Electron Densities with Energies of Electrostatic Interactions. Crystals 2019, 9, 668. https://doi.org/10.3390/cryst9120668
Kumar P, Cabaj MK, Dominiak PM. Intermolecular Interactions in Ionic Crystals of Nucleobase Chlorides—Combining Topological Analysis of Electron Densities with Energies of Electrostatic Interactions. Crystals. 2019; 9(12):668. https://doi.org/10.3390/cryst9120668
Chicago/Turabian StyleKumar, Prashant, Małgorzata Katarzyna Cabaj, and Paulina Maria Dominiak. 2019. "Intermolecular Interactions in Ionic Crystals of Nucleobase Chlorides—Combining Topological Analysis of Electron Densities with Energies of Electrostatic Interactions" Crystals 9, no. 12: 668. https://doi.org/10.3390/cryst9120668
APA StyleKumar, P., Cabaj, M. K., & Dominiak, P. M. (2019). Intermolecular Interactions in Ionic Crystals of Nucleobase Chlorides—Combining Topological Analysis of Electron Densities with Energies of Electrostatic Interactions. Crystals, 9(12), 668. https://doi.org/10.3390/cryst9120668