Crystallographic Insights into Uranyl Sulfate Minerals Formation: Synthesis and Crystal Structures of Three Novel Cesium Uranyl Sulfates

 , , and
, , and 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Synthesis
2.2. Chemical Analysis
2.3. Single-Crystal X-Ray Diffraction Study
3. Results
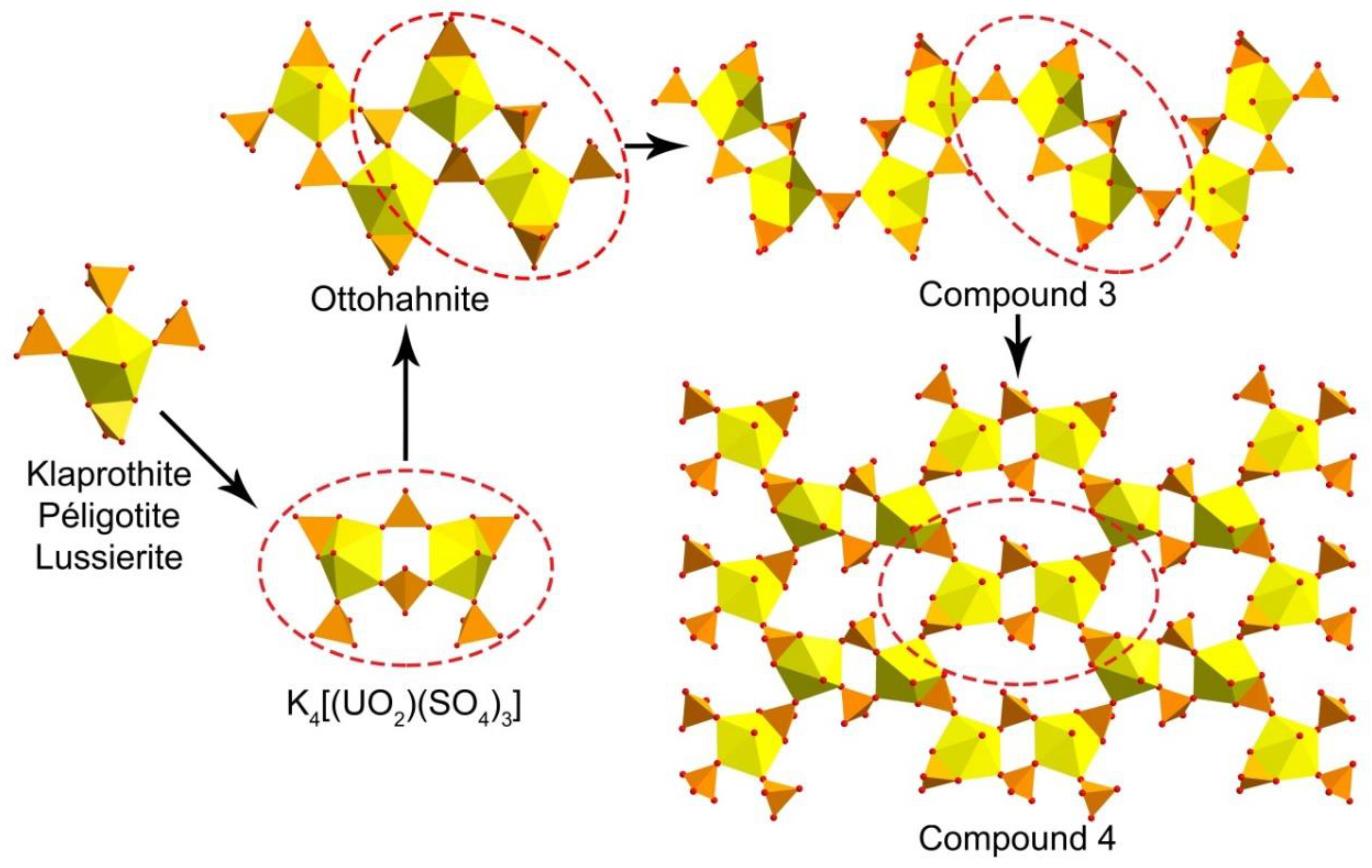
3.1. Structure Descriptions
3.2. Topological Analysis
3.3. Structural Complexity
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Finch, R.J.; Ewing, R.C. The corrosion of uraninite under oxidizing conditions. J. Nucl. Mater. 1992, 190, 133–156. [Google Scholar]
- Finch, R.J.; Murakami, T. Systematics and paragenesis of uranium minerals. In Uranium: Mineralogy, Geochemistry and the Environment; Burns, P.C., Ewing, R.C., Eds. Rev. Mineral. Geochem. 1999, 38, 91–179. [Google Scholar]
- Plášil, J. Oxidation-hydration weathering of uraninite: The current state-of-knowledge. J. Geosci. 2014, 59, 99–114. [Google Scholar]
- Krivovichev, S.V.; Plášil, J. Mineralogy and crystallography of uranium. In Uranium: From Cradle to Grave; Burns, P.C., Sigmon, G.E., Eds. MAC Short Courses 2013, 43, 15–119. [Google Scholar]
- Plášil, J. Uranyl-oxide hydroxy-hydrate minerals: Their structural complexity and evolution trends. Eur. J. Mineral. 2018, 30, 237–251. [Google Scholar]
- Walker, T.L. Schoepite, a new uranium mineral from Kasolo, Belgian Congo. Amer. Miner. 1923, 8, 67–69. [Google Scholar]
- Plášil, J. The crystal structure of uranyl-oxide mineral schoepite, [(UO2)4O(OH)6](H2O)6, revisited. J. Geosci. 2018, 63, 65–73. [Google Scholar]
- Serezhkina, L.B.; Grigor’ev, M.S.; Makarov, A.S.; Serezhkin, V.N. Synthesis and Structure of Cesium-Containing Zippeite. Radiochemistry 2015, 57, 20–25. [Google Scholar]
- Nipruk, O.V.; Knyazev, A.V.; Chernorukov, G.N.; Pykhova, Y.P. Synthesis and study of hydrated uranium(VI) oxides, UO3·nH2O. Radiochemistry 2011, 53, 146–150. [Google Scholar]
- CrysAlisPro Software System, version 1.171.38.46; Rigaku Oxford Diffraction: Oxford, UK, 2015.
- Sheldrick, G.M. SHELXT—Integrated space-group and crystal structure determination. Acta Crystallogr. 2015, A71, 3–8. [Google Scholar]
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Crystallogr. 2015, C71, 3–8. [Google Scholar]
- Dolomanov, O.V.; Bourhis, L.J.; Gildea, R.J.; Howard, J.A.K.; Puschmann, H. OLEX2: A complete structure solution, refinement and analysis program. J. Appl. Cryst. 2009, 42, 339–341. [Google Scholar]
- Burns, P.C.; Miller, M.L.; Ewing, R.C. U6+ minerals and inorganic phases: A comparison and hierarchy of structures. Can. Mineral. 1996, 34, 845–880. [Google Scholar]
- Krivovichev, S.V. Structural Crystallography of Inorganic Oxysalts; Oxford University Press: Oxford, UK, 2008. [Google Scholar]
- Grechishnikova, E.V.; Virovets, A.V.; Peresypkina, E.V.; Serezhkina, L.B. Synthesis and crystal structure of the (C2N4H7O)[UO2(SO4)(OH)]·0.5H2O. Zh. Neorg. Khim. (Russ.) 2005, 50, 1800–1805. [Google Scholar]
- Nazarchuk, E.V.; Charkin, D.O.; Siidra, O.I.; Gurzhiy, V.V. Synthesis and Crystal Structures of New Layered Uranyl Compounds Containing Dimers [(UO2)2O8] of Edge-Linked Pentagonal Bipyramids. Radiochemistry 2018, 60, 498–506. [Google Scholar]
- Unruh, D.K.; Baranay, M.; Pressprich, L.; Stoffer, M.; Burns, P.C. Synthesis and characterization of uranyl chromate sheet compounds containing edge-sharing dimers of uranyl pentagonal bipyramids. J. Solid State Chem. 2012, 186, 158–164. [Google Scholar]
- Ok, K.M.; Baek, J.; Halasyamani, P.S.; O’Hare, D. New Layered Uranium Phosphate Fluorides: Syntheses, Structures, Characterizations, and Ion-Exchange Properties of A(UO2)F(HPO4)·xH2O (A = Cs+, Rb+, K+; x = 0−1). Inorg. Chem. 2006, 45, 10207–10214. [Google Scholar]
- Plášil, J.; Hauser, J.; Petříček, V.; Meisser, N.; Mills, S.J.; Škoda, R.; Fejfarová, K.; Čejka, J.; Sejkora, J.; Hloušek, J.; et al. Crystal structure and formula revision of deliensite, Fe[(UO2)2(SO4)2(OH)2](H2O)7. Mineral. Mag. 2012, 76, 2837–2860. [Google Scholar]
- Kampf, A.R.; Kasatkin, A.V.; Čejka, J.; Marty, J. Plášilite, Na(UO2)(SO4)(OH)·2H2O, a new uranyl sulfate mineral from the Blue Lizard mine, San Juan County, Utah, USA. J. Geosci. 2015, 60, 1–10. [Google Scholar]
- Gurzhiy, V.V.; Plášil, J. Structural complexity of natural uranyl sulfates. Acta Crystallogr. 2019, B75, 39–48. [Google Scholar]
- Demartin, F.; Diella, V.; Donzelli, S.; Gramaccioli, C.M.; Pilati, T. The importance of accurate crystal structure determination of uranium minerals. I. Phosphuranylite KCa(H3O)3(UO2)7(PO4)4O4·8H2O. Acta Crystallogr. 1991, B47, 439–446. [Google Scholar]
- Krivovichev, S.V. Combinatorial topology of salts of inorganic oxoacids: Zero-, one- and two-dimensional units with corner-sharing between coordination polyhedra. Crystallogr. Rev. 2004, 10, 185–232. [Google Scholar]
- Fedoseev, A.M.; Budantseva, N.A.; Grigor’ev, M.S.; Bessonov, A.A.; Astafurova, L.N.; Lapitskaya, T.S.; Krupa, J.C. Sulfate Compounds of Hexavalent Neptunium and Plutonium. Radiochim. Acta 1999, 86, 17–22. [Google Scholar]
- Almond, P.M.; Peper, S.; Bakker, E.; Albrecht-Schmitt, T.E. Variable Dimensionality and New Uranium Oxide Topologies in the Alkaline-Earth Metal Uranyl Selenites AE[(UO2)(SeO3)2] (AE=Ca, Ba) and Sr[(UO2)(SeO3)2] · 2H2O. J. Solid State Chem. 2002, 168, 358–366. [Google Scholar]
- Norquist, A.J.; Doran, M.B.; O’Hare, D. The effects of linear diamine chain length in uranium sulfates. Solid State Sci. 2003, 5, 1149–1158. [Google Scholar]
- Krivovichev, S.V. Topological complexity of crystal structures: Quantitative approach. Acta Crystallogr. A 2012, 68, 393–398. [Google Scholar]
- Krivovichev, S.V. Structural complexity and configurational entropy of crystalline solids. Acta Crystallogr. B 2016, 72, 274–276. [Google Scholar]
- Krivovichev, S.V. Ladders of information: What contributes to the structural complexity in inorganic crystals. Z. Kristallogr. 2018, 233, 155–161. [Google Scholar]
- Krivovichev, V.G.; Krivovichev, S.V.; Charykova, M.V. Selenium Minerals: Structural and Chemical Diversity and Complexity. Minerals 2019, 9, 455. [Google Scholar]
- Gurzhiy, V.V.; Kuporev, I.V.; Kovrugin, V.M.; Murashko, M.N.; Kasatkin, A.V.; Plášil, J. Crystal Chemistry and Structural Complexity of Natural and Synthetic Uranyl Selenites. Crystals 2019, 9, 639. [Google Scholar]
- Gurzhiy, V.V.; Tyumentseva, O.S.; Krivovichev, S.V.; Krivovichev, V.G.; Tananaev, I.G. Mixed uranyl sulfate-selenates: variable composition and crystal structures. Cryst. Growth Des. 2016, 16, 4482–4492. [Google Scholar]
- Gurzhiy, V.V.; Krivovichev, S.V.; Tananaev, I.G. Dehydration-driven evolution of topological complexity in ethylamonium uranyl selenates. J. Solid State Chem. 2017, 247, 105–112. [Google Scholar]
- Gurzhiy, V.V.; Tyumentseva, O.S.; Britvin, S.N.; Krivovichev, S.V.; Tananaev, I.G. Ring opening of azetidine cycle: First examples of 1-azetidinepropanamine molecules as a template in hybrid organic-inorganic compounds. J. Molec. Struct. 2018, 1151, 88–96. [Google Scholar]
- Gurzhiy, V.V.; Tyumentseva, O.S.; Izatulina, A.R.; Krivovichev, S.V.; Tananaev, I.G. Chemically Induced Polytypic Phase Transitions in the Mg[(UO2)(TO4)2(H2O)](H2O)4 (T = S, Se) System. Inorg. Chem. 2019, 58, 14760–14768. [Google Scholar]
- Blatov, V.A.; Shevchenko, A.P.; Proserpio, D.M. Applied topological analysis of crystal structures with the program package ToposPro. Cryst. Growth Des. 2014, 14, 3576–3586. [Google Scholar]
- Kampf, A.R.; Plášil, J.; Kasatkin, A.V.; Marty, J.; Čejka, J. Klaprothite, péligotite and ottohahnite, three new minerals with bidentate UO7–SO4 linkages from the Blue Lizard mine, San Juan County, Utah, USA. Miner. Mag. 2017, 81, 753–779. [Google Scholar]
- Kampf, A.R.; Olds, T.A.; Plášil, J.; Nash, B.P.; Marty, J. Lussierite, a new sodium uranyl sulfate mineral with bidentate UO7–SO4 linkage from the Blue Lizard mine, San Juan County, Utah, USA. Miner. Mag. 2019, 1–25. [Google Scholar] [CrossRef]
- Plášil, J.; Meisser, N.; Čejka, J. The crystal structure of Na6[(UO2)(SO4)4](H2O)4: X-ray and Raman spectroscopy study. Canad. Miner. 2015, 54, 5–20. [Google Scholar]
- Burns, P.C.; Hayden, L.A. A uranyl sulfate cluster in Na10[(UO2)(SO4)4](SO4)2·3H2O. Acta Crystallogr. C 2002, 58, i121–i123. [Google Scholar]
- Hayden, L.A.; Burns, P.C. The sharing of an edge between a uranyl pentagonal bipyramid and sulfate tetrahedron in the structure of KNa5[(UO2)(SO4)4](H2O). Canad. Miner. 2002, 40, 211–216. [Google Scholar]
- Hayden, L.A.; Burns, P.C. A novel uranyl sulfate cluster in the structure of Na6(UO2)(SO4)4(H2O)2. J. Solid State Chem. 2002, 163, 313–318. [Google Scholar]
- Mikhailov, Y.N.; Kokh, L.A.; Kuznetsov, V.G.; Grevtseva, T.G.; Sokol, S.K.; Ellert, G.V. Synthesis and crystal structure of potassium trisulfatouranylate K4(UO2(SO4)3. Koord. Khimiya 1977, 3, 500–513. (In Russian) [Google Scholar]

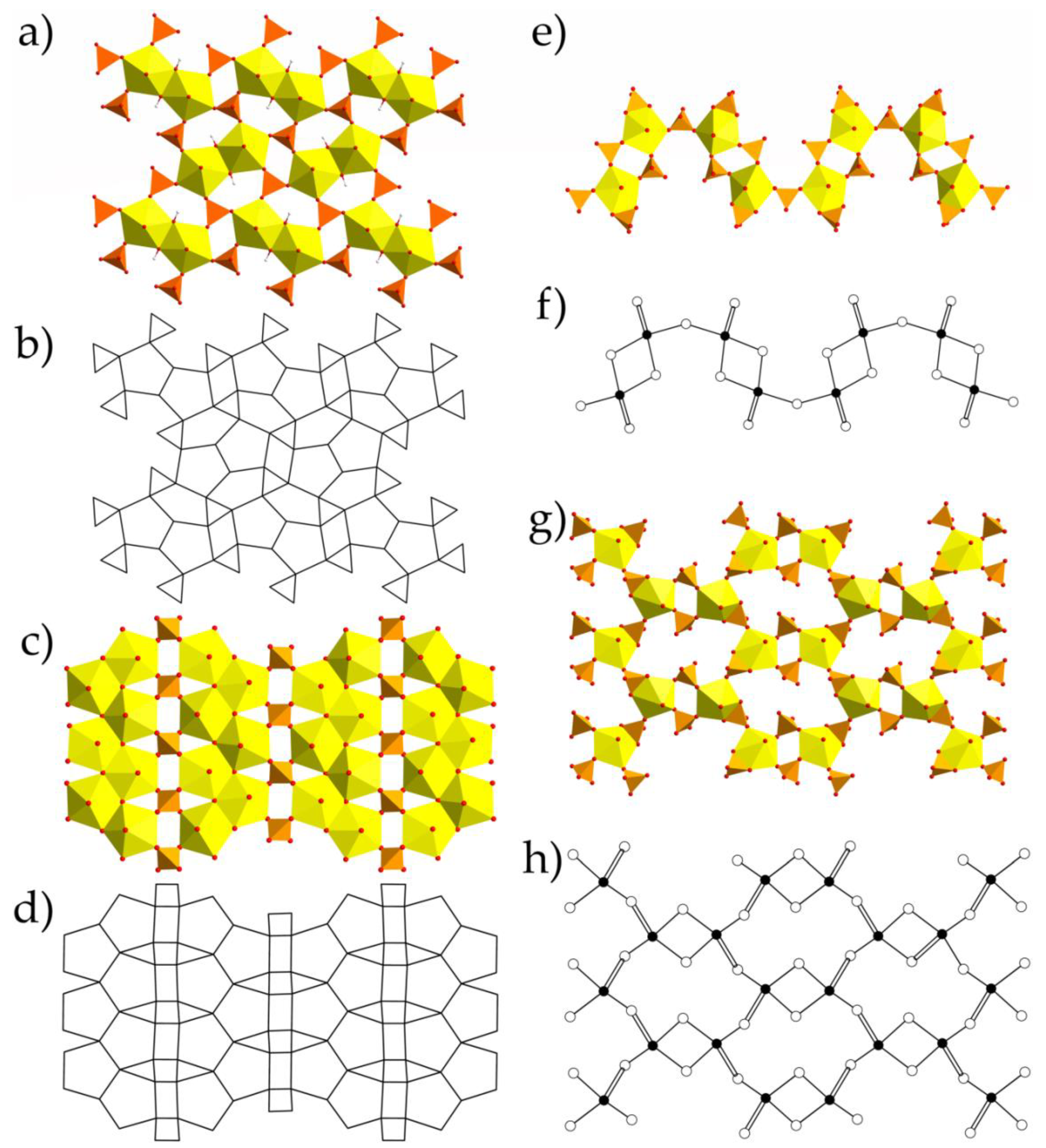


{kind=link}
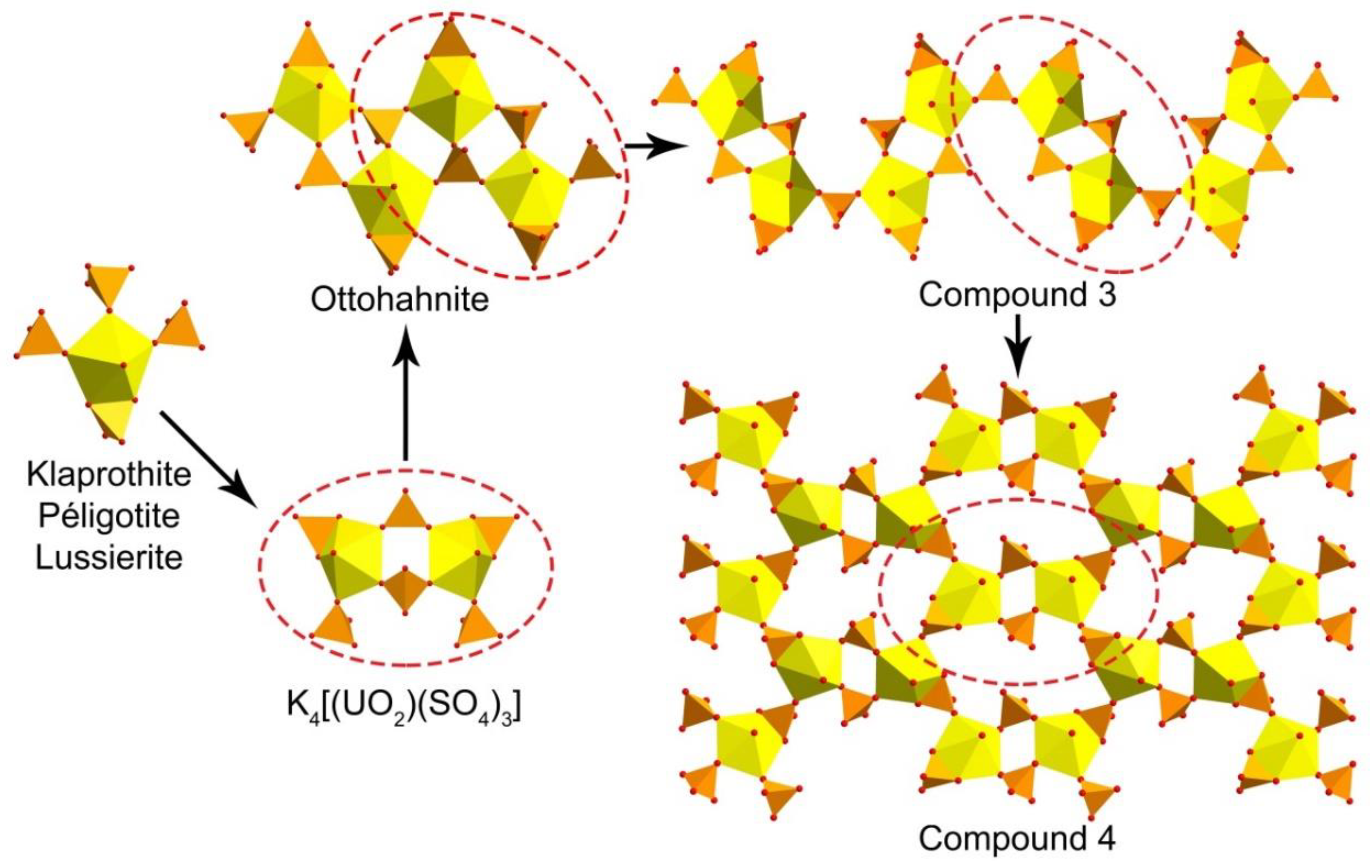
{kind=link}
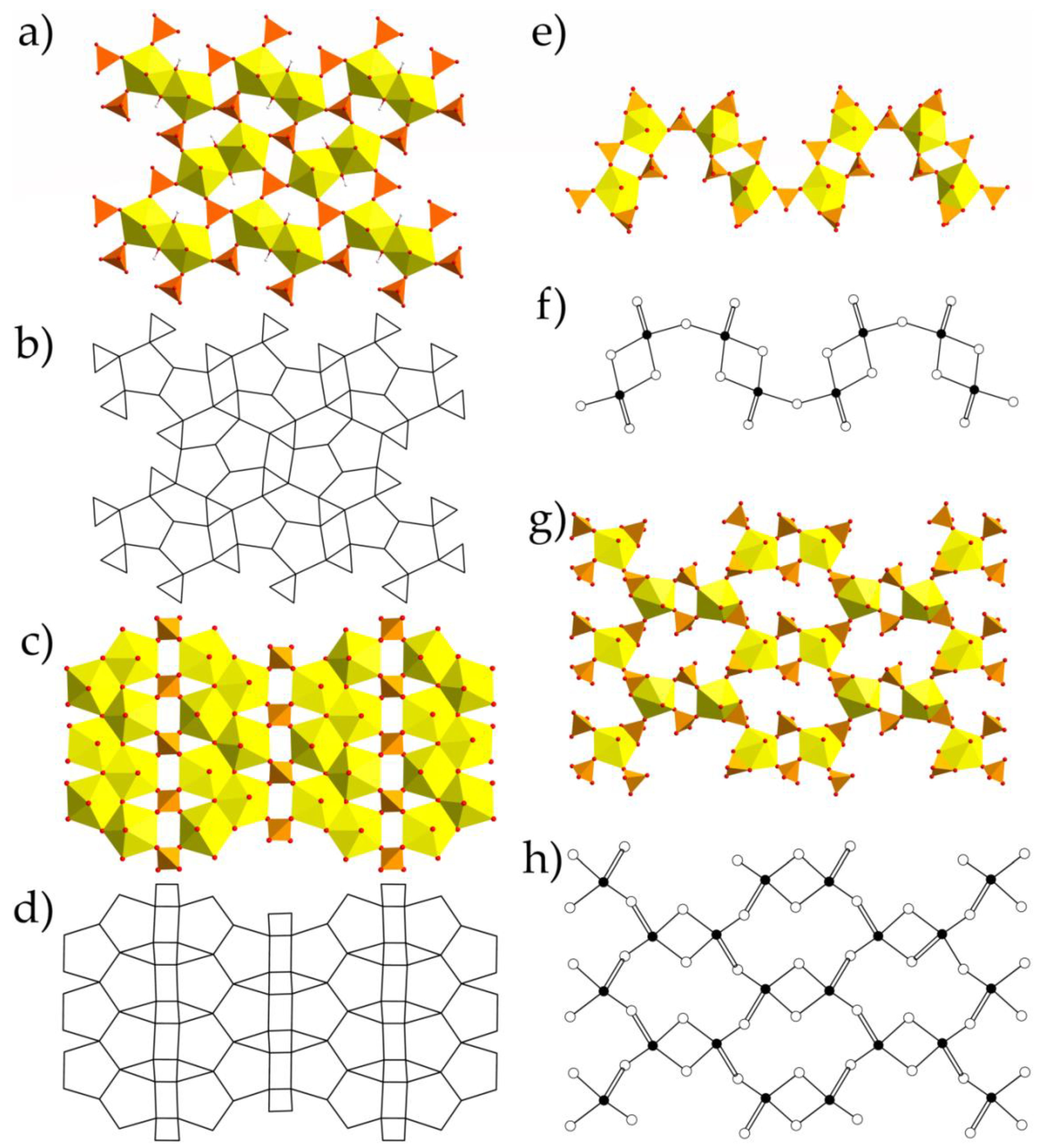
{kind=link}
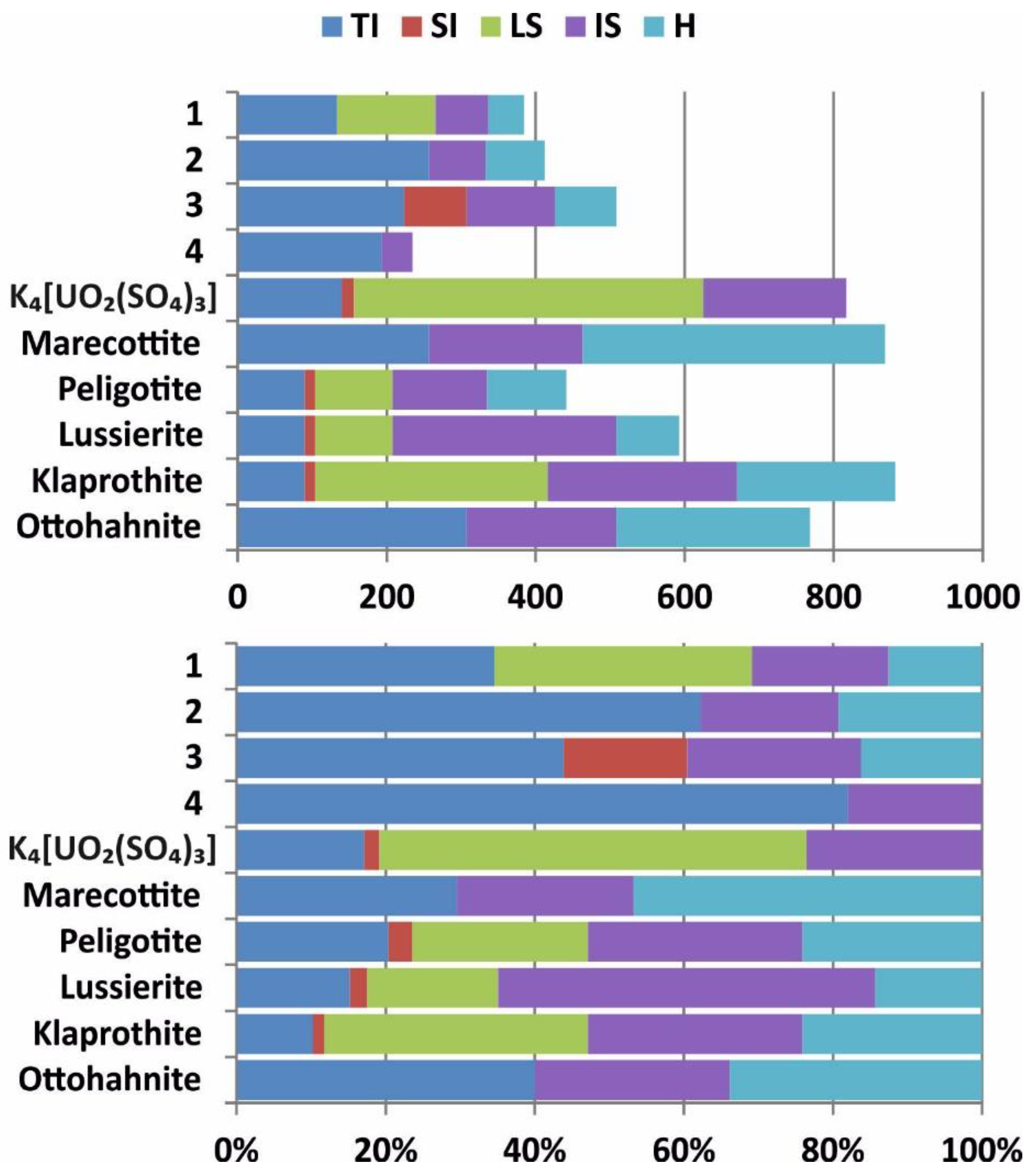
{kind=link}
| Compound | 1 | 3 | 4 |
|---|---|---|---|
| Crystal System | Orthorhombic | Triclinic | Monoclinic |
| a (Å) | 9.2021(3) | 7.5829(3) | 10.8351(5) |
| b (Å) | 13.2434(5) | 14.4441(11) | 9.0317(5) |
| c (Å) | 12.5610(3) | 14.6458(14) | 11.8494(6) |
| α (°) | 90 | 93.737(7) | 90 |
| β (°) | 90 | 99.535(5) | 110.7510(10) |
| γ (°) | 90 | 99.614(5) | 90 |
| V (Å3) | 1530.77(8) | 1552.6(2) | 1084.35(10) |
| Molecular weight | 520.51 | 1871.87 | 727.97 |
| Space group | Pnma | P–1 | P21/n |
| μ (mm−1) | 26.156 | 17.779 | 22.004 |
| Temperature (K) | 293(2) | ||
| Z | 8 | 2 | 4 |
| Dcalc (g/cm3) | 4.517 | 4.004 | 4.459 |
| Crystal size (mm3) | 0.06 × 0.04 × 0.02 | 0.04 × 0.03 × 0.005 | 0.08 × 0.05 × 0.02 |
| Diffractometer | Bruker Kappa Apex II Duo | ||
| Radiation | MoKα | ||
| Total reflections | 13617 | 8814 | 27773 |
| Unique reflections | 1824 | 5151 | 2493 |
| Angle range 2θ (°) | 4.47–55.00 | 3.84–50.00 | 4.38–55.00 |
| Reflections with |Fo| ≥ 4σF | 1560 | 3806 | 2308 |
| Rint | 0.0503 | 0.0784 | 0.0358 |
| Rσ | 0.0201 | 0.0949 | 0.0154 |
| R1 (|Fo| ≥ 4σF) | 0.0261 | 0.0829 | 0.0131 |
| wR2 (|Fo| ≥ 4σF) | 0.0607 | 0.2050 | 0.0278 |
| R1 (all data) | 0.0336 | 0.1077 | 0.0157 |
| wR2 (all data) | 0.0648 | 0.2215 | 0.0287 |
| S | 1.054 | 1.068 | 1.040 |
| ρmin, ρmax, e/Å3 | −1.097, 2.145 | −4.179, 3.819 | −0.604, 0.692 |
| CSD | 1965819 | 1965817 | 1965818 |
| Bond | Bond | ||
|---|---|---|---|
| U1–O1 | 1.768(5) | S1–O6 | 1.481(5) |
| U1–O2 | 1.768(5) | S1–O7 | 1.426(6) |
| < U1–OUr > | 1.768 | < S1–O > | 1.465 |
| U1–O3 | 2.351(4) | ||
| U1–OH4 | 2.330(5) | < Cs1–O > CN * = 8 | 3.313 |
| U1–OH4 | 2.322(5) | < Cs2–O > CN * = 10 | 3.302 |
| U1–O5 | 2.421(5) | ||
| U1–O6 | 2.358(5) | Angle | |
| < U1–Oeq > | 2.356 | U1–O4–U1 | 113.0(2) |
| S1–O5–U1 | 140.3(3) | ||
| S1–O3 | 1.478(4) | S1–O6–U1 | 140.3(3) |
| S1–O5 | 1.473(5) |
| Bond | Bond | ||
|---|---|---|---|
| U1–O1 | 1.770(19) | S3–O14 | 1.50(2) |
| U1–O2 | 1.80(2) | < S3–O > | 1.47 |
| < U1–OUr > | 1.785 | ||
| U1–O3 | 2.336(19) | S4–O17 | 1.51(2) |
| U1–O4 | 2.40(2) | S4–O18 | 1.48(2) |
| U1–O5 | 2.428(17) | S4–O19 | 1.43(2) |
| U1–O6 | 2.347(19) | S4–O20 | 1.42(3) |
| U1–O7 | 2.35(2) | < S4–O > | 1.46 |
| < U1–Oeq > | 2.37 | ||
| S5–O21 | 1.48(3) | ||
| U2–O15 | 1.76(2) | S5–O22 | 1.43(3) |
| U2–O16 | 1.77(2) | S5–O23 | 1.46(3) |
| < U2–OUr > | 1.765 | S5–O24 | 1.43(3) |
| U2–O14 | 2.349(19) | < S5–O > | 1.45 |
| U2–O17 | 2.42(2) | ||
| U2–O21 | 2.35(3) | < Cs1–O > CN * = 12 | 3.26 |
| U2–O22 | 2.38(3) | < Cs2–O > CN * = 10 | 3.32 |
| U2–O22A | 2.26(10) | < Cs3–O > CN * = 9 | 3.33 |
| < U2–Oeq > | 2.35 | < Cs4–O > CN * = 10 | 3.33 |
| < Cs5–O > CN * = 9 | 3.27 | ||
| S1–O3 | 1.492(19) | < Cs6–O > CN * = 10 | 3.28 |
| S1–O7 | 1.50(2) | ||
| S1–O8 | 1.42(2) | Angle | |
| S1–O9 | 1.43(3) | U1–O3–S1 | 138.4(13) |
| < S1–O > | 1.46 | U1–O4–S2 | 100.9(9) |
| U1–O5–S2 | 99.6(9) | ||
| S2–O4 | 1.505(19) | U1–O6–S3 | 141.8(12) |
| S2–O5 | 1.510(19) | U1–O7–S1 | 140.0(14) |
| S2–O10 | 1.42(2) | U2–O14–S3 | 135.7(12) |
| S2–O11 | 1.42(2) | U2–O17–S4 | 100.5(10) |
| < S2–O > | 1.46 | U2–O18–S4 | 101.3(11) |
| U2–O21–S5 | 146.0(16) | ||
| S3–O6 | 1.497(19) | U2–O22–S5 | 146(2) |
| S3–O12 | 1.494(19) | U2–O22A–S5 | 157(7) |
| S3–O13 | 1.39(2) |
| Bond | Bond | ||
|---|---|---|---|
| U1–O1 | 1.775(2) | S2–O7 | 1.477(2) |
| U1–O2 | 1.768(2) | S2–O8 | 1.430(3) |
| < U1–OUr > | 1.772 | S2–O9 | 1.486(2) |
| U1–O4 | 2.301(2) | S2–O10 | 1.492(2) |
| U1–O5 | 2.319(2) | < S2–O > | 1.471 |
| U1–O7 | 2.322(2) | ||
| U1–O9 | 2.478(2) | < Cs1–O > CN * = 10 | 3.286 |
| U1–O10 | 2.482(2) | < Cs2–O > CN * = 10 | 3.311 |
| < U1–Oeq > | 2.380 | ||
| Angle | |||
| S1–O3 | 1.446(2) | S1–O4–U1 | 142.45(14) |
| S1–O4 | 1.502(2) | S1–O5–U1 | 133.36(14) |
| S1–O5 | 1.502(2) | S2–O7–U1 | 148.09(16) |
| S1–O6 | 1.440(2) | S2–O9–U1 | 99.54(11) |
| < S1–O > | 1.473 | S2–O10–U1 | 99.22(11) |
| Compound | Complexity Parameters of the Crystal Structure | Structural Complexity of the U-S Unit | Topological Complexity of the U-S Unit | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Sp. Gr. | ν | IG | IG,total | Layer/Rod Gr. | ν | IG | IG,total | Layer/Rod Gr. | ν | IG | IG,total | |
| 1 | Pnma | 100 | 3.844 | 384.386 | p21/a | 40 | 3.322 | 132.877 | p21/a | 40 | 3.322 | 132.877 |
| 2 | P–1 | 78 | 5.285 | 412.261 | p–1 | 54 | 4.755 | 256.764 | p–1 | 54 | 4.755 | 256.764 |
| 3 | P–1 | 92 | 5.524 | 508.168 |  –1 –1 | 62 | 4.954 | 307.160 | amam | 62 | 3.599 | 223.160 |
| 4 | P–1 | 141 | 6.161 | 868.677 | p–1 | 54 | 4.755 | 256.764 | p–1 | 54 | 4.755 | 256.764 |
| K4[UO2(SO4)3] | Pnma | 176 | 4.641 | 816.860 | pm | 36 | 4.337 | 156.117 | pmm2 | 36 | 3.892 | 140.117 |
| Marecottite | P21/n | 60 | 3.907 | 234.413 | p21/a | 52 | 3.700 | 192.423 | p21/a | 52 | 3.700 | 192.423 |
| Peligotite | P–1 | 82 | 5.382 | 441.319 | p–1 | 23 | 4.524 | 104.042 | pm | 23 | 3.915 | 90.042 |
| Lussierite | Cc | 104 | 5.700 | 592.846 | ||||||||
| Klaprothite | P21/c | 164 | 5.382 | 882.639 | ||||||||
| Ottohahnite | P–1 | 126 | 6.000 | 768.000 | p–1 | 62 | 4.954 | 307.160 | p–1 | 62 | 4.954 | 307.160 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tyumentseva, O.S.; Kornyakov, I.V.; Britvin, S.N.; Zolotarev, A.A.; Gurzhiy, V.V. Crystallographic Insights into Uranyl Sulfate Minerals Formation: Synthesis and Crystal Structures of Three Novel Cesium Uranyl Sulfates. Crystals 2019, 9, 660. https://doi.org/10.3390/cryst9120660
Tyumentseva OS, Kornyakov IV, Britvin SN, Zolotarev AA, Gurzhiy VV. Crystallographic Insights into Uranyl Sulfate Minerals Formation: Synthesis and Crystal Structures of Three Novel Cesium Uranyl Sulfates. Crystals. 2019; 9(12):660. https://doi.org/10.3390/cryst9120660
Chicago/Turabian StyleTyumentseva, Olga S., Ilya V. Kornyakov, Sergey N. Britvin, Andrey A. Zolotarev, and Vladislav V. Gurzhiy. 2019. "Crystallographic Insights into Uranyl Sulfate Minerals Formation: Synthesis and Crystal Structures of Three Novel Cesium Uranyl Sulfates" Crystals 9, no. 12: 660. https://doi.org/10.3390/cryst9120660
APA StyleTyumentseva, O. S., Kornyakov, I. V., Britvin, S. N., Zolotarev, A. A., & Gurzhiy, V. V. (2019). Crystallographic Insights into Uranyl Sulfate Minerals Formation: Synthesis and Crystal Structures of Three Novel Cesium Uranyl Sulfates. Crystals, 9(12), 660. https://doi.org/10.3390/cryst9120660