Cyclodextrin-Driven Formation of Double Six-Ring (D6R) Silicate Cage: NMR Spectroscopic Characterization from Solution to Crystals


Abstract

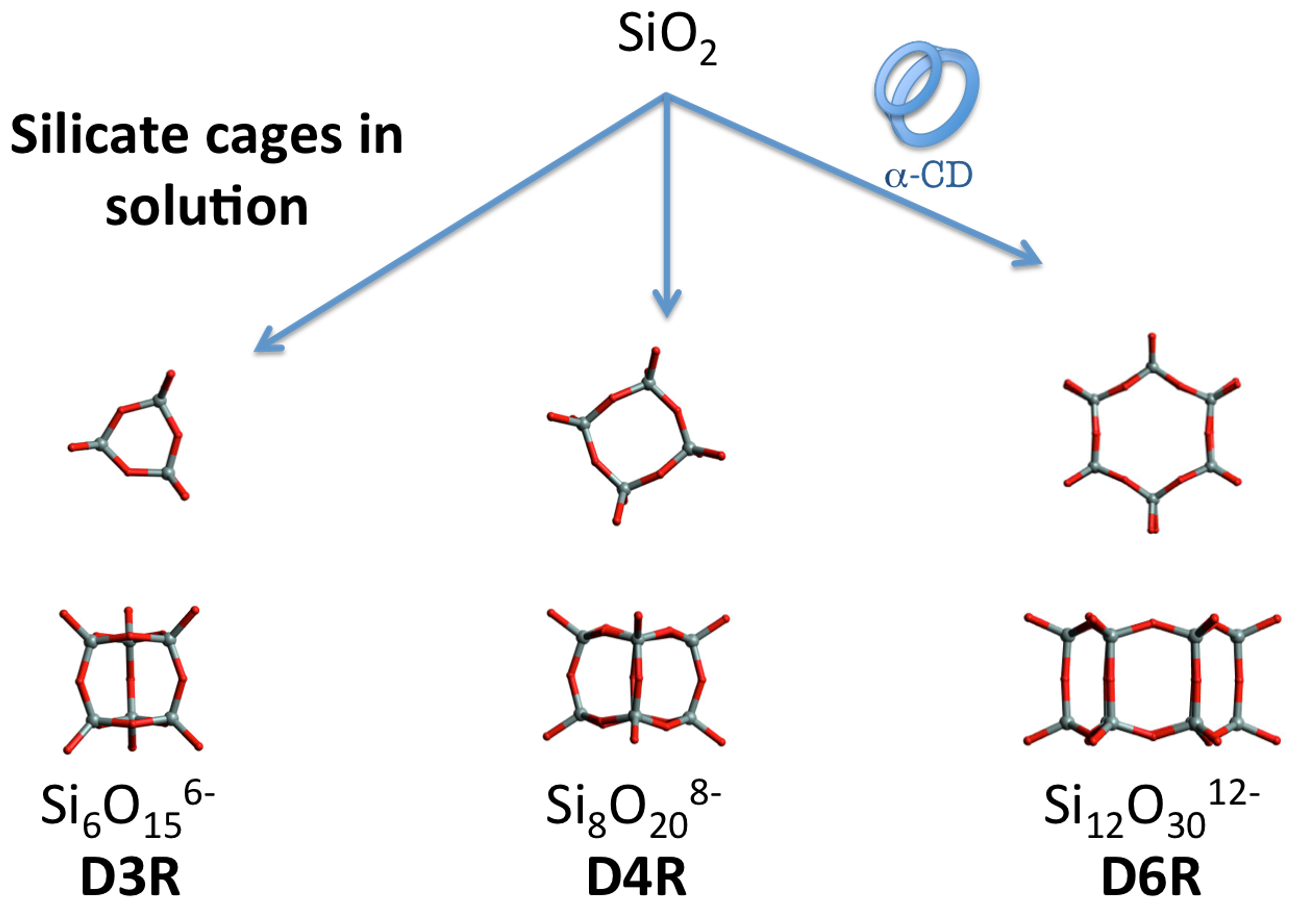
1. Introduction
2. Materials and Methods
2.1. Chemicals
2.2. Synthesis of K12Si12O30·2α-CD·36H2O (D6R@α-CD)
2.3. Nuclear Magnetic Resonance (NMR) Study in Solution
- (i)
- kinetics: 1 α-CD:6 TEOS:6 KOH:300 D2O
- (ii)
- effect of KCl: 1 α-CD:6 TEOS:6 KOH:y D2O:x KCl (y = 600–1000, x = 15–60)
- (iii)
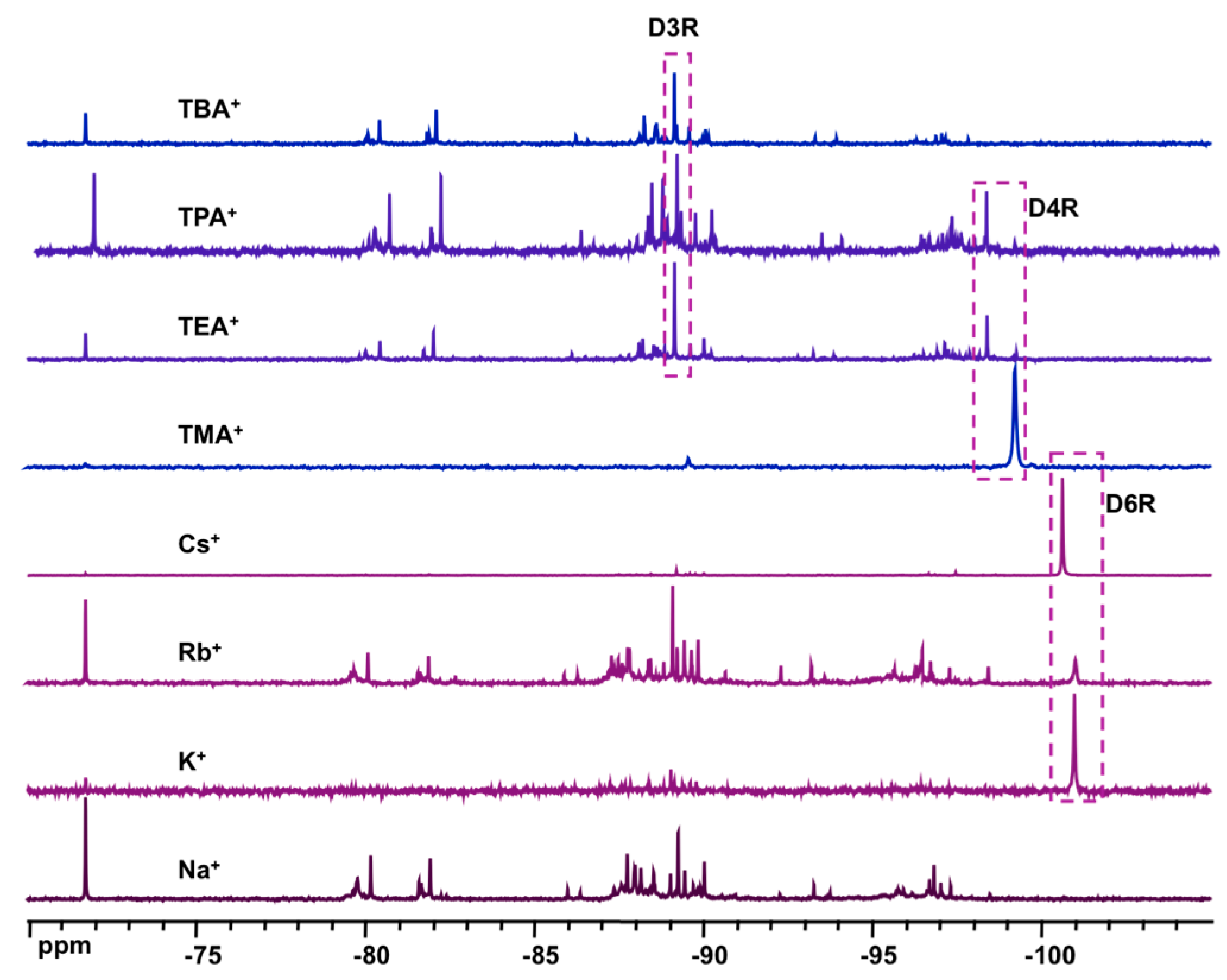
- effect of base: 1 α-CD:6 TEOS:6 XOH:200 D2O (X = Na, K, Rb, Cs, TMA, TEA, TPA, TBA)
- (iv)
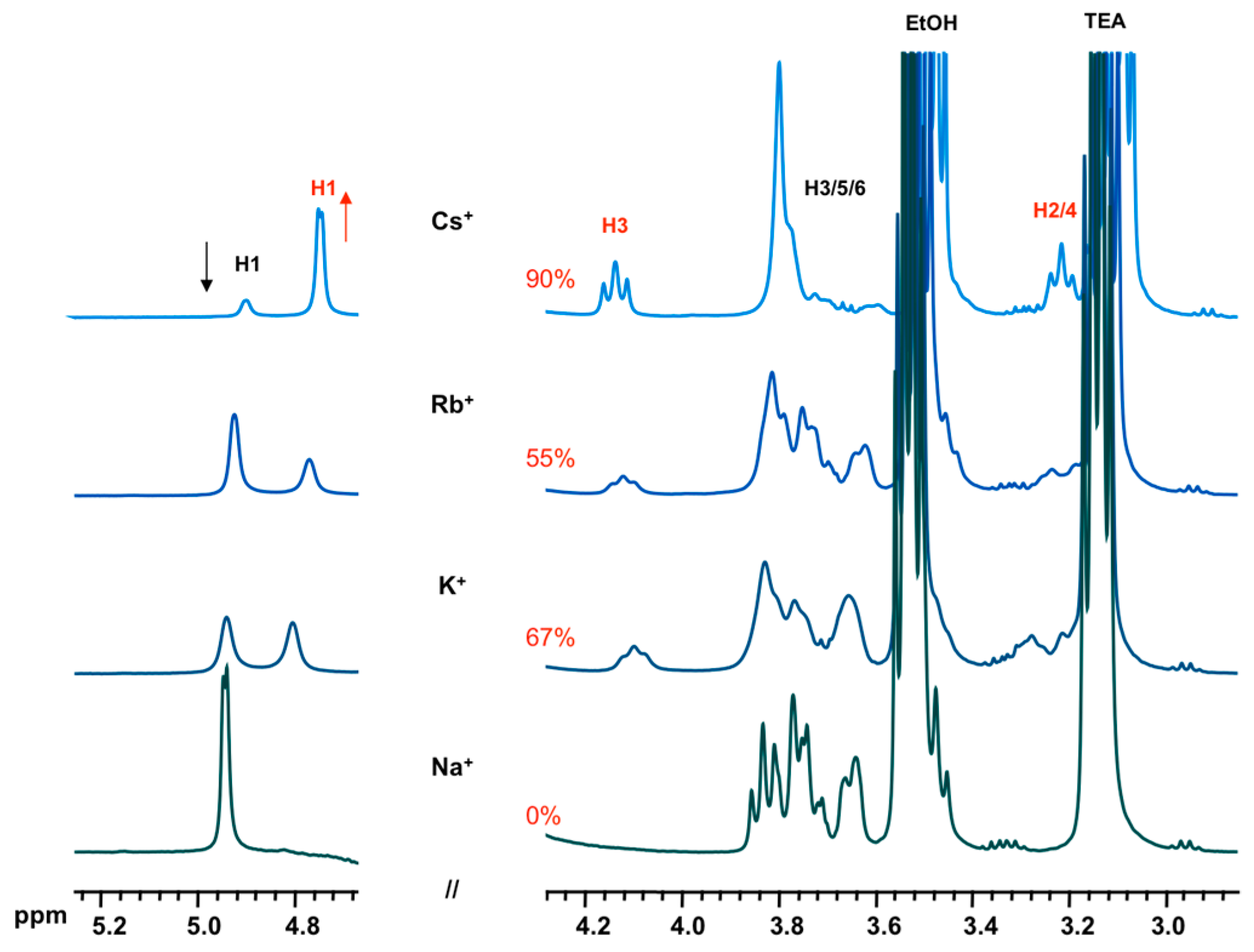
- effect of cation: 1 α-CD:6 TEOS:6 TEAOH:900 D2O:40 XCl (X = Na, K, Rb, Cs)
2.4. Characterization Techniques
2.4.1. NMR Spectroscopy
2.4.2. Fourier Transform Infrared (FT-IR) and Thermogravimetric Analysis (TGA)
2.4.3. Single Crystal X-ray Diffraction (XRD)
3. Results and Discussions
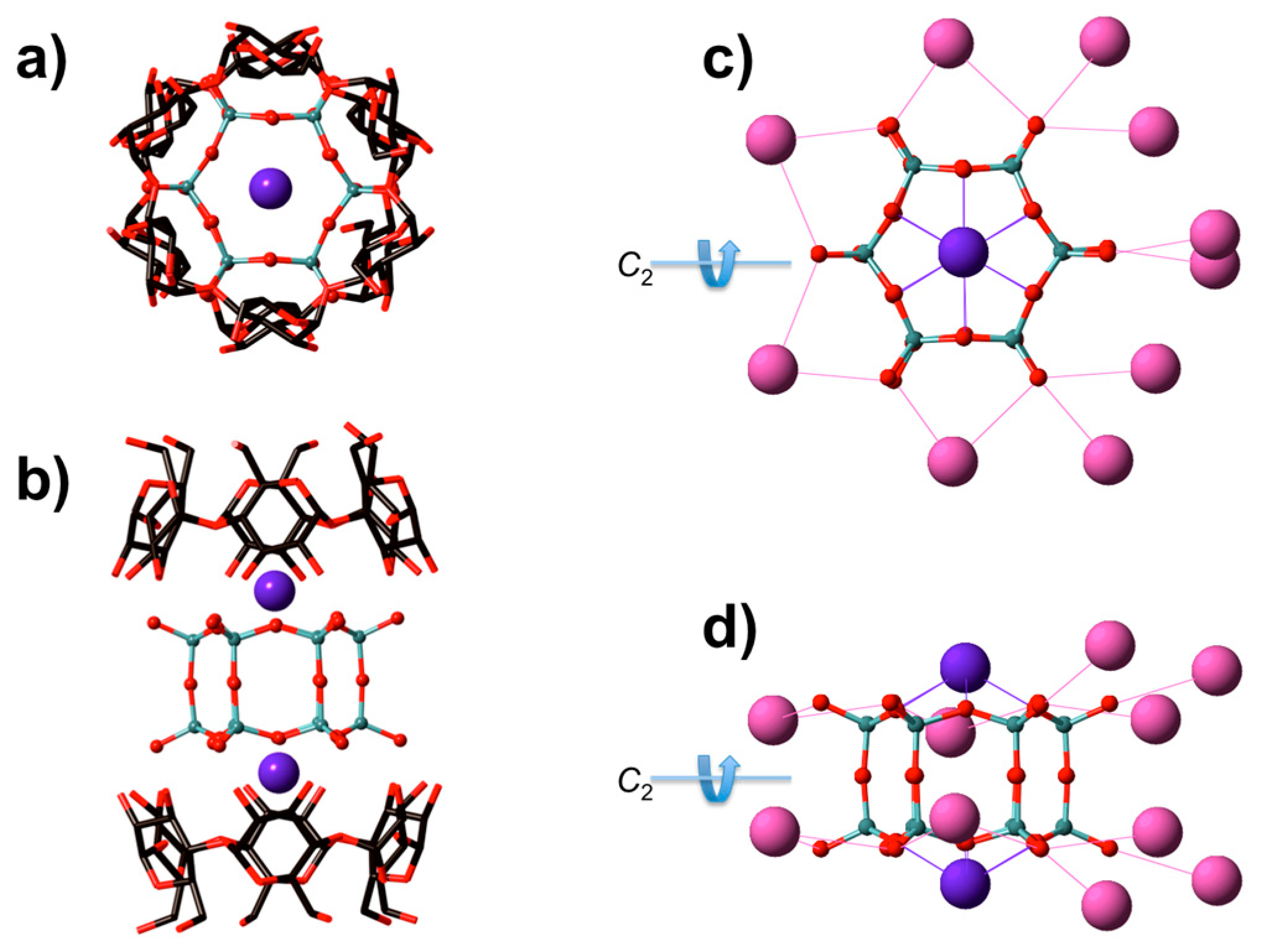
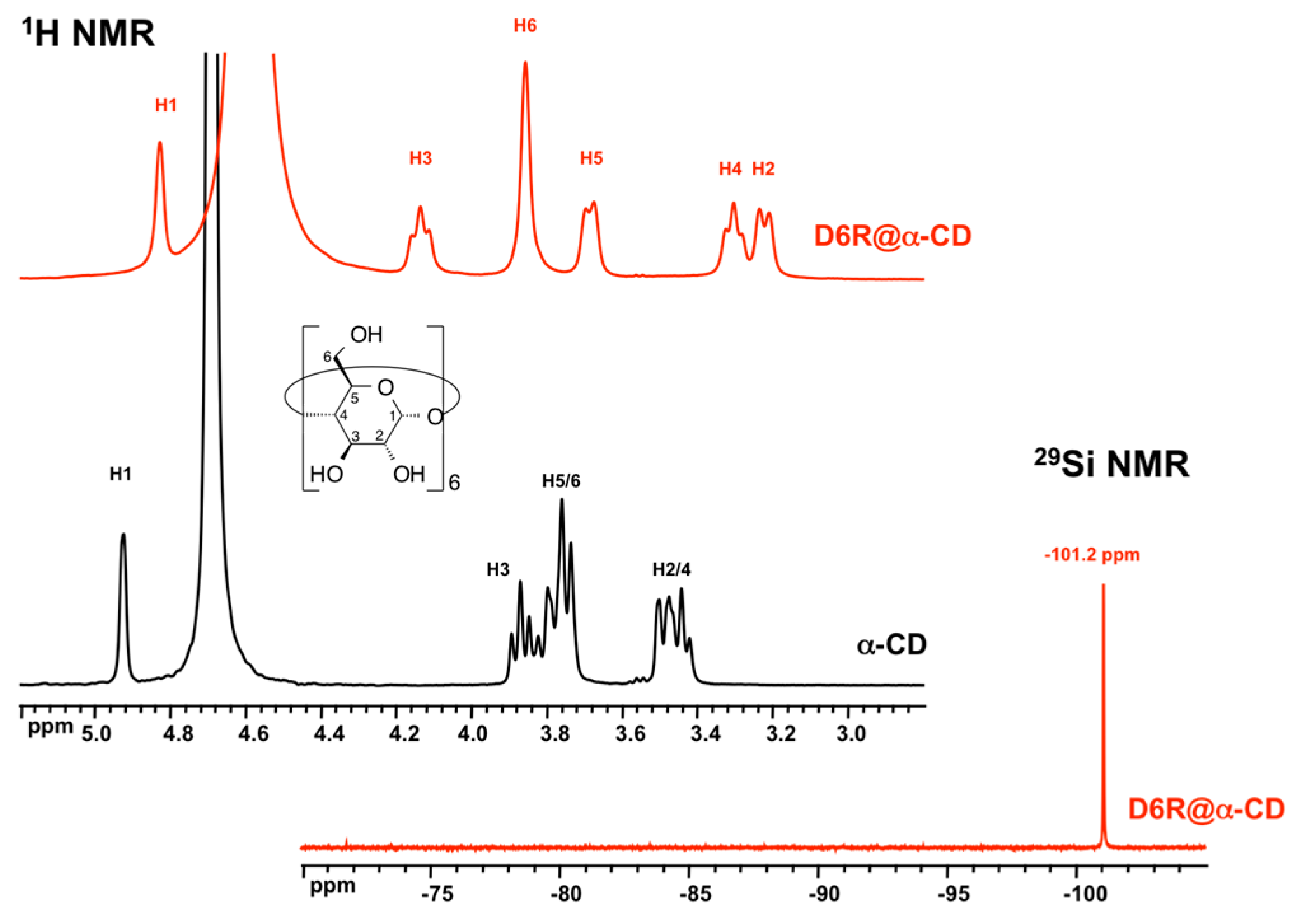
3.1. XRD Structure and NMR Characterization of D6R@α-CD
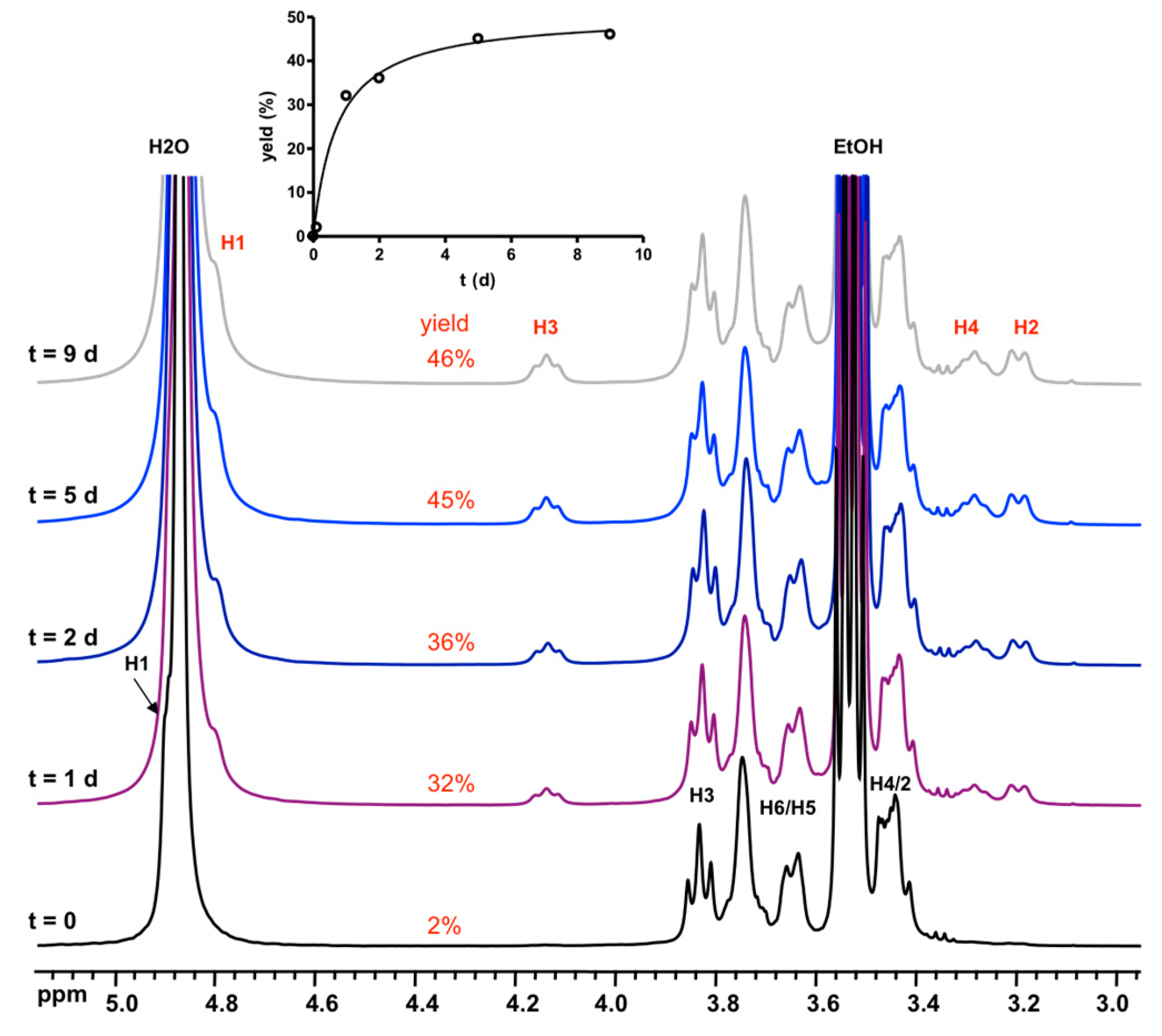
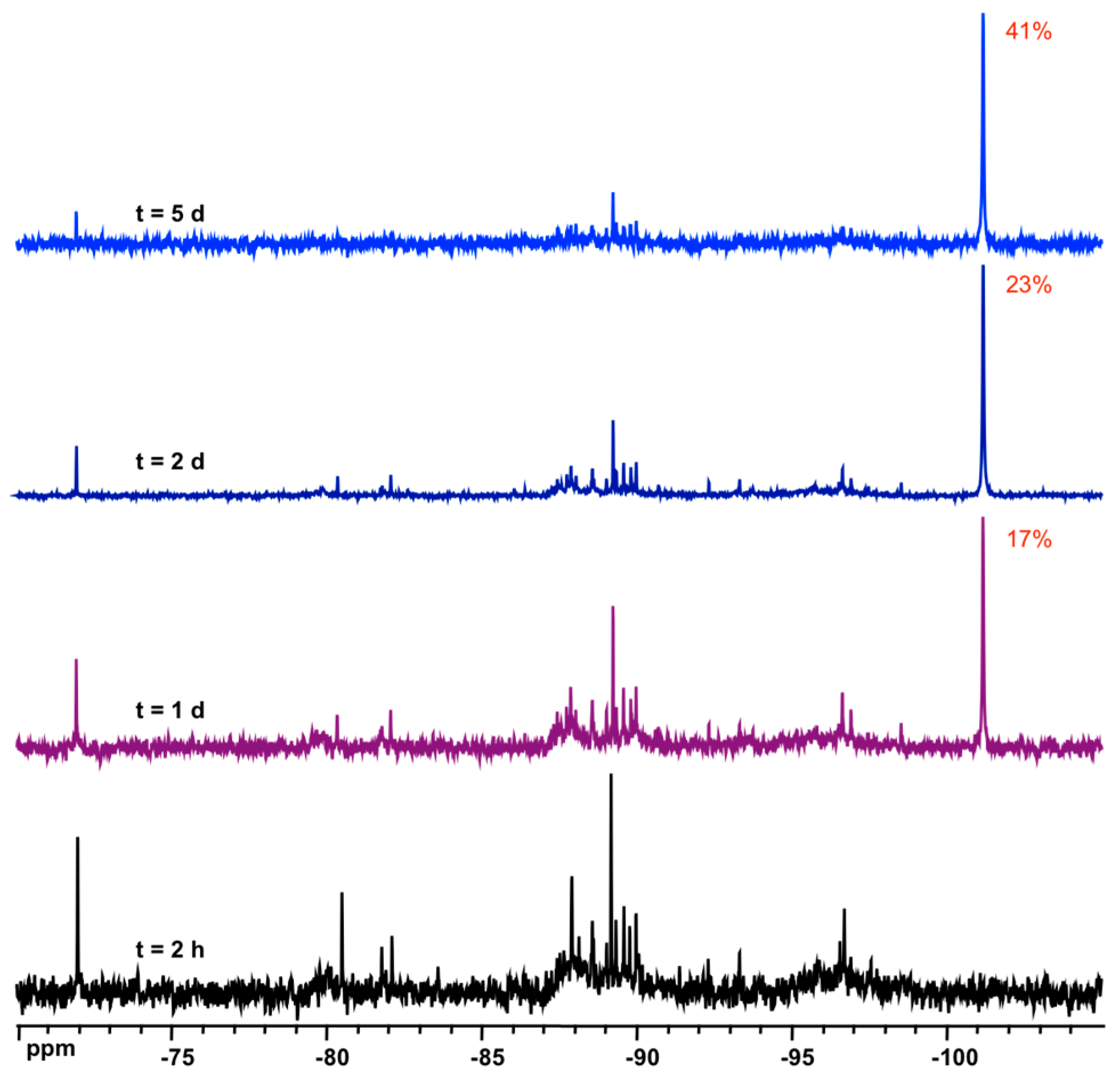
3.2. Formation and Stability of D6R@α-CD in Solution
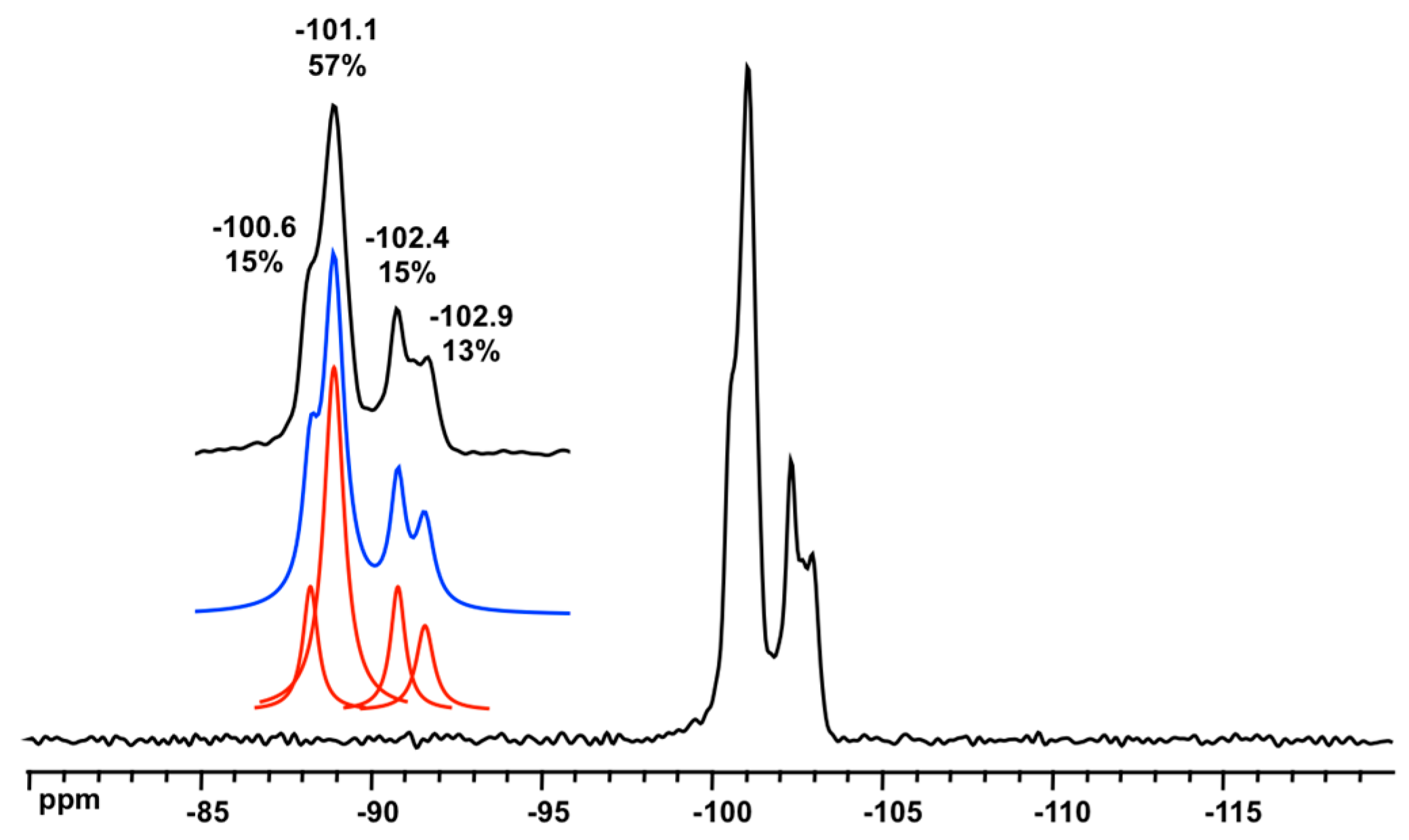
3.3. Solid State NMR Characterization
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Li, Y.; Li, L.; Yu, J.H. Applications of Zeolites in Sustainable Chemistry. Chem 2017, 3, 928–949. [Google Scholar] [CrossRef]
- Zecchina, A.; Bordiga, S.; Vitillo, J.G.; Ricchiardi, G.; Lamberti, C.; Spoto, G.; Bjorgen, M.; Lillerud, K.P. Liquid hydrogen in protonic chabazite. J. Am. Chem. Soc. 2005, 127, 6361–6366. [Google Scholar] [CrossRef] [PubMed]
- Chassaing, S.; Beneteau, V.; Pale, P. Green catalysts based on zeolites for heterocycle synthesis. Curr. Opin. Green Sustain. Chem. 2018, 10, 35–39. [Google Scholar] [CrossRef]
- Ennaert, T.; Van Aelst, J.; Dijkmans, J.; De Clercq, R.; Schutyser, W.; Dusselier, M.; Verboekend, D.; Sels, B.F. Potential and challenges of zeolite chemistry in the catalytic conversion of biomass. Chem. Soc. Rev. 2016, 45, 584–611. [Google Scholar] [CrossRef] [PubMed]
- Rangnekar, N.; Mittal, N.; Elyassi, B.; Caro, J.; Tsapatsis, M. Zeolite membranes—A review and comparison with MOFs. Chem. Soc. Rev. 2015, 44, 7128–7154. [Google Scholar] [CrossRef] [PubMed]
- Barrer, R.M. Zeolites and their synthesis. Zeolites 1981, 1, 130–140. [Google Scholar] [CrossRef]
- Database of Zeolite Structures. Available online: http://www.iza-structure.org/databases/ (accessed on 6 December 2018).
- Haouas, M.; Taulelle, F. Revisiting the identification of structural units in aqueous silicate solutions by two-dimensional silicon-29 INADEQUATE. J. Phys. Chem. B 2006, 110, 3007–3014. [Google Scholar] [CrossRef]
- Knight, C.T.G.; Balec, R.J.; Kinrade, S.D. The structure of silicate anions in aqueous alkaline solutions. Angew. Chem.-Int. Edit. 2007, 46, 8148–8152. [Google Scholar] [CrossRef]
- Haouas, M. Nuclear Magnetic Resonance Spectroscopy for In Situ Monitoring of Porous Materials Formation under Hydrothermal Conditions. Materials 2018, 11, 1416. [Google Scholar] [CrossRef]
- Benner, K.; Klufers, P.; Schuhmacher, J. A molecular composite constructed in aqueous alkaline solution from a double six-ring silicate and alpha-cyclodextrin. Angew. Chem.-Int. Edit. 1997, 36, 743–745. [Google Scholar] [CrossRef]
- Assaf, K.I.; Ural, M.S.; Pan, F.F.; Georgiev, T.; Simova, S.; Rissanen, K.; Gabel, D.; Nau, W.M. Water Structure Recovery in Chaotropic Anion Recognition: High-Affinity Binding of Dodecaborate Clusters to -Cyclodextrin. Angew. Chem.-Int. Edit. 2015, 54, 6852–6856. [Google Scholar] [CrossRef] [PubMed]
- Moussawi, M.A.; Haouas, M.; Floquet, S.; Shepard, W.E.; Abramov, P.A.; Sokolov, M.N.; Fedin, V.P.; Cordier, S.; Ponchel, A.; Monflier, E.; et al. Nonconventional Three-Component Hierarchical Host-Guest Assembly Based on Mo-Blue Ring-Shaped Giant Anion, gamma-Cyclodextrin, and Dawson-type Polyoxometalate. J. Am. Chem. Soc. 2017, 139, 14376–14379. [Google Scholar] [CrossRef] [PubMed]
- Prochowicz, D.; Kornowicz, A.; Lewinski, J. Interactions of Native Cyclodextrins with Metal Ions and Inorganic Nanoparticles: Fertile Landscape for Chemistry and Materials Science. Chem. Rev. 2017, 117, 13461–13501. [Google Scholar] [CrossRef] [PubMed]
- Falaise, C.; Moussawi, M.A.; Floquet, S.; Abramov, P.A.; Sokolov, M.N.; Haouas, M.; Cadot, E. Probing Dynamic Library of Metal-Oxo Building Blocks with gamma-Cyclodextrin. J. Am. Chem. Soc. 2018, 140, 11198–11201. [Google Scholar] [CrossRef] [PubMed]
- Wiebcke, M.; Felsche, J. The tetraethylammonium cation in a double three-ring silicate heteronetwork clathrate and a polyhedral clathrate hydrate: Low-temperature single-crystal X-ray diffraction studies on (NEt4)6Si6O15, 40.8H2O and NEt4OH,9H2O. Microporous Mesoporous Mater. 2001, 43, 289–297. [Google Scholar] [CrossRef]
- Wiebcke, M.; Grube, M.; Koller, H.; Engelhardt, G.; Felsche, J. Structural Links Between Zeolite-Type and Clathrate Hydrate-Type Materials—Redetermination of the Crystal-Structure of (N(CH3)4)8Si8O20.65H2O by Single-Crystal X-Ray-Diffraction and Variable-Temperature MAS NMR-Spectroscopy. Microporous Mater. 1993, 2, 55–63. [Google Scholar] [CrossRef]
- Enkelmann, D.D.; Hofmann, D.W.M.; Merz, K. Deuterium Shifts the Equilibrium: How Heavy Water Can Influence Organic Multicomponent Crystal Formation. Cryst. Growth Des. 2017, 17, 4726–4729. [Google Scholar] [CrossRef]
- Sasmal, D.K.; Dey, S.; Das, D.K.; Bhattacharyya, K. Deuterium isotope effect on femtosecond solvation dynamics in methyl beta-cyclodextrins. J. Chem. Phys. 2009, 131, 44509. [Google Scholar] [CrossRef] [PubMed]
- Dolomanov, O.V.; Bourhis, L.J.; Gildea, R.J.; Howard, J.A.K.; Puschmann, H. OLEX2: A complete structure solution, refinement and analysis program. J. Appl. Crystallogr. 2009, 42, 339–341. [Google Scholar] [CrossRef]
- Sheldrick, G.M. SHELXT—Integrated space-group and crystal-structure determination. Acta Crystallogr. Sect. A 2015, 71, 3–8. [Google Scholar] [CrossRef]
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Crystallogr. Sect. C-Struct. Chem. 2015, 71, 3–8. [Google Scholar] [CrossRef]
- Spek, A.L. PLATON SQUEEZE: A tool for the calculation of the disordered solvent contribution to the calculated structure factors. Acta Crystallogr. Sect. C-Struct. Chem. 2015, 71, 9–18. [Google Scholar] [CrossRef]
- Engelhardt, G.; Michel, D. High-Resolution Solid-State NMR of Silicates and Zeolites; John Wiley & Sons: New York, NY, USA, 1987; pp. 75–105. [Google Scholar]
- Falcone, J.S.; Bass, J.L.; Krumrine, P.H.; Brensinger, K.; Schenk, E.R. Characterizing the Infrared Bands of Aqueous Soluble Silicates. J. Phys. Chem. A 2010, 114, 2438–2446. [Google Scholar] [CrossRef]
- van Tendeloo, L.; Haouas, M.; Martens, J.A.; Kirschhock, C.E.A.; Breynaert, E.; Taulelle, F. Zeolite synthesis in hydrated silicate ionic liquids. Faraday Discuss. 2015, 179, 437–449. [Google Scholar] [CrossRef]
- Hoebbel, D.; Garzo, G.; Engelhardt, G.; Ebert, R.; Lippmaa, E.; Alla, M. On the Constitution of Silicate Anions in Tetraethylammonium Silicates and their Aqueous Solutions. Z. Anorg. Allg. Chem. 1980, 465, 15–33. [Google Scholar] [CrossRef]
- Caratzoulas, S.; Vlachos, D.G.; Tsapatsis, M. On the role of tetramethylammonium cation and effects of solvent dynamics on the stability of the cage-like silicates Si6O156- and Si8O208- in aqueous solution. A molecular dynamics study. J. Am. Chem. Soc. 2006, 128, 596–606. [Google Scholar] [CrossRef]
- Lebihan, M.T.; Kalt, A.; Wey, R. Structural study of KHSi2O5 and H2Si2O5. Bull. Soc. Fr. Mineral. Cristallogr. 1971, 94, 15. [Google Scholar]
- Schmidmair, D.; Kahlenberg, V.; Perfler, L.; Tobbens, D.M. Structural, spectroscopic and computational studies on the monoclinic polymorph (form I) of potassium hydrogen disilicate (KHSi2O5). Mineral. Mag. 2014, 78, 609–622. [Google Scholar] [CrossRef]
- Magi, M.; Lippmaa, E.; Samoson, A.; Engelhardt, G.; Grimmer, A.R. Solid-state high-resolution Si-29 chemical shifts in silicates. J. Phys. Chem. 1984, 88, 1518–1522. [Google Scholar] [CrossRef]
- Engelhardt, G.; Radeglia, R. A semi-empirical quantum-chemical rationalization of the correlation between SiOSi angles and Si-29 NMR chemical shifts of silica polymorphs and framework aluminosilicates (Zeolites). Chem. Phys. Lett. 1984, 108, 271–274. [Google Scholar] [CrossRef]
- Verlooy, P.L.H.; Robeyns, K.; Van Meervelt, L.; Lebedev, O.I.; Van Tendeloo, G.; Martens, J.A.; Kirschhock, C.E.A. Synthesis and characterization of the new cyclosilicate hydrate (hexamethyleneimine)(4)center dot Si8O16(OH)(4) center dot 12H(2)O. Microporous Mesoporous Mater. 2010, 130, 14–20. [Google Scholar] [CrossRef]
- Wiebcke, M.; Emmer, J.; Felsche, J.; Hoebbel, D.; Engelhardt, G. TMPA4Si8O20.34H2O and DDBO4Si8O20.32H2O are heteronetwork clathrates with 1,1,4,4-tetramethylpiperazinium (TMPA) and 1,4-dimethyl-1,4-diazoniabicyclo2.2.2octane (DDBO) guest cations. Z. Anorg. Allg. Chem. 1994, 620, 757–765. [Google Scholar] [CrossRef]
- Wiebcke, M.; Koller, H. Single crystal X-ray diffraction and variable temperature MAS NMR study on the heterogeneous network clathrate Na(N(CH3)4)7Si8O20.54H2O. Acta Crystallogr. Sect. B-Struct. Commun. 1992, 48, 449–458. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Empirical Formula | K12Si12C72H192O126 (K12Si12O30·2α-CD·36H2O) |
|---|---|
| Formula weight | 3880.44 |
| Temperature/K | 230(2) |
| Crystal system | Orthorhombic |
| Space group | C2221 |
| a/Å | 14.841(4) |
| b/Å | 21.855(6) |
| c/Å | 41.91(1) |
| α/° | 90 |
| β/° | 90 |
| γ/° | 90 |
| Volume/Å3 | 16,081(7) |
| Z | 4 |
| ρcalc/gcm−3 | 1.603 |
| μ/mm−1 | 0.515 |
| F(000) | 7104 |
| Crystal size/mm3 | 0.2 × 0.13 × 0.08 |
| Radiation | Mo Kα (λ = 0.71073) |
| 2Θ range for data collection/° | 5.0 to 50.1 |
| Index ranges | −17 ≤ h ≤ 17, −30 ≤ k ≤ 30, −49 ≤ l ≤ 49 |
| Reflections collected | 69910 |
| Independent reflections | 14,190, Rint = 0.0697, Rsigma = 0.0523 |
| Data/restraints/parameters | 14,190/6/891 |
| Goodness of fit on F2 | 1.048 |
| Final R indexes [I > = 2σ (I)] | R1 = 0.0778, wR2 = 0.2185 |
| Final R indexes [all data] | R1 = 0.0844, wR2 = 0.2245 |
| Largest diff. peak/hole/eÅ−3 | 1.69/−1.54 |
| Flack parameter | 0.13(9) |
| H1 | H2 | H3 | H4 | H5 | H6 | |
| α-CD | 4.92 | 3.49 | 3.87 | 3.44 | 3.75 | 3.78 |
| D6R-α-CD | 4.83 | 3.22 | 4.14 | 3.30 | 3.69 | 3.86 |
| Δδ(ppm) | +0.09 | +0.27 | −0.27 | +0.14 | +0.06 | −0.08 |
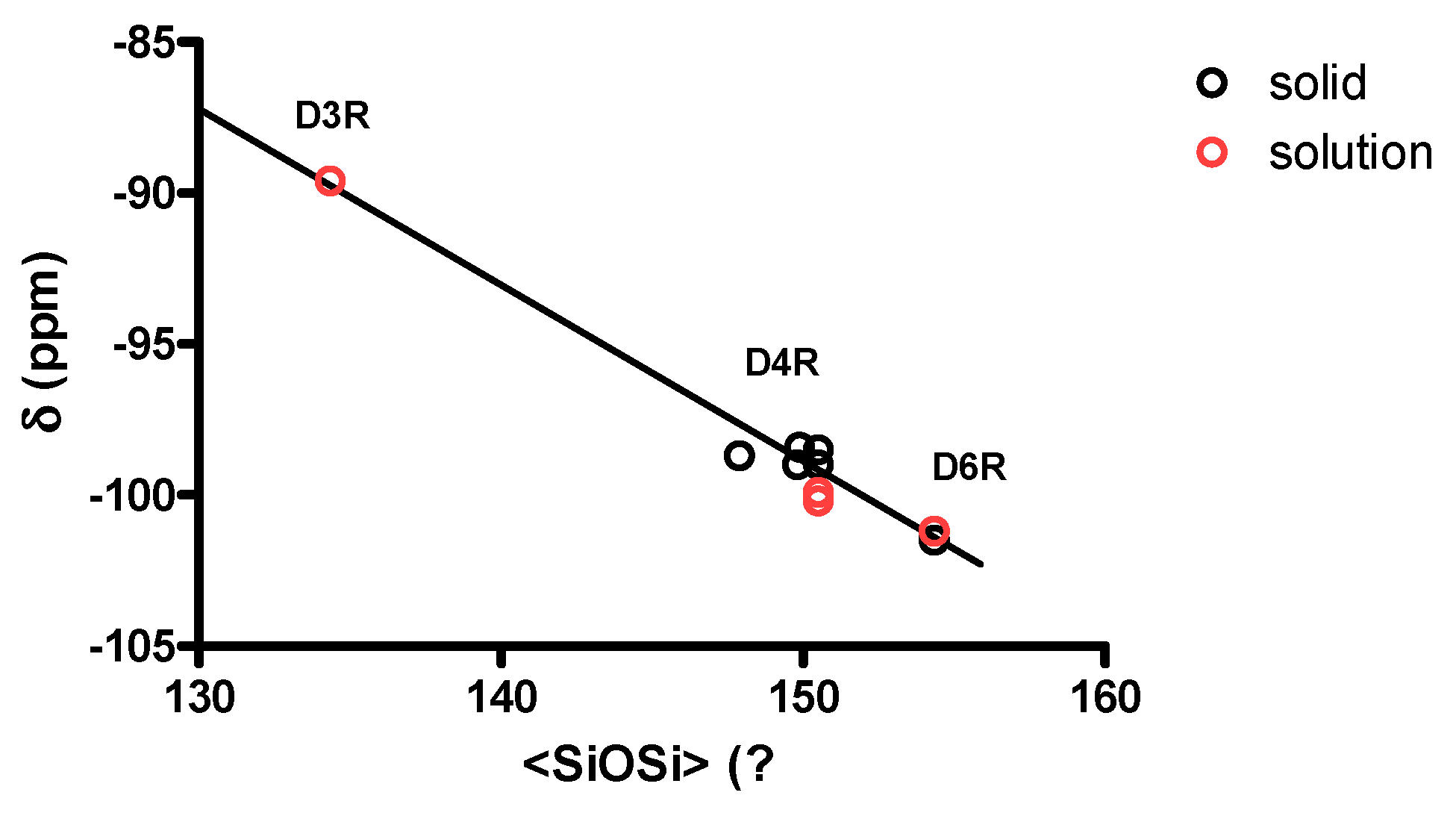
| Environment | Mean SiOSi (°) | Average SiOSi (°) | 29Si (ppm) | References |
|---|---|---|---|---|
| D3R/3R 1 | 131.0 (×6) 2 | 134.3 | −89.6 | [16] |
| D3R/4R | 141.3 (×3) | [16] | ||
| D4R/4R | from 147.9 to 150.5 | −98.4 to −100.2 | [17,33,34,35] | |
| D6R/6R | 144.8 (×12) | 154.3 | −101.2 to −101.5 | this work |
| D6R/4R | 173.3 (×6) | this work |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Haouas, M.; Falaise, C.; Martineau-Corcos, C.; Cadot, E. Cyclodextrin-Driven Formation of Double Six-Ring (D6R) Silicate Cage: NMR Spectroscopic Characterization from Solution to Crystals. Crystals 2018, 8, 457. https://doi.org/10.3390/cryst8120457
Haouas M, Falaise C, Martineau-Corcos C, Cadot E. Cyclodextrin-Driven Formation of Double Six-Ring (D6R) Silicate Cage: NMR Spectroscopic Characterization from Solution to Crystals. Crystals. 2018; 8(12):457. https://doi.org/10.3390/cryst8120457
Chicago/Turabian StyleHaouas, Mohamed, Clément Falaise, Charlotte Martineau-Corcos, and Emmanuel Cadot. 2018. "Cyclodextrin-Driven Formation of Double Six-Ring (D6R) Silicate Cage: NMR Spectroscopic Characterization from Solution to Crystals" Crystals 8, no. 12: 457. https://doi.org/10.3390/cryst8120457
APA StyleHaouas, M., Falaise, C., Martineau-Corcos, C., & Cadot, E. (2018). Cyclodextrin-Driven Formation of Double Six-Ring (D6R) Silicate Cage: NMR Spectroscopic Characterization from Solution to Crystals. Crystals, 8(12), 457. https://doi.org/10.3390/cryst8120457