Determination of the Full 207Pb Chemical Shift Tensor of Anglesite, PbSO4, and Correlation of the Isotropic Shift to Lead–Oxygen Distance in Natural Minerals



Abstract
1. Introduction
- (i)
- Data acquisition and evaluation is rather time-consuming when compared to static or MAS NMR of polycrystalline samples;
- (ii)
- tensor eigenvector determination in the crystal frame requires knowledge of the exact orientation of the crystal on the goniometer axis;
- (iii)
- single crystals of sufficient size must be available because of the comparatively poor signal-to-noise ratio of NMR spectroscopy.
2. Single-Crystal NMR of Pb with Spin
2.1. General Definitions
2.2. Single-Crystal NMR of Anglesite, PbSO
3. Results and Discussion
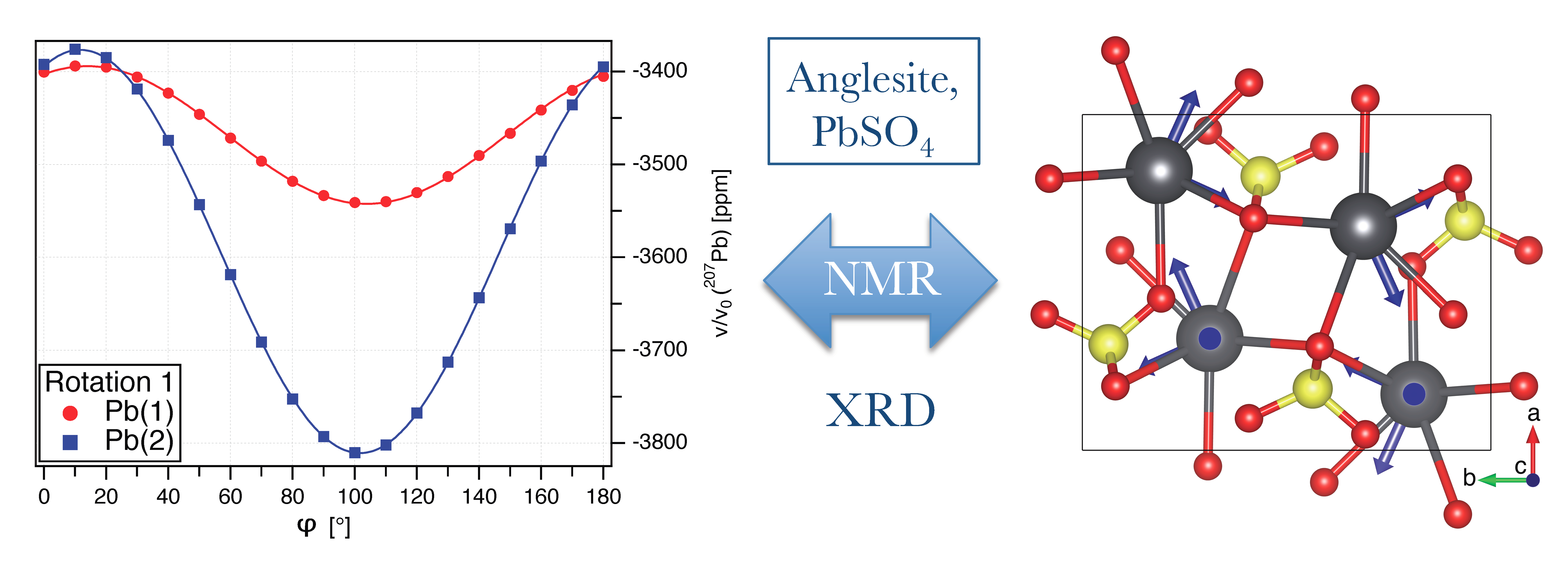
3.1. Pb NMR of Anglesite: Single Crystal
3.2. Pb NMR of Anglesite: Magic-Angle Spinning
3.3. Correlation of Pb Isotropic Shifts and Pb–O Distances
4. Conclusions
- (i)
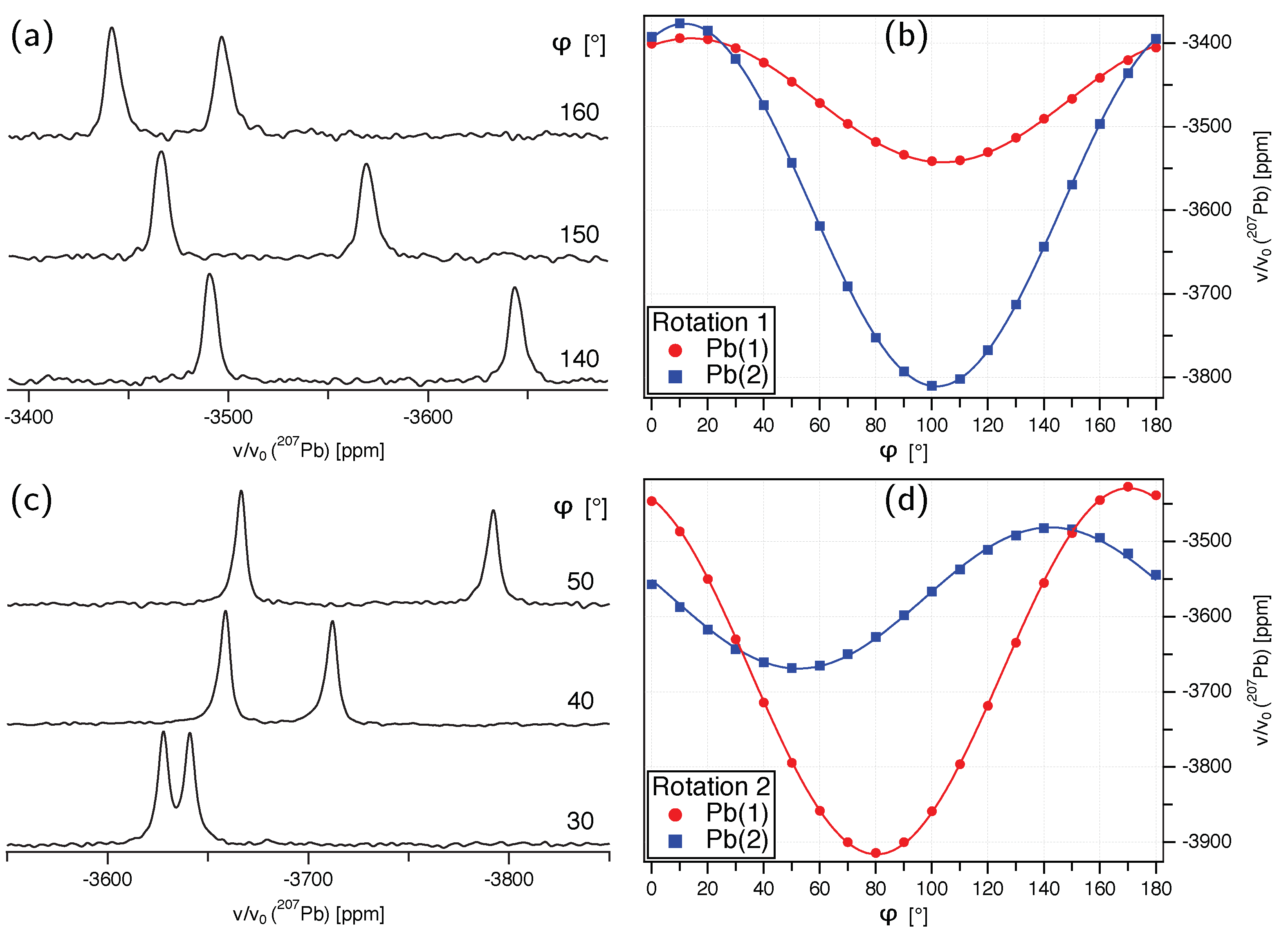
- The presumed extensive effort for data acquisition and evaluation is clearly manageable for the anglesite system. Despite the relatively low natural abundance of Pb of 22.1%, the spectra for the rotation pattern shown in Figure 3b were acquired with only eight scans and 60 s recycle delay, meaning the absolute measurement time was only about 2.5 hours. It can be seen from Figure 3a that the signal-to-noise ratio of the single-crystal spectra is satisfactory under these conditions. In contrast to static spectra of polycrystalline material (see, for example, Reference [6]), spectral intensity is focused on only few resonance lines, making detection less demanding. Our total experimental time was, of course, longer than 2.5 hours, with reorientation of the crystal and especially the subsequent necessary tuning and matching of the probe causing additional delays (the procedure of reorient/tune/match, however, may in principle be automated, considerably lessening the work load of the operator [56]). To further improve the efficiency of both data acquisition and processing, we utilised crystallographic symmetry relations [12,13,14] between two groups of magnetically inequivalent Pb atoms, reducing the necessary data for determining the full CS tensor to two rotation patterns, about two nonparallel rotation axes. A generous estimate of the optimised total time for data acquisition would therefore be 7–8 h. It took three hours to record the MAS spectrum of a polycrystalline sample shown in Figure 5a, yet, for Pb spectra of systems with large , two such MAS spectra are usually required to unequivocally assign the isotropic peak. In terms of pure experimental time, therefore, single-crystal NMR compares not unfavourably to MAS NMR, although the necessary effort for data processing and evaluation is obviously more extensive. At the end of this process, however, the full CS tensor is gained, and not only the value of available from fast MAS experiments.
- (ii)
- The necessity to know the orientation of the rotation axis in the crystal frame is generally solvable in a number of ways, including optical orientation using well-developed crystal faces. The most precise method is certainly the use of X-ray diffraction on the mounted crystal on which NMR measurements are to be performed. However, the large crystals needed for NMR tend to absorb most of the X-ray beam, in particular when compounds with high lead content, such as anglesite, are being investigated. For these systems, resolving the orientation of the rotation axis by relying only on NMR data is a very useful approach, as shown here and in earlier work [15,16,17].
- (iii)
- For the scarce availability of single crystals of sufficient size for NMR experiments, no general solution appears to exist. For the area of Pb-NMR, however, a considerable number of compounds also occur as natural minerals (see Table 2). For many lead compounds, comparatively large single crystals are therefore available from mineral collections.
5. Materials and Methods
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Ewald, P.P.; Fifty Years of X-ray Diffraction. International Union of Crystallography. 1999. Available online: http://www.iucr.org/publ/50yearsofxraydiffraction (accessed on 14 January 2019).
- Harris, R.K. NMR crystallography: The use of chemical shifts. Solid State Sci. 2004, 6, 1025–1037. [Google Scholar] [CrossRef]
- Martineau, C.; Senker, J.; Taulelle, F. NMR Crystallography. Ann. Rep. NMR Spectrosc. 2014, 82, 1–57. [Google Scholar] [CrossRef]
- Bryce, D.L. NMR crystallography: Structure and properties of materials from solid-state nuclear magnetic resonance observables. IUCrJ 2017, 4, 350–359. [Google Scholar] [CrossRef] [PubMed]
- Available online: https://www.iucr.org/iucr/commissions/nmr-crystallography (accessed on 14 January 2019).
- Neue, G.; Dybowski, C.; Smith, M.L.; Hepp, M.A.; Perry, D.L. Determination of 207Pb2+ chemical shift tensors from precise powder lineshape analysis. Solid State Nucl. Magn. Reson. 1996, 6, 241–250. [Google Scholar] [CrossRef]
- Herzfeld, J.; Berger, A.E. Sideband intensities in NMR spectra of samples spinning at the magic angle. J. Chem. Phys. 1980, 73, 6021–6030. [Google Scholar] [CrossRef]
- Hodgkinson, P.; Emsley, L. The reliability of the determination of tensor parameters by solid-state nuclear magnetic resonance. J. Chem. Phys. 1997, 107, 4808–4816. [Google Scholar] [CrossRef]
- Volkoff, G.M.; Petch, H.E.; Smellie, D.W.L. Nuclear electric quadrupole interactions in single crystals. Can. J. Phys. 1952, 30, 270–289. [Google Scholar] [CrossRef]
- Vosegaard, T.; Hald, E.; Langer, V.; Skov, H.J.; Daugaard, P.; Bildsøe, H.; Jakobsen, H.J. Improved hardware and software for single-crystal NMR spectroscopy. J. Magn. Reson. 1998, 135, 126–132. [Google Scholar] [CrossRef]
- Hou, G.; Paramasivam, S.; Byeon, I.J.L.; Gronenborn, A.M.; Polenova, T. Determination of relative tensor orientations by γ-encoded chemical shift anisotropy/heteronuclear dipolar coupling 3D NMR spectroscopy in biological solids. Phys. Chem. Chem. Phys. 2010, 12, 14873–14883. [Google Scholar] [CrossRef]
- Weil, J.A. Use of symmetry-related crystal sites for measuring tensor properties in magnetic resonance. J. Magn. Reson. 1973, 10, 391–393. [Google Scholar] [CrossRef]
- Berglund, B.; Tegenfeldt, J. The determination of quadrupole coupling tensors from single-crystal NMR data. J. Magn. Reson. 1978, 30, 451–455. [Google Scholar] [CrossRef]
- Tesche, B.; Zimmermann, H.; Poupko, R.; Haeberlen, U. The Single-Rotation Method of Determining Quadrupole Coupling Tensors in Monoclinic Crystals. Error Analysis and Application to Bullvalene. J. Magn. Reson. Ser. A 1993, 140, 68–77. [Google Scholar] [CrossRef]
- Bräuniger, T.; Poupko, R.; Luz, Z.; Zimmermann, H.; Haeberlen, U. Quantification of the orientational disorder in ortho-dichlorotetramethylbenzene: A single crystal deuterium nuclear magnetic resonance and x-ray study of the site populations. J. Chem. Phys. 2001, 115, 8049–8059. [Google Scholar] [CrossRef]
- Zeman, O.E.O.; Moudrakovski, I.L.; Hoch, C.; Hochleitner, R.; Schmahl, W.W.; Karaghiosoff, K.; Bräuniger, T. Determination of the 31P and 207Pb Chemical Shift Tensors in Pyromorphite, Pb5(PO4)3Cl, by Single-Crystal NMR Measurements and DFT Calculations. Z. Anorg. Allg. Chem. 2017, 643, 1635–1641. [Google Scholar] [CrossRef]
- Zeman, O.E.O.; Hoch, C.; Hochleitner, R.; Bräuniger, T. NMR interaction tensors of 51V and 207Pb in vanadinite, Pb5(VO4)3Cl, determined from DFT calculations and single-crystal NMR measurements, using only one general rotation axis. Solide State Nucl. Magn. Reson. 2018, 89, 11–20. [Google Scholar] [CrossRef] [PubMed]
- Hafner, S.; Raymond, M. The nuclear quadrupole coupling tensors of Al27 in kyanite. Am. Mineral. 1967, 52, 1632–1642. [Google Scholar]
- Bryant, P.L.; Harwell, C.R.; Wu, K.; Fronczek, F.R.; Hall, R.W.; Butler, L.G. Single-crystal 27Al NMR of andalusite and calculated electric field gradients: The first complete NMR assignment for a 5-coordinated aluminum site. J. Phys. Chem. A 1999, 103, 5246–5252. [Google Scholar] [CrossRef]
- Antao, S.M. Structural trends for celestite (SrSO4), anglesite (PbSO4), and barite (BaSO4): Confirmation of expected variations within the SO4 groups. Am. Mineral. 2012, 97, 661–665. [Google Scholar] [CrossRef]
- Momma, K.; Izumi, F. VESTA 3 for three-dimensional visualization of crystal, volumetric and morphology data. J. Appl. Cryst. 2011, 44, 1272–1276. [Google Scholar] [CrossRef]
- Fayon, F.; Farnan, I.; Bessada, C.; Coutures, J.; Massiot, D.; Coutures, J.P. Empirical Correlations between 207Pb NMR Chemical Shifts and Structure in Solids. J. Am. Chem. Soc. 1997, 119, 6837–6843. [Google Scholar] [CrossRef]
- Mason, H.E.; Hirner, J.J.; Xu, W.; Parise, J.B.; Phillips, B.L. Solid-state NMR spectroscopy of Pb-rich apatite. Magn. Reson. Chem. 2009, 47, 1062–1070. [Google Scholar] [CrossRef] [PubMed]
- Dmitrenko, O.; Bai, S.; Beckmann, P.A.; van Bramer, S.; Vega, A.J.; Dybowski, C. 207Pb The Relationship between 207Pb NMR Chemical Shift and Solid-State Structure in Pb(II) Compounds. J. Phys. Chem. A 2008, 112, 3046–3052. [Google Scholar] [CrossRef] [PubMed]
- Senocrate, A.; Moudrakovski, I.; Kim, G.Y.; Yang, T.Y.; Gregori, G.; Grätzler, M.; Maier, J. The Nature of Ion Conduction in Methylammonium Lead Iodide: A Multimethod Approach. Angew. Chem. Int. Ed. 2017, 56, 7755–7759. [Google Scholar] [CrossRef] [PubMed]
- Hanrahan, M.P.; Men, L.; Rosales, B.A.; Vela, J.; Rossini, A.J. Sensitivity-Enhanced 207Pb Solid-State NMR Spectroscopy for the Rapid, Non-Destructive Characterization of Organolead Halide Perovskites. Chem. Mater. 2018, 30, 7005–7015. [Google Scholar] [CrossRef]
- Senocrate, A.; Moudrakovski, I.; Maier, J. Short-range ion dynamics in methylammonium lead iodide by multinuclear solid state NMR and 127I NQR. Phys. Chem. Chem. Phys. 2018, 20, 20043–20055. [Google Scholar] [CrossRef] [PubMed]
- Charpentier, T. The PAW/GIPAW approach for computing NMR parameters: A new dimension added to NMR study of solids. Solide State Nucl. Magn. Reson. 2011, 40, 1–20. [Google Scholar] [CrossRef]
- Alkan, F.; Dybowski, C. Chemical-shift tensors of heavy nuclei in network solids: A DFT/ZORA investigation of 207Pb chemical-shift tensors using the bond-valence method. Phys. Chem. Chem. Phys. 2015, 17, 25014–25026. [Google Scholar] [CrossRef]
- Mehring, M. Principles of High Resolution NMR in Solids; Springer: New York, NY, USA, 1983; ISBN 3-540-11852-7. [Google Scholar]
- Haeberlen, U. High Resolution NMR in Solids: Selective Averaging. In Advances in Magnetic Resonance; Waugh, J., Ed.; Academic Press: New York, NY, USA, 1976; ISBN 0-12-025561-8. [Google Scholar]
- Laws, D.D.; Bitter, H.-M.L.; Jerschow, A. Solid-State NMR Spectroscopic Methods in Chemistry. Angew. Chem. Int. Ed. 2002, 41, 3096–3129. [Google Scholar] [CrossRef]
- Anet, F.A.L.; O’Leary, D.J. The shielding tensor. Part I: Understanding its symmetry properties. Concepts Magn. Reson. 1991, 3, 193–214. [Google Scholar] [CrossRef]
- Rose, M.E. Elementary Theory of Angular Momentum; John Wiley & Sons: New York, NY, USA, 1957; ISBN 0-486-68480-6. [Google Scholar]
- Millot, Y.; Man, P.P. Active and passive rotations with Euler angles in NMR. Concepts Magn. Reson. 2012, 40A, 215–252. [Google Scholar] [CrossRef]
- Greer, G.J.; Michaelis, V.K.; Katz, M.J.; Leznoff, D.B.; Schreckenbach, G.; Kroeker, S. Characterising lone-pair activity of lead(II) by 207Pb solid-state NMR spectroscopy: coordination polymers of [N(CN)2]− and [Au(CN)2]− with terpyridine ancillary ligands. Chem. Eur. J. 2011, 17, 3609–3618. [Google Scholar] [CrossRef] [PubMed]
- Bielecki, A.; Burum, D.P. Temperature dependence of 207Pb MAS spectra of solid lead nitrate: An accurate, sensitive thermometer for variable-temperature MAS. J. Magn. Reson. A 1995, 116, 215–220. [Google Scholar] [CrossRef]
- Van Gorkom, L.C.M.; Hook, J.M.; Logan, M.B.; Hanna, J.V.; Wasylishen, R.E. Solid-state lead-207 NMR of lead(II) nitrate: Localized heating effects at high magic angle spinning speeds. Magn. Reson. Chem. 1995, 33, 791–795. [Google Scholar] [CrossRef]
- Milder, T.; Ernst, H.; Freude, D. 207Pb NMR detection of spinning-induced temperature gradients in MAS rotors. Solid State Nucl. Magn. Reson. 1995, 5, 269–271. [Google Scholar] [CrossRef]
- Hoppe, R. Effective coordination numbers (ECoN) and mean Active ionic radii (MEFIR). Z. Kristallog. Cryst. Mater. 1979, 150, 23–52. [Google Scholar] [CrossRef]
- Leciejewicz, J. On the crystal structure of tetragonal (red) PbO. Acta Crystallogr. 1961, 14, 1304. [Google Scholar] [CrossRef]
- Hill, R.J. Refinement of the structure of orthorhombic PbO (massicot) by Rietveld analysis of neutron powder diffraction data. Acta Crystallogr. 1985, C41, 1281–1284. [Google Scholar] [CrossRef]
- Boucher, M.L.; Peacor, D.R.Z. The crystal structure of alamosite, PbSiO3. Krist 1968, 126, 98–111. [Google Scholar] [CrossRef]
- Gavarri, J.R.; Weigel, D.J. Oxydes de plomb. I. Structure cristalline du minium Pb3O4, á température ambiante (293 K). Solid State Chem. 1975, 13, 252–257. [Google Scholar] [CrossRef]
- Nelmes, R.J.; Kuhs, W.F. The crystal structure of tetragonal PbTiO3 at room temperature and at 700 K. Solid State Commun. 1985, 54, 721–723. [Google Scholar] [CrossRef]
- Okudera, H. Relationships among channel topology and atomic displacements in the structures of Pb5(BO4)3Cl with B = P (pyromorphite), V (vanadinite), and As (mimetite). Am. Mineral. 2013, 98, 1573–1579. [Google Scholar] [CrossRef]
- Moreau, J.M.; Galez, P.; Peigneux, J.P.; Korzhik, M.V. Structural characterization of PbWO4 and related new phase Pb7W8O(32−x). J. Alloy. Compd. 1996, 238, 46–48. [Google Scholar] [CrossRef]
- Nolle, A. 207Pb Magnetic Shielding Anisotropy in Pb(NO3)2, PbCO3, PbCrO4, PbMoO4 and PbWO4 by Fourier Transform NMR. Z. Naturforsch. 1977, 32a, 964–967. [Google Scholar] [CrossRef]
- Sleight, A.W. Accurate cell dimensions for ABO4 molybdates and tungstates. Acta Crystallogr. 1972, B28, 2899–2902. [Google Scholar] [CrossRef]
- Effenberger, H.; Pertlik, F. Four monazite type structures: Comparison of SrCrO4, SrSeO4, PbCrO4(crocoite), and PbSeO4. Z. Kristallogr. 1986, 176, 75–83. [Google Scholar] [CrossRef]
- Basso, R.; Lucchetti, G.; Zefiro, L.; Palenzona, A. Rosiaite, PbSb2O6, a new mineral from the Cetine mine, Siena, Italy. Eur. J. Mineral. 1996, 8, 487–492. [Google Scholar] [CrossRef]
- Yoko, T.; Tadanaga, K.; Miyaji, F.; Sakka, S. A 207Pb MAS-NMR study of Pb-containing glasses. J. Non-Cryst. Solids 1992, 150, 192–196. [Google Scholar] [CrossRef]
- Chevrier, G.; Giester, G.; Heger, G.; Jarosch, D.; Wildner, M.; Zemann, J. Neutron single-crystal refinement of cerussite, PbCO3, and comparison with other aragonite-type carbonates. Z. Kristallog. Cryst. Mater. 1992, 199, 67–74. [Google Scholar] [CrossRef]
- Brusset, H.; Gillier-Pandraud, H.; Mahe, R. Structure cristalline du metaniobate de plomb rhomboedrique. C. R. Acad. Sci. C 1966, 263, 217–219. [Google Scholar]
- Zhao, P.; Prasad, S.; Huang, J.; Fitzgerald, J.J.; Shore, J.S. Lead-207 NMR Spectroscopic Study of Lead-Based Electronic Materials and Related Lead Oxides. J. Phys. Chem. B 1999, 103, 10617–10626. [Google Scholar] [CrossRef]
- Pecher, O.; Halat, D.M.; Lee, J.; Liu, Z.; Griffith, K.J.; Braun, M.; Grey, C.P. Enhanced efficiency of solid-state NMR investigations of energy materials using an external automatic tuning/matching (eATM) robot. J. Magn. Reson. 2017, 275, 127–136. [Google Scholar] [CrossRef] [PubMed]
- Kunwar, A.C.; Turner, G.L.; Oldfield, E. Solid-state spin-echo Fourier transform NMR of 39K and 67Zn salts at high field. J. Magn. Reson. 1986, 69, 124–127. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Single-Crystal NMR | MAS NMR | |
|---|---|---|
| ppm | ppm | |
| ppm | ppm | |
| ppm | ppm | |
| 0.00, 0.00 | ||
| 90.00, 65.59 | ||
| 90.00, 155.59 | ||
| ppm | ppm | |
| ppm | ||
| ppm | ppm |
| Mineral | Space Group (Reference) | Pb Coord. No. | Pb–O Mean Bond Length (nm) | Pb–O Shortest Bond Length (nm) | (ppm) | NMR Reference |
|---|---|---|---|---|---|---|
| red PbO [Litharge] | [41] | 4 | 0.2309 | 0.2309 | 1939 ± 5 | [22] |
| yellow PbO [Massicot] | [42] | 4 | 0.2358 | 0.2221 | 1515 ± 5 | [22] |
| PbSiO: Pb(1) [Alamosite] Pb(2) [Alamosite] Pb(3) | [43] | 5 5 6 | 0.2518 0.2492 0.2633 | 0.2291 0.2256 0.2299 | ± 5 93 ± 5 −366 ± 5 | [22] [22] [22] |
| PbO: Pb(1) [Minium] Pb(2) | [44] | 6 4 | 0.2176 0.2374 | 0.2131 0.2215 | −1105 ± 1 795 ± 2 | [22] [22] |
| PbTiO [Macedonite] | [45] | 12 (8+4) | 0.2846 | 0.2517 | −1395.1 ± 8.4 | [6] |
| Pb(VO)Cl: Pb(1) [Vanadinite] Pb(2) | [46] | 9 6 | 0.2719 0.2664 | 0.2483 0.2324 | −1619 ± 2 −1729 ± 9 | [17] [17] |
| PbWO [Stolzite] | [47] | 8 | 0.2596 | 0.2579 | −2003 | [48] |
| PbMoO [Wulfenite] | [49] | 8 | 0.2657 | 0.2608 | −2009 ± 2 | [22] |
| PbCrO [Crocoite] | [50] | 8 | 0.2634 | 0.2532 | −2236.2 ± 2.9 | [6] |
| PbSbO [Rosiaite] | [51] | 6 | 0.2554 | 0.2554 | −2581 ± 3 | [52] |
| PbCO [Cerussite] | [53] | 9 | 0.2692 | 0.2594 | −2630 ± 2 | [22] |
| Pb(PO)Cl: Pb(1) [Pyromorphite] Pb(2) | [46] | 9 6 | 0.2710 0.2761 | 0.2556 0.2353 | −2810 ± 14 −2172 ± 11 | [16] [16] |
| PbNbO: Pb(1) [Changbaiite] Pb(2) [Changbaiite] Pb(3) | [54] | 6 6 (3+3) 12 (6+6) | 0.2577 0.2962 0.2738 | 0.2560 0.2571 0.2343 | −2829 −2829 134 | [55] [55] [55] |
| PbSO [Anglesite] | [20] | 10 | 0.2785 | 0.2612 | −3615 ± 3 | this work |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zeman, O.E.O.; Steinadler, J.; Hochleitner, R.; Bräuniger, T. Determination of the Full 207Pb Chemical Shift Tensor of Anglesite, PbSO4, and Correlation of the Isotropic Shift to Lead–Oxygen Distance in Natural Minerals. Crystals 2019, 9, 43. https://doi.org/10.3390/cryst9010043
Zeman OEO, Steinadler J, Hochleitner R, Bräuniger T. Determination of the Full 207Pb Chemical Shift Tensor of Anglesite, PbSO4, and Correlation of the Isotropic Shift to Lead–Oxygen Distance in Natural Minerals. Crystals. 2019; 9(1):43. https://doi.org/10.3390/cryst9010043
Chicago/Turabian StyleZeman, Otto E. O., Jennifer Steinadler, Rupert Hochleitner, and Thomas Bräuniger. 2019. "Determination of the Full 207Pb Chemical Shift Tensor of Anglesite, PbSO4, and Correlation of the Isotropic Shift to Lead–Oxygen Distance in Natural Minerals" Crystals 9, no. 1: 43. https://doi.org/10.3390/cryst9010043
APA StyleZeman, O. E. O., Steinadler, J., Hochleitner, R., & Bräuniger, T. (2019). Determination of the Full 207Pb Chemical Shift Tensor of Anglesite, PbSO4, and Correlation of the Isotropic Shift to Lead–Oxygen Distance in Natural Minerals. Crystals, 9(1), 43. https://doi.org/10.3390/cryst9010043