
Quasicrystals and Other Aperiodic Structures in Mineralogy



Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
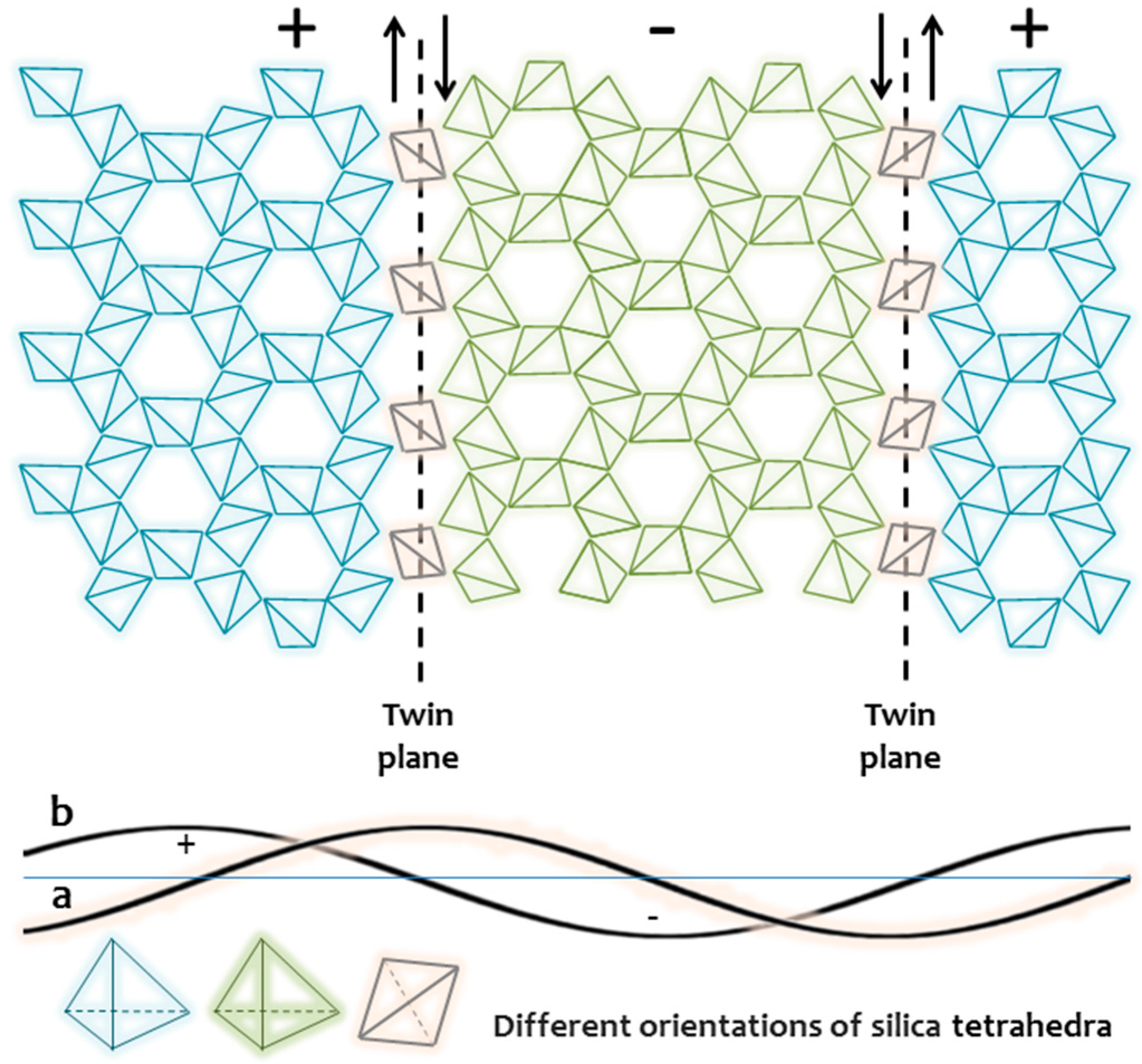{kind=link}
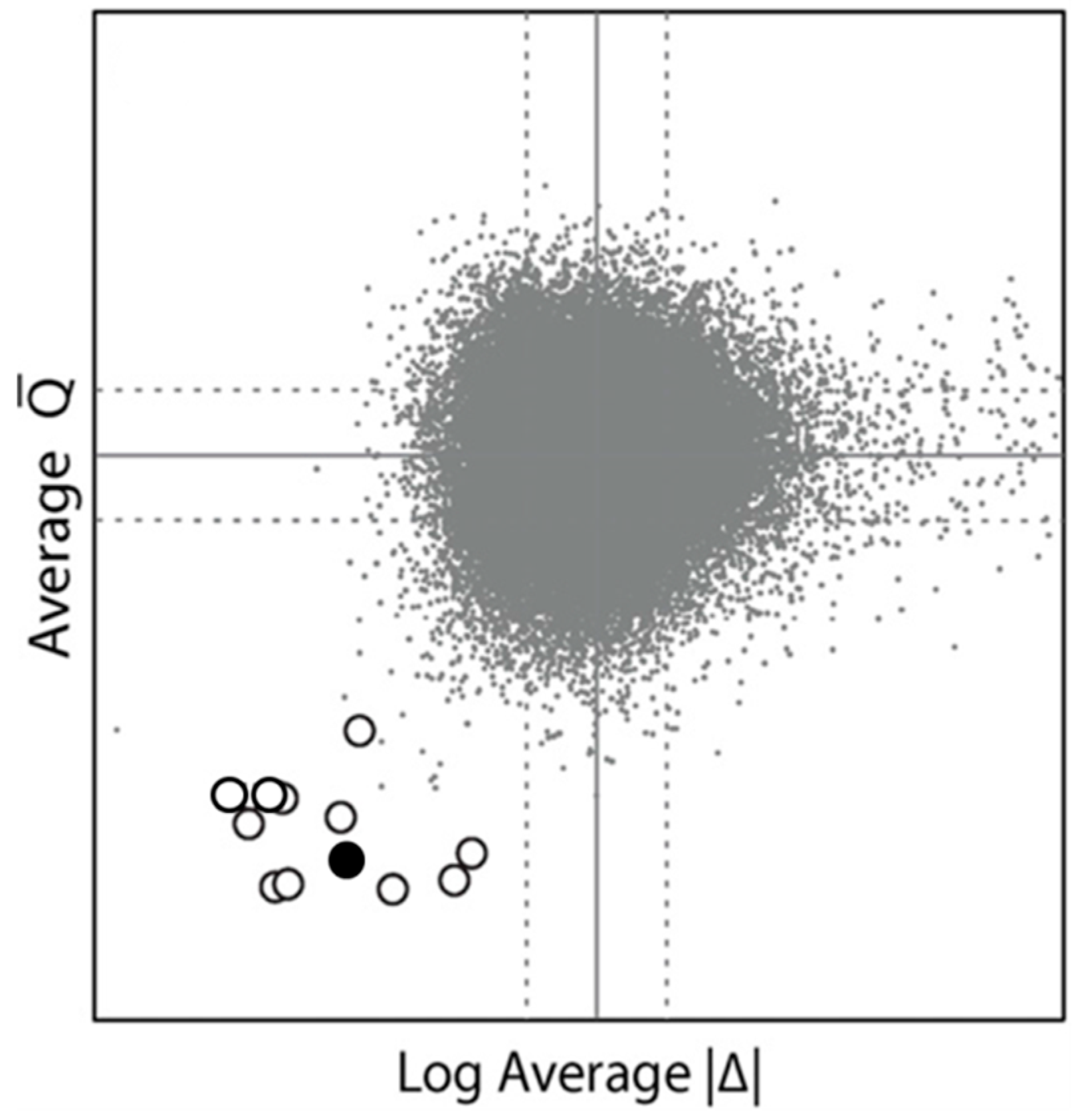{kind=link}
{kind=link}
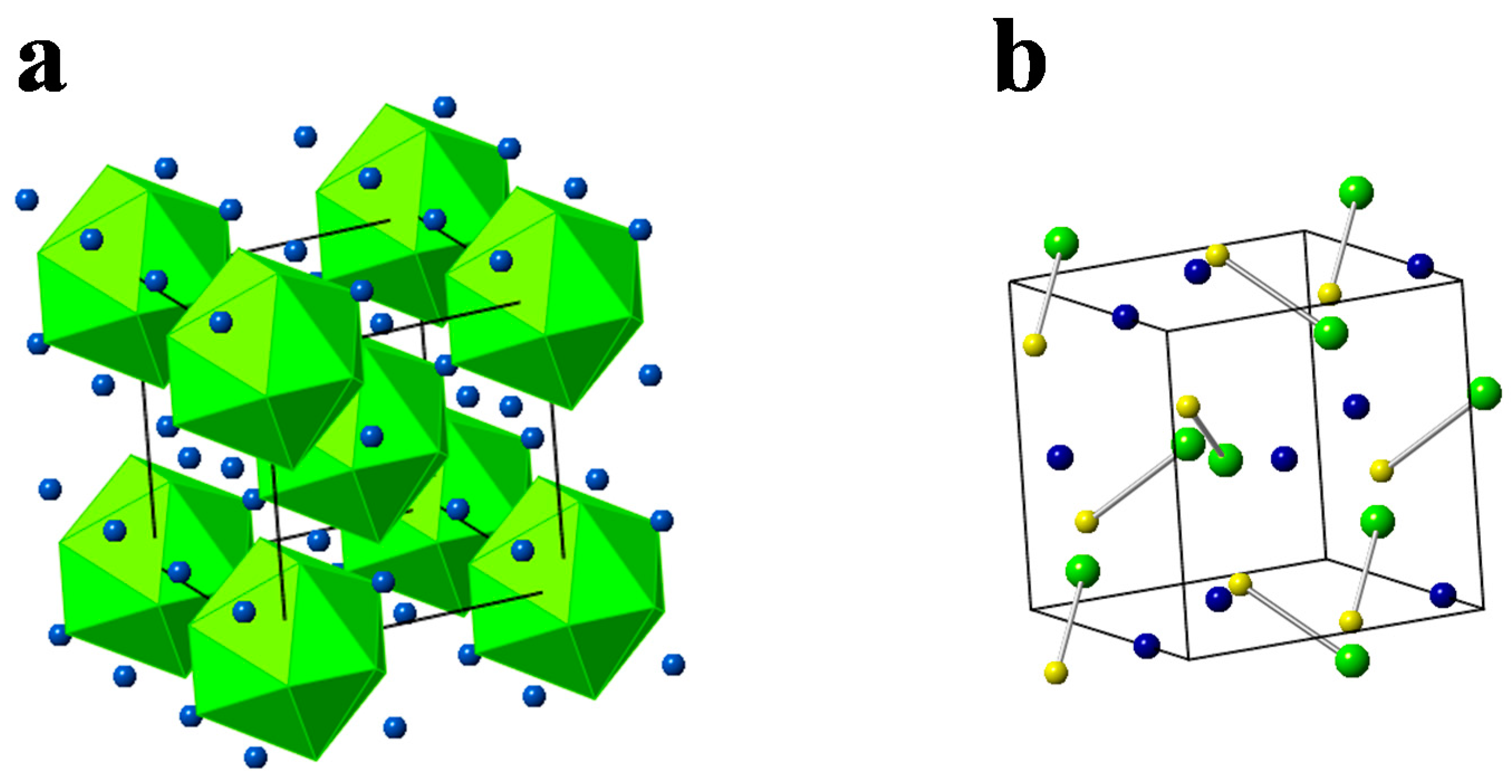{kind=link}
{kind=link}
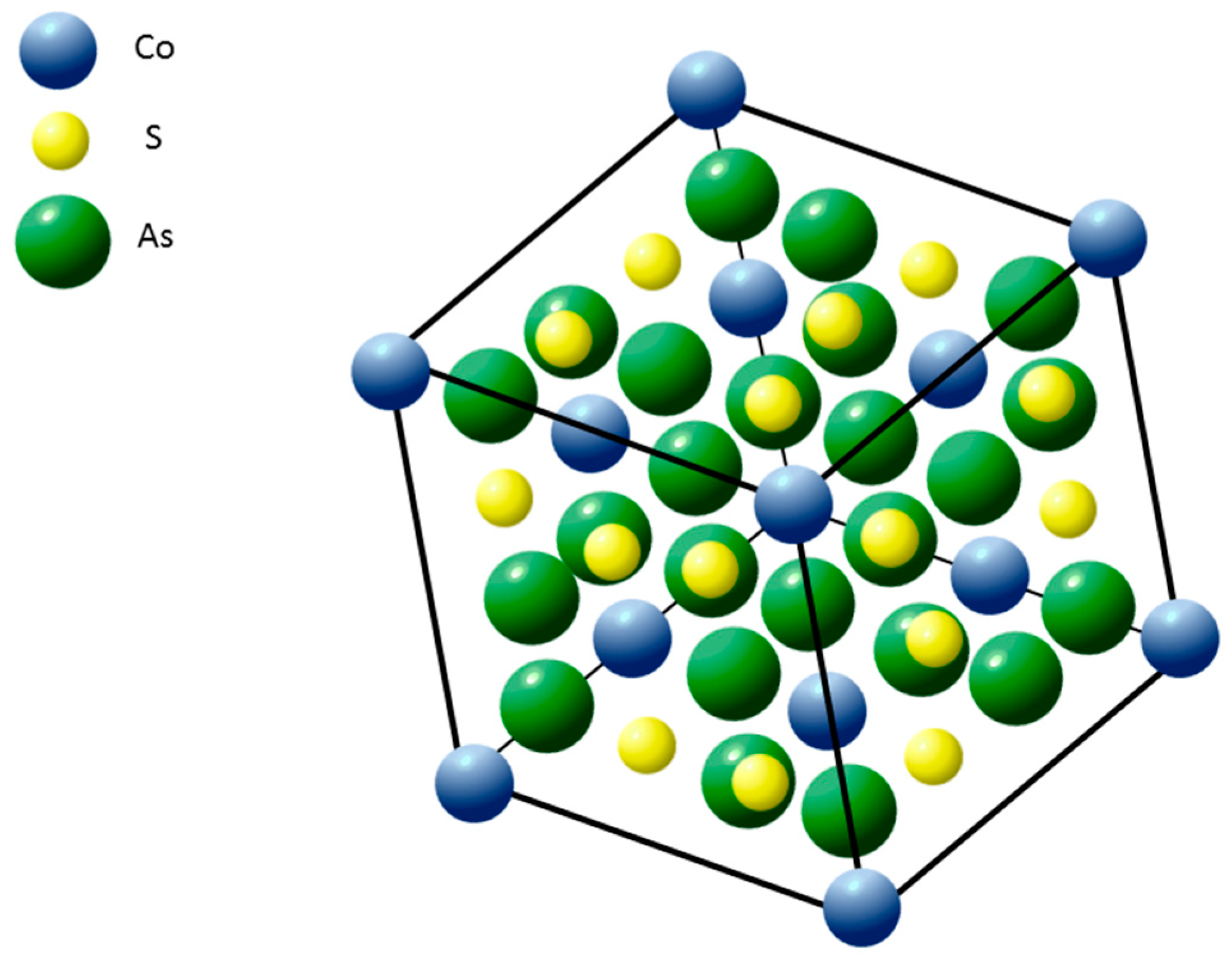{kind=link}
1. Introduction
2. Before Crystallography
3. The Crystallographic Paradigm and “Dissident” Minerals
4. Quasicrystals and Minerals
5. Conclusions
Acknowledgements
Author Contributions
Conflicts of Interest
References
- Romé de L’Isle, J.B. Cristallographie, ou Descriptions des Formes Propres à Tous les Corps du Règne Mineral, Dans l’état de Combinaison Saline, Pierreuse, ou Métallique, 2ème ed.; L’Imprimerie de Monsieur: Paris, France, 1783. (In French) [Google Scholar]
- Haüy, R.J. Traité de Minéralogie; des Mines, G., Ed.; Chez Louis: Paris, France, 1801; Volume IV et V. (In French) [Google Scholar]
- Bravais, A. Mémoire sur les Sytèmes Formés par des Points Distribués Régulièrement sur un Plan ou dans L’espace. J. de l’École Polytechnique Paris. 1850, 19, 1–128. (In French) [Google Scholar]
- Fedorov, E.S. Symmetrie der Regelmässigen Systeme von Figuren. In Verhandlungen der Russisch-Kaiserlichen Mineralogischen Gesellschaft zu St. Petersburg; St. Petersburg: Saint Petersburg, Russia, 1891, 2, 1–145. (In German)
- Schoenflies, A. Kristallsysteme und Kristallstructur; B.G. Teubner: Leipzig, Germany, 1891. (In German) [Google Scholar]
- Barlow, W. Probable Nature of the Internal Symmetry of Crystals. Nature 1883, 29, 186–188. [Google Scholar] [CrossRef]
- Friedrich, W.; Knipping, P.; Laue, M. Interferenz-Erscheinungen bei Röntgenstrahlen. Annalen der Physik 1913, 346, 971–988. [Google Scholar] [CrossRef]
- Bragg, W.H.; Bragg, W.L. The Reflexion of X-rays by Crystals. Proc. R. Soc. Lond. A 1913, 88, 428–438. [Google Scholar] [CrossRef]
- Shechtman, D.S.; Blech, I.; Gratias, D.; Cahn, J.W. Metallic phase with long-range orientational order and no translational symmetry. Phys. Rev. Lett. 1984, 53, 1951–1954. [Google Scholar] [CrossRef]
- International Union of Crystallography. Report of the Executive Committe for 1991. Acta Cryst. 1992, A48, 922–946. [Google Scholar]
- Bindi, L.; Steinhardt, P.J.; Yao, N.; Lu, P.J. Icosahedrite, Al63Cu24Fe13, the first natural quasicrystal. Am. Min. 2011, 96, 928–931. [Google Scholar] [CrossRef]
- Bindi, L.; Yao, N.; Lin, C.; Hollister, L.S.; Andronicos, C.L.; Distler, V.V.; Eddy, M.P.; Kostin, A.; Kryachko, V.; MacPherson, G.J.; et al. Natural quasicrystal with decagonal Symmetry. Sci. Rep. 2015, 9111. [Google Scholar] [CrossRef] [PubMed]
- Bindi, L.; Yao, N.; Lin, C.; Hollister, L.S.; Andronicos, C.L.; Distler, V.V.; Eddy, M.P.; Kostin, A.; Kryachko, V.; MacPherson, G.J. Decagonite, Al71Ni24Fe5, a quasicrystal with decagonal symmetry from the Khatyrka CV3 carbonaceous chondrite. Am. Min. 2015, 100, 2340–2343. [Google Scholar] [CrossRef]
- Boisen, M.B., Jr.; Gibbs, G.V. Mathematical Crystallography. In Reviews in Mineralogy; Ribbe, P.H., Ed.; Mineralogical Society of America: Chantilly, VA, USA, 1985; Volume 15, pp. 191–194. [Google Scholar]
- Gévay, G.; Szederkény, T. Quasicrystals and their spontaneous formation possibilities in the nature. Acta Miner. Petrogr. 1988, XXIX, 5–12. [Google Scholar]
- Asimow, P.D.; Lin, C.; Bindi, L.; Ma, C.; Tschauner, O.; Hollister, L.S.; Steinhard, P.J. Shock synthesis of quasicrystals with implications for their origin in asteroid collisions. PNAS 2016, 113, 7077–7081. [Google Scholar] [CrossRef] [PubMed]
- Pliny the Elder. Natural History, Volume X: Book 37; Eichholz, D.E., Translator; Loeb Classical Library 419; Harvard University Press: Cambridge, MA, USA, 1962. [Google Scholar]
- Agricola, G. De Natura Fossilium (Textbook of Mineralogy); Translated from the first Latin of 1546 by Bandy, M.C. and Bandy M.; Courier Corporation: New York, NY, USA, 2004. [Google Scholar]
- Palissy, B. Discours admirables; La Rocque, Aurèle, Translator; Urbana University of Illinois Press: Urbana, IL, USA, 1957. [Google Scholar]
- Steno, N. De Solido intra Solidum Naturaliter Contento. Dissertationis Prodromus, 1ª ed.; Sub Signu Stellae. Florencia 1669; Sequeiros, L., Translator; ECT. AEPECT. 2002, 10, 243–283. (In Spanish)
- Romé de L’Isle, J.B. Essai de Cristallographie: ou, Description des figures Géométriques, Propres à Différens Corps du Regne Mineral Connus Vulgairement sous le nom de Cristaux; Chez Didot Jeune: Paris, France, 1772. (In French) [Google Scholar]
- Démeste, J. Lettres du Docteur Démeste au Docteur Bernard. Sur la Chymie, la Docimasie, la Cristallographie, la Lithologie, la Minéralogie et la Physique en général; Chez: Didot, Imprimeur de Monsieur, et Clousier, Imprimeur & Libraire: Paris, France, 1779; Volume 2. (In French) [Google Scholar]
- Romé de L’Isle, J.B. Catalogue Systématique et Raisonné des Curiosités de la Nature et de l’art qui Composent le Cabinet de M. Dávila; Chez Briasson: Paris, France, 1767; Volume 2. (In French) [Google Scholar]
- Rao, K.R.M.; Rao, P.H. Elasticity and piezomagnetism in pentagonal and icosahedral quasi-crystals: A group-theoretical study. J. Phys. Condens. Matter 1992, 4, 5997. [Google Scholar] [CrossRef]
- Rao, K.R.M.; Rao, P.H.; Chaitanya, B.S.K. Piezoelectricity in quasicrystals: A group-theoretical study. Pramana J. Phys. 2007, 68, 481–487. [Google Scholar]
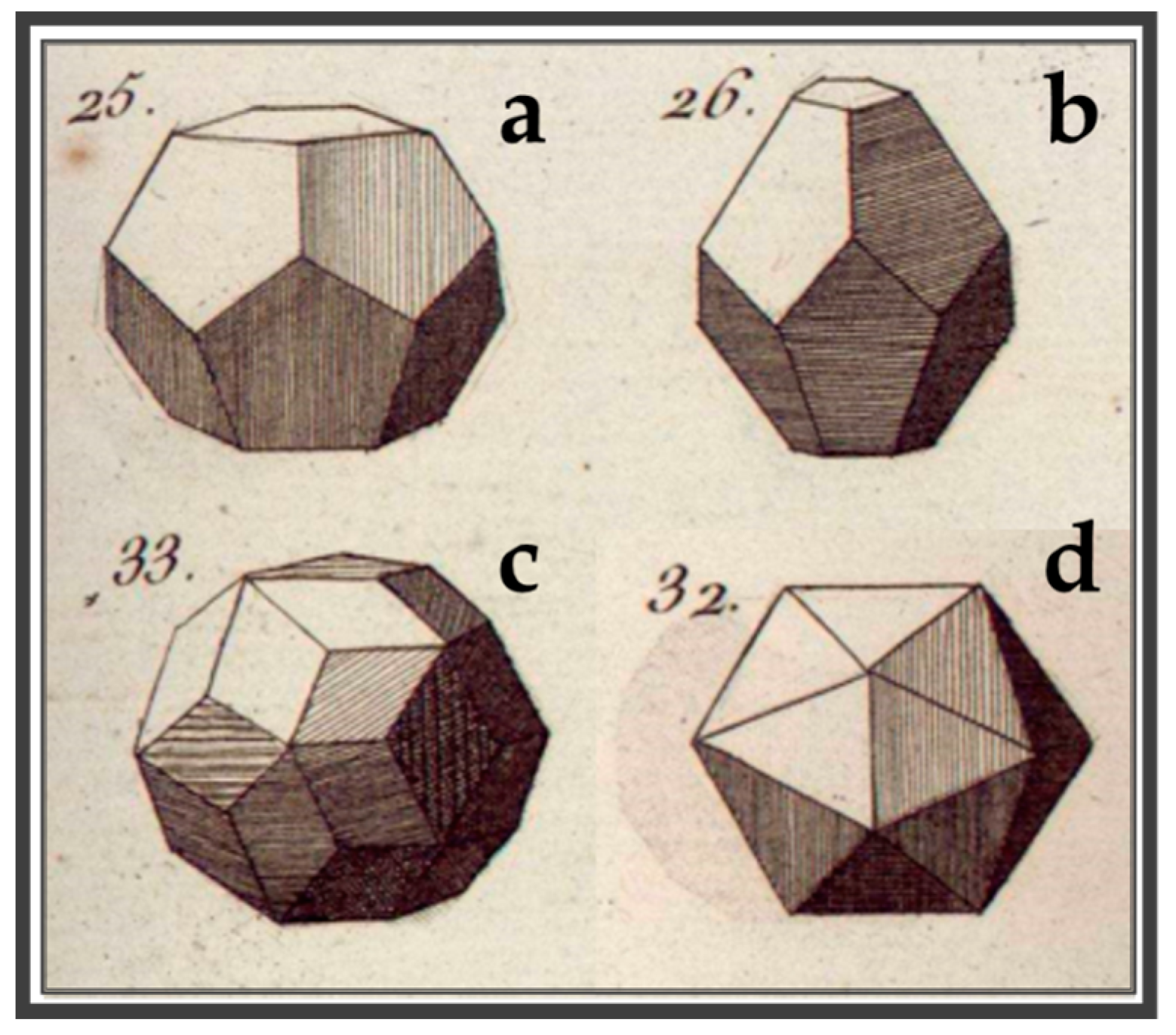
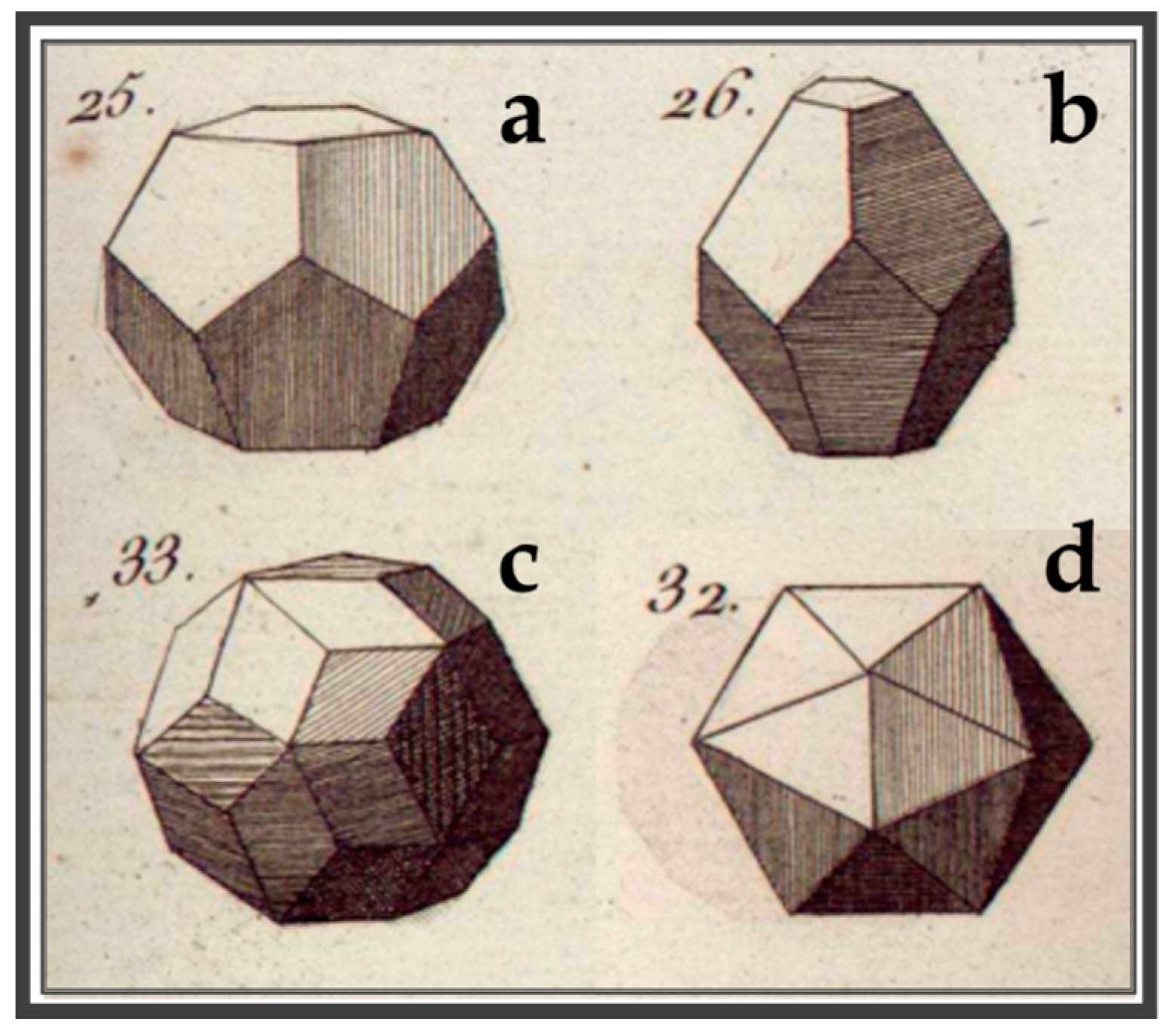
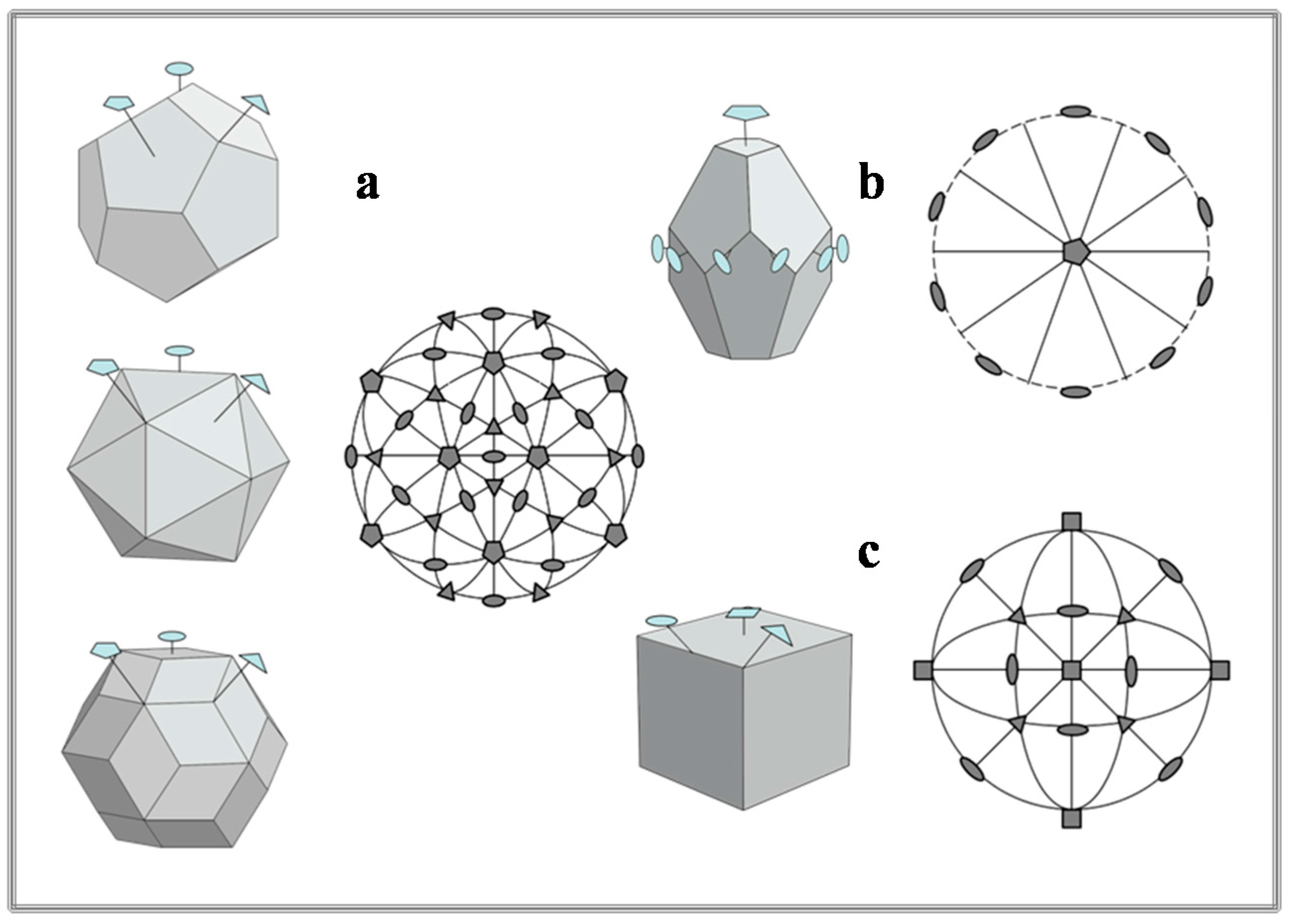
- Pina, C.M.; López-Acevedo, V. Eighteenth-century forms of quasicrystals. Acta Cryst. 2016, A72, 81–84. [Google Scholar] [CrossRef] [PubMed]
- Tsai, A.P.; Inoue, A.; Masumoto, T. A stable quasicrystal in Al-Cu-Fe system. Jpn. J. Appl. Phys. 1987, 26, L1505–L1507. [Google Scholar] [CrossRef]
- Jamshidi, A.L.C.L.; Nascimento, L.; Rodbari, R.J.; Barbosa, G.F.; Machado, F.L.A.; Pacheco, J.G.A.; Barbosa, C.M.B.M.J. Use alloy quasicrystalline Al62.2Cu25.3Fe12.5 for steam reforming of methanol. Chem. Eng. Process Technol. 2014, 5, 187. [Google Scholar]
- Van Blaaderen, A. Materials science: Quasicrystals from nanocrystals. Nature 2009, 461, 892–893. [Google Scholar] [CrossRef] [PubMed]
- Mackay, A. Quasicrystals turn to the sixth-dimension. Nature 1990, 344, 21. [Google Scholar] [CrossRef] [PubMed]
- Des Cloizeaux, A. Manuel de Minéralogie; Dunod: Pairs, France, 1862. (In French) [Google Scholar]
- De Lapparent, A. Cours de Minéralogie; Librairie F. Savy: Pairs, France, 1884. (In French) [Google Scholar]
- De Lapparent, A. Cours de Minéralogie, 3ème ed; Masson et Cie.: Pairs, France, 1899. (In French) [Google Scholar]
- Hessel, J.F.C. Krystallometrie oder Krystallonomie und Krystallographie. In Gehler’s Physikalisches Wörterbuch; W. Engelmann: Leipzig, Germany, 1830; pp. 1023–1340. (In German) [Google Scholar]
- Hessel, J.F.C. Krystallometrie, Oder Krystallonomie und Krystallographie; W. Engelmann: Leipzig, Germany, 1897. (In German) [Google Scholar]
- Bragg, W.H.; Bragg, W.L. X rays and Crystal Structure; G. Bell and Sons Ltd.: London, UK, 1915. [Google Scholar]
- Bragg, W.L. The History of W-ray Analysis; Longman Green and Company: London, UK, 1943. [Google Scholar]
- Authier, A. Early Days of X-ray Crystallography. In International Union of Crystallography. Texts on Crystallography; OUP: Oxford, UK, 2013. [Google Scholar]
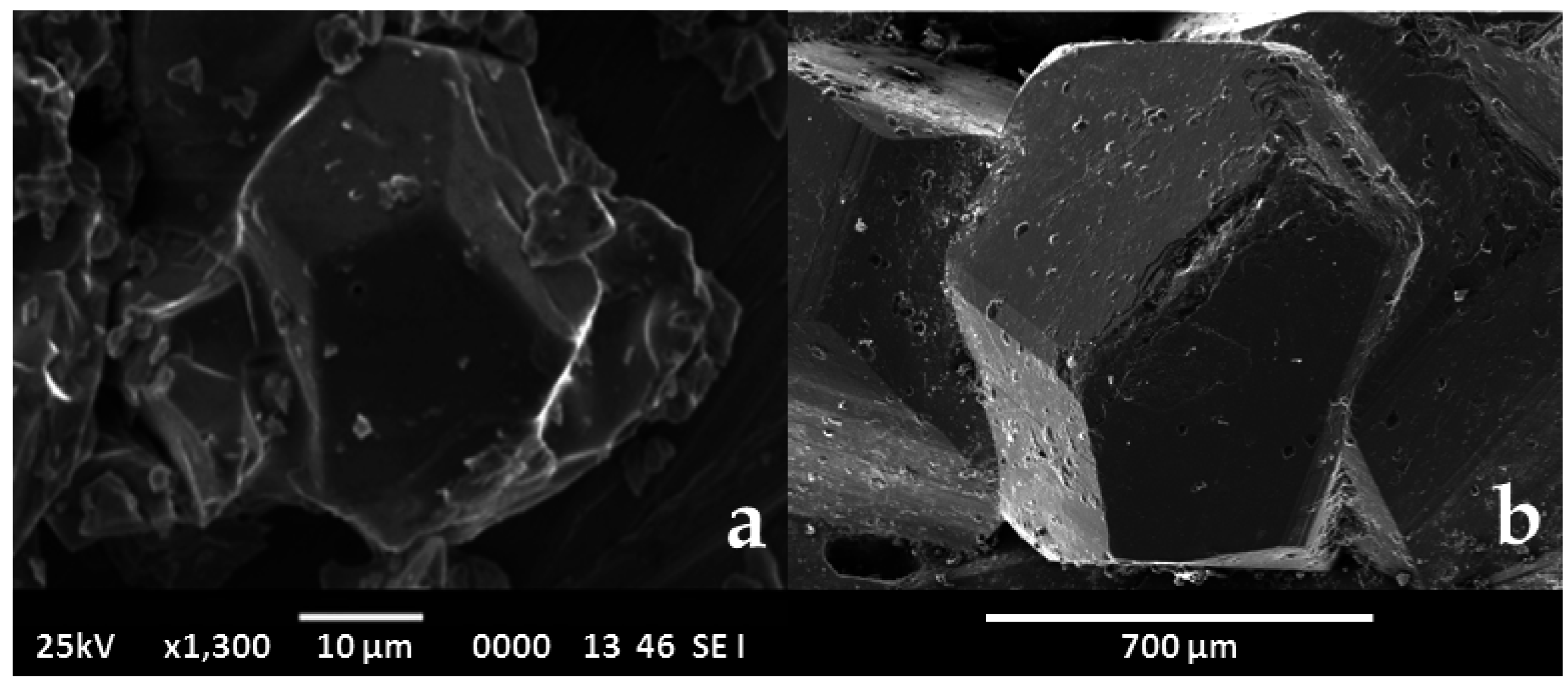
- Penfield, S.L.; Ford, W.E. On calaverite. Am J. Sci. 1901, 4–12, 225–246. [Google Scholar] [CrossRef]
- Goldschmidt, V.M.; Palache, C.; Peacock, M. Über calaverite. Neues Jahrbuch. Miner. Geol. Paleo. 1931, 63, 1–15. [Google Scholar]
- Sueno, S.; Kimata, M.; Ohmasa, M. The atom splitting and modulated structure in calaverite. In Modulated Structures; Cowley, J.M., Cohen, J.B., Salamon, M.B., Wuensch, B.J., Eds.; AIP Conference Proceedings: New York, NY, USA, 1979; pp. 331–349. [Google Scholar]
- Van Tendeloo, G.; Gregoriades, P.; Amenlinckx, S. Electron microscopic studies of modulated structures in (Au, Ag)Te2: Part I Calaverite AuTe2. J. Solid State Chem. 1983, 50, 321–334. [Google Scholar] [CrossRef]
- Maciá-Barber, E. Thermoelectric Materials: Advances and Applications; Pan Stanford Publishing Pte. Ltd.: Singapore, 2015. [Google Scholar]
- Gómez Herrero, A. Estructuras Moduladas en Calcogenuros Ternarios. Ph.D Thesis, Complutense University of Madrid, Mayo, Spain, 2012. [Google Scholar]
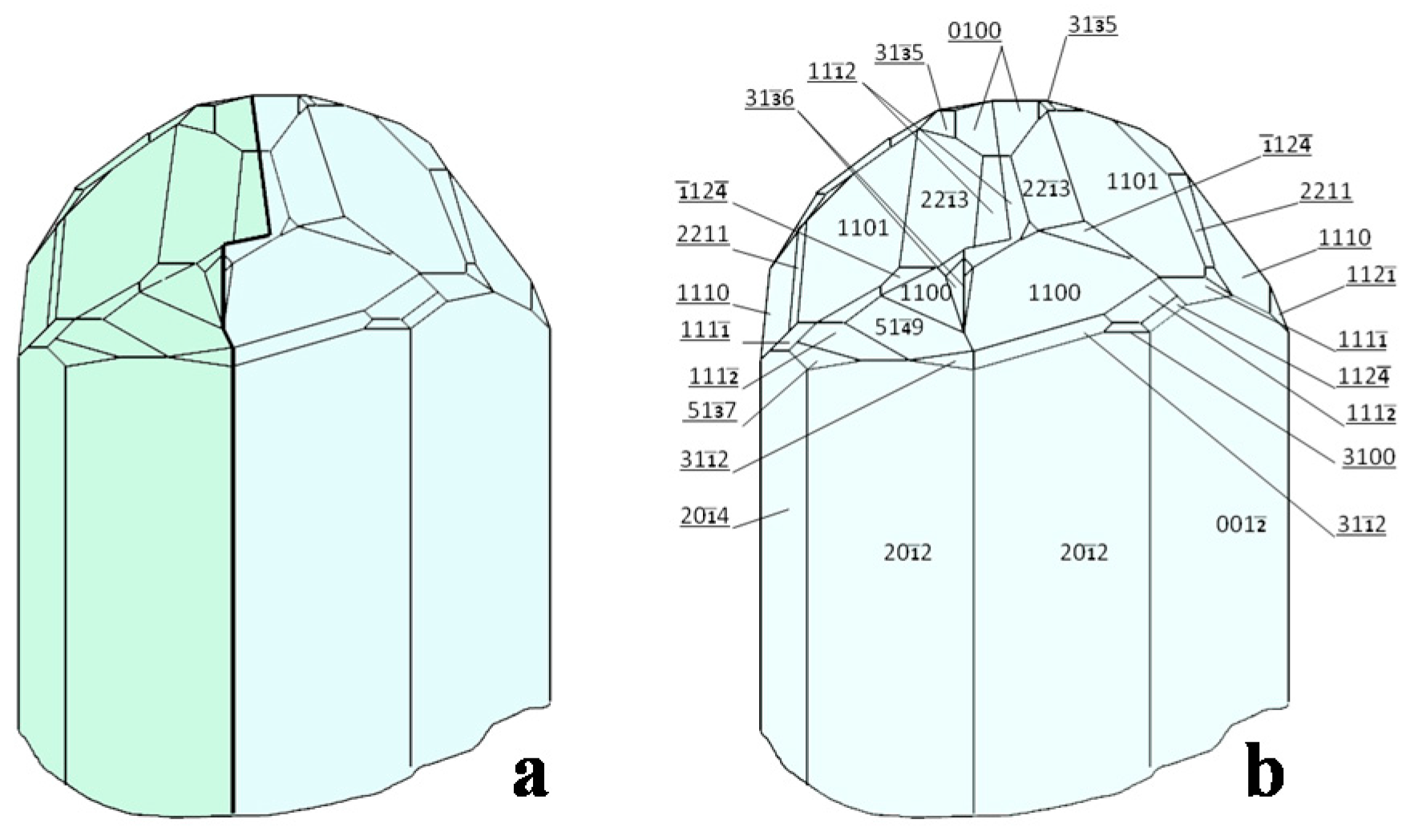
- Janner, A.; Dam, B. The Morphology of Calaverite (AuTe2) from Data of 1931. Solution of an Old Problem of Rational Indices. Acta Cryst. 1989, A45, 115–123. [Google Scholar] [CrossRef]
- Chapuis, G. Crystallographic excursion in superspace. Cryst. Eng. 2003, 6, 187–195. [Google Scholar] [CrossRef]
- Putnis, A.; McConnell, J.D.C. Principles of Mineral Behaviour; Springer: New York, NY, USA, 1980. [Google Scholar]
- Putnis, A. Introduction to Mineral Sciences; Cambridge University Press: Cambridge, UK, 1992. [Google Scholar]
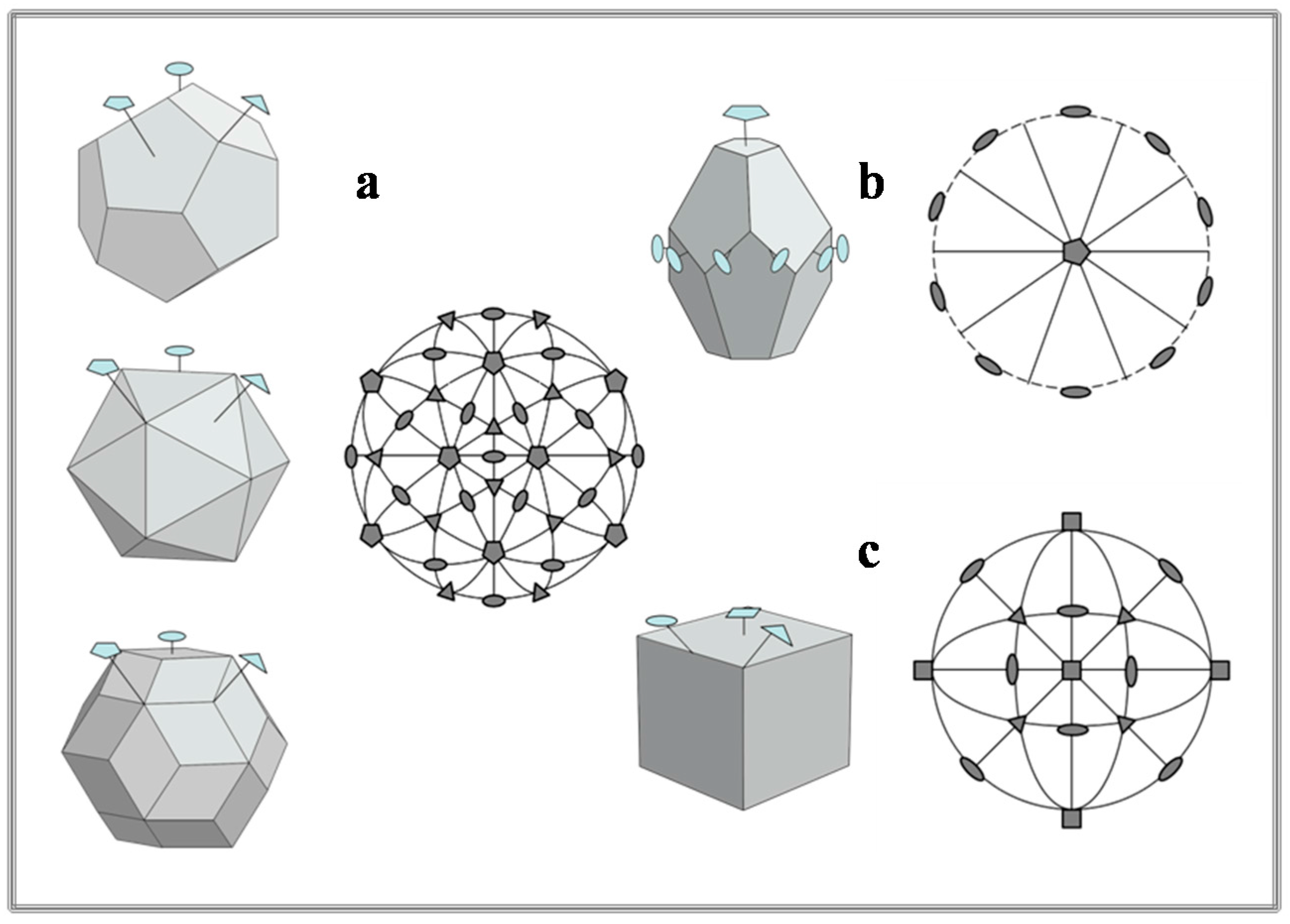
- Abe, E.; Yan, Y.; Pennycook, S.J. Quasicrystals as cluster aggregates. Nat. Mater. 2004, 3, 759–767. [Google Scholar] [CrossRef] [PubMed]
- Steinhardt, P.J. Quasicrystals: A brief history of the impossible. Rend. Lincei 2013, 24, 85–91. [Google Scholar] [CrossRef]
- ICDD. Available online: http://www.icdd.com/ (accessed on 15 September 2016).
- Lu, P.J.; Deffeyes, K.; Steinhardt, P.J.; Yao, N. Identifying and Indexing Icosahedral Quasicrystals from Powder Diffraction Patterns. Phys. Rev. Lett. 2001, 87, 55071–55074. [Google Scholar] [CrossRef] [PubMed]
- Levine, D.; Steinhardt, P.J. Quasicrystals. I. Definition and structure. Phys. Rev. B. 1986, 34, 596–616. [Google Scholar]
- Bindi, L.; Steinhardt, P.J.; Yao, N.; Lu, P.J. Natural Quasicrystals. Science 2009, 324, 1306–1309. [Google Scholar] [CrossRef] [PubMed]
- Bindi, L.; Eiler, J.M.; Guan, Y.; Hollister, L.S.; MacPherson, G.; Steinhardt, P.J.; Yao, N. Evidence for the extraterrestrial origin of a natural quasicrystal. PNAS 2012, 109, 1396–1401. [Google Scholar] [CrossRef] [PubMed]
- Perkins, R. Natural Quasicrystals May be the Result of Collisions between Objects in the Asteroid Belt. California Institute of Technology, 2016. Available online: http://phys.org/news/2016-06-natural-quasicrystals-result-collisions-asteroid.html (accessed on 15 September 2016).
- Mindat.org. Available online: http://www.mindat.org/ (accessed on 15 September 2016).
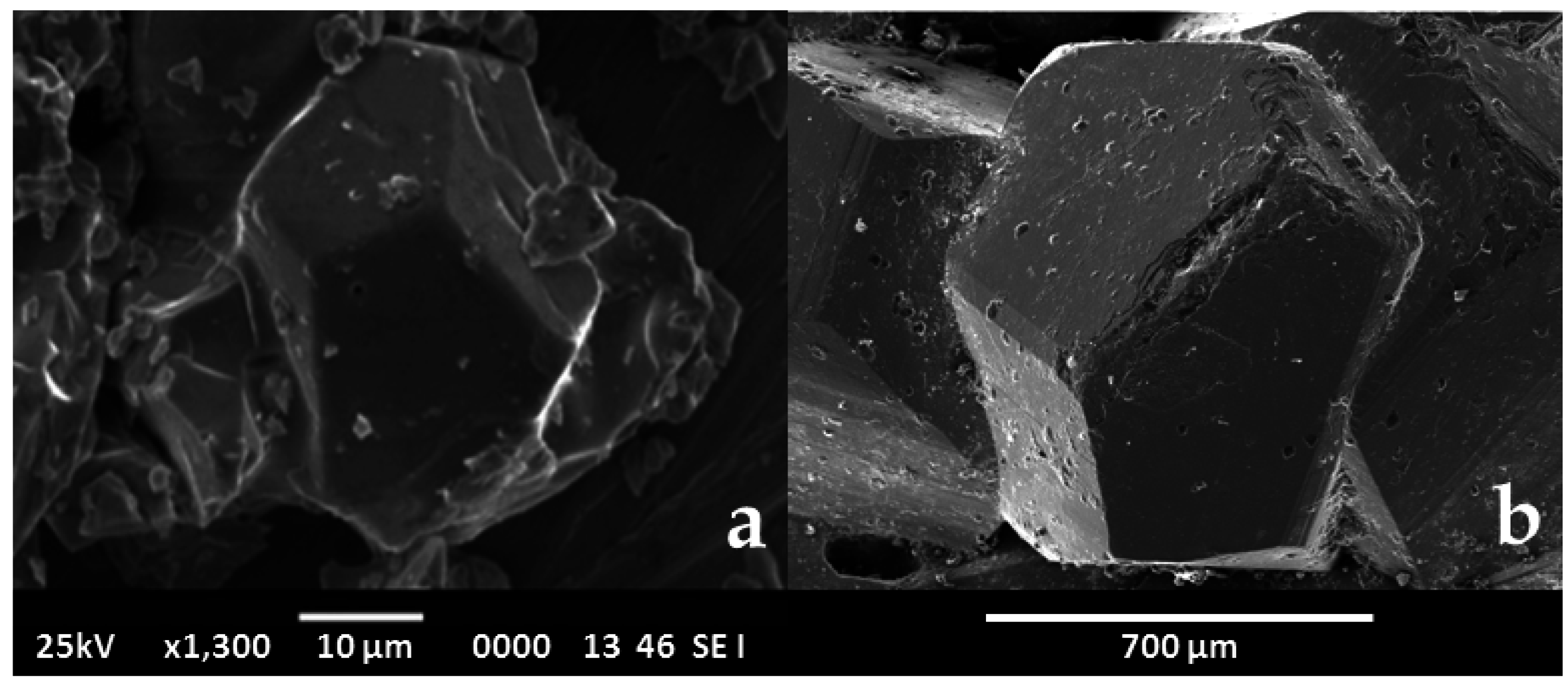
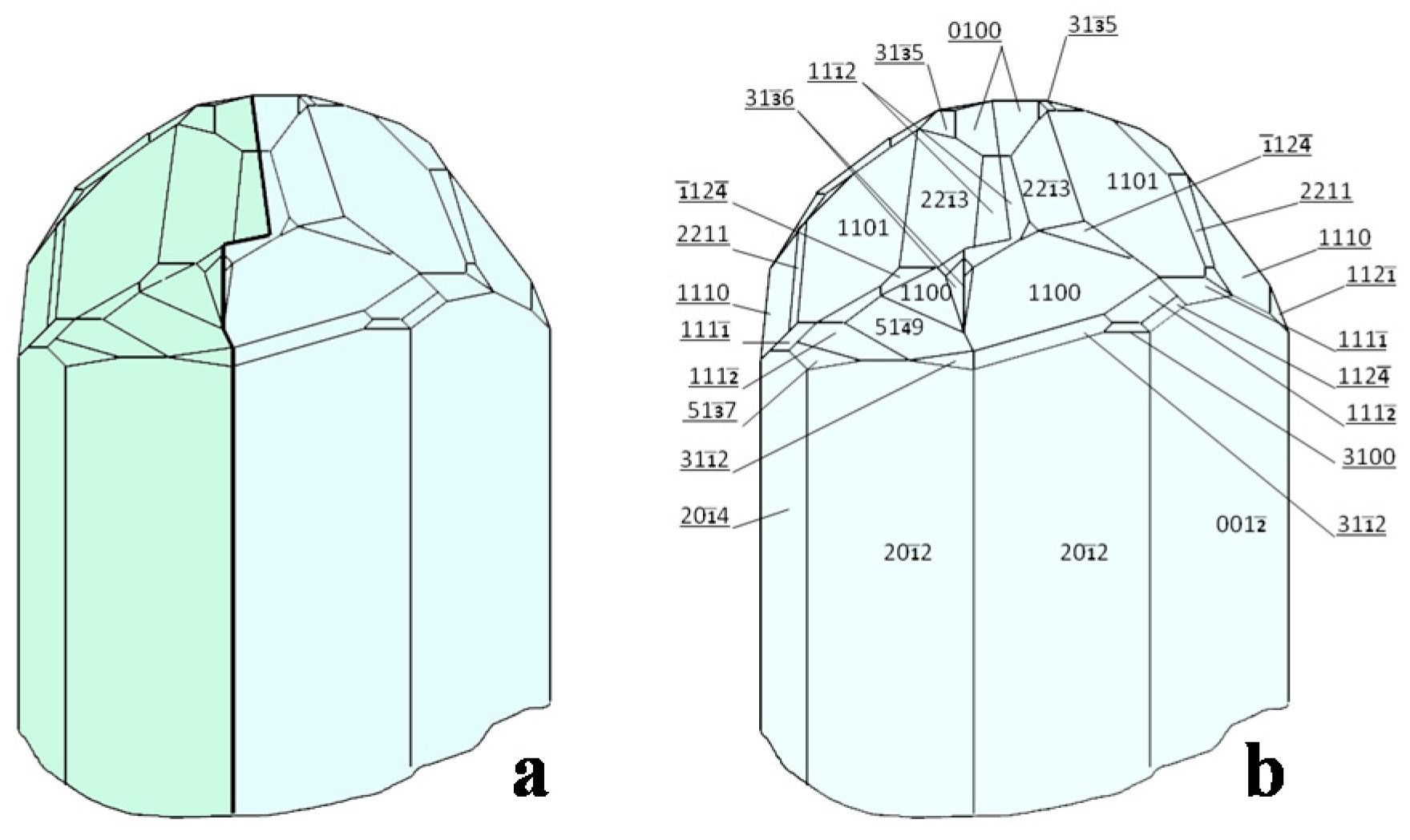
- Oftedal, I. The crystal structure of skutterudite and related minerals. A preliminary paper. Norsk Geol. Tidsskr. 1926, 8, 250–257. [Google Scholar]
- Oftedal, I. The crystal structure of skutterudite and smaltite-cloantite. Z. Kristallogr. 1928, A66, 517–546. [Google Scholar]
- Mandel, N.; Donohue, J. The refinement of the crystal structure of skutterudite, CoAs3. Acta Cryst. 1971, B27, 2288. [Google Scholar] [CrossRef]
- Aleksandrov, K.S.; Beznosikov, B.V. Crystal Chemistry and Prediction of Compounds with a Structure of Skutterudite Type. Crystallogr. Rep. 2007, 52, 28–36. [Google Scholar] [CrossRef]
- Keppens, V.; Mandrus, D.; Sales, B.C.; Chakoumakos, B.C.; Dai, P.; Coldea, R.; Maple, M.B.; Gajewski, D.A.; Freeman, E.J.; Bennington, S. Localized vibrational modes in metallic solids. Nature 1998, 395, 876–878. [Google Scholar]
- Rull-Bravo, M.; Moure, A.; Fernández, J.F.; Martín-González, M. Skutterudites as thermoelectric materials. RSC Adv. 2015, 5, 41653–41667. [Google Scholar] [CrossRef]
- Frank, F.C. Supercooling of liquids. Proc. R. Soc. Lond. A 1952, 215, 43–46. [Google Scholar] [CrossRef]
- Leocmach, M.; Tanaka, H. Roles of icosahedral and crystal-like order in the hard spheres glass transition. Nat. Commun. 2012, 974. [Google Scholar] [CrossRef] [PubMed]
- Hirata, A.; Kang, L.J.; Fujita, T.; Klumov, B.; Matsue, K.; Kotani, M.; Yavari, A.R.; Chen, M.W. Geometric Frustration of Icosahedron in Metallic Glasses. Science 2013, 341, 376–379. [Google Scholar] [CrossRef] [PubMed]
- Steinhardt, P.J.; Nelson, D.R.; Ronchetti, M. Bond-orientational order in liquids and glasses. Phys. Rev. B 1983, 28, 784–805. [Google Scholar] [CrossRef]
- Sadoc, J.F.; Mosseri, R. Geometrical Frustration; Cambridge Univ. Press: Cambridge, UK, 1999. [Google Scholar]
- Tarjus, G.; Kivelson, S.A.; Nussinov, Z.; Viot, P. The frustration-based approach of supercooled liquids and the glass transition: A review and critical assessment. J. Phys. Condens. Matter. 2005, 17, R1143–R1182. [Google Scholar] [CrossRef]
- Hannis, S.; Bide, T.; Minks, A. Cobalt. BGS. Natural Environment Research Council. Available online: http://www.bgs.ac.uk/mineralsuk/search/downloadSearch.cfc?method=viewDownloads (accessed on 15 September 2016).
- Slack, J.F.; Causey, J.D.; Eppinger, R.G.; Gray, J.E.; Johnson, C.A.; Lund, K.I.; Schulz, K.J. Co-Cu-Au Deposits in Metasedimentary Rocks. A Preliminary Report. USGS 2010, 2010–1212. [Google Scholar]
- Gyese, R.F., Jr; Kerr, P.F. The crystal structures of ordered and disordered cobaltite. Am. Min. 1965, 50, 1002–1014. [Google Scholar]
- Bayliss, P. A further crystal structure refinement of cobaltite. Am. Min. 1982, 67, 1048–1057. [Google Scholar]
- Fleet, M.E.; Burns, P.C. Structure and twinning of cobaltite. Can. Mineral. 1990, 28, 719–723. [Google Scholar]
- Pauling, L. Apparent icosahedral symmetry is due to directed multiple twinning of cubic crystals. Nature 1985, 317, 512514. [Google Scholar] [CrossRef]
- Pauling, L. The nonsense about quasicrystals. Sci. News 1986, 129, 3. [Google Scholar] [CrossRef]
- Pauling, L. So-called icosahedral and decagonal quasicrystals are twins of an 820-atom cubic crystal. Phys. Rev. Lett. 1987, 58, 365–368. [Google Scholar] [CrossRef] [PubMed]
- Pauling, L. Interpretation of so-called icosahedral and decagonal quasicrystals of alloys showing apparent icosahedral symmetry elements as twins of an 820-atom cubic crystal. Comput. Math. Appl. 1989, 17, 337–339. [Google Scholar] [CrossRef]
- The Pauling Blog. Oregon Experience. Available online: https://paulingblog.wordpress.com/tag/quasicrystals/ (accessed on 15 September 2016).
- Mackay, A.L. Pauling’s model not universally accepted. Nature 1986, 319, 103–104. [Google Scholar] [CrossRef]
- Bancel, P.A.; Heiney, P.A.; Horn, P.M.; Steinhardt, P.J. Comment on a paper by Linus Pauling. Proc. Natl. Acad. Sci. USA 1989, 86, 8600–8601. [Google Scholar] [CrossRef] [PubMed]
- Vegas, A. FeLiPO4: Dissection of a crystal structure. The parts and the whole. In Inorganic 3D Structures; Vegas, A., Ed.; Springer: Heidelberg, Germany, 2011; pp. 67–92. [Google Scholar]
- Bevan, D.J.M.; Martin, R.L.; Vegas, A. Rationalisation of the substructures derived from the three fluorite-related [Li6(MVLi)N4] polymorphs: An analysis in terms of the “Bärnighausen Trees” and of the “Extended Zintl-Klemm Concept. In Inorganic 3D Structures; Vegas, A., Ed.; Springer: Heidelberg, Germany, 2011; pp. 93–133. [Google Scholar]
- Vegas, A. Concurrent pathways in the phase transitions of alloys and oxides: Towards an Unified Vision of Inorganic Solids. In Inorganic 3D Structures; Vegas, A., Ed.; Springer: Heidelberg, Germany, 2011; pp. 133–198. [Google Scholar]




© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pina, C.M.; López-Acevedo, V. Quasicrystals and Other Aperiodic Structures in Mineralogy. Crystals 2016, 6, 137. https://doi.org/10.3390/cryst6110137
Pina CM, López-Acevedo V. Quasicrystals and Other Aperiodic Structures in Mineralogy. Crystals. 2016; 6(11):137. https://doi.org/10.3390/cryst6110137
Chicago/Turabian StylePina, Carlos M., and Victoria López-Acevedo. 2016. "Quasicrystals and Other Aperiodic Structures in Mineralogy" Crystals 6, no. 11: 137. https://doi.org/10.3390/cryst6110137
APA StylePina, C. M., & López-Acevedo, V. (2016). Quasicrystals and Other Aperiodic Structures in Mineralogy. Crystals, 6(11), 137. https://doi.org/10.3390/cryst6110137