1. Introduction
Native cyclodextrins (CDs) are cyclic oligosaccharides with low chemical toxicity [
1,
2]. They are formed by several α-
d-glucopyranose units, linked by α(1→4)
O-glycosidic bonds. A CD molecule has a hollow truncated cone-like shape where the primary hydroxy groups sit on the narrow side and the secondary ones on the wide side of the truncated cone. The architecture of CD molecules favors the partition of the structure into an outer surface and an inner cavity, which are hydrophilic and hydrophobic in character, respectively [
3]. This molecular feature boosted the interest of using CDs as “molecular cages” [
4] in both the solid and solution states, particularly in the pharmaceutical field where CDs are known to improve the aqueous solubility, and thus bioavailability, of poorly soluble active pharmaceutical ingredients [
3]. The formation of CD inclusion complexes in the solid state, particularly with organic molecules, has been pioneered by Saenger [
5,
6,
7] and Harata [
8]. Three packing types have been described for both CD hydrates and inclusion complexes: two of these belong to the cage type and are known as herringbone- and brick-type packing, and the third one is the channel type [
7].The packing preference of CD molecules for one packing type is closely related to the size and shape of the guest molecule [
7].
The work presented here is part of ongoing investigations of cyclodextrin inclusion complex and hydrate formation at both ambient- and high-pressure crystallization conditions using water as crystallization medium [
9]. α-CD (
Figure 1), the smallest natural cyclic oligosaccharide with six sugar units, was chosen for studying inclusion complex formation with small molecules (here defined as molecules with a molecular weight <500 Daltons), particularly to compare and contrast the effects of non-ambient (low-temperature and high-pressure) crystallization conditions on complex formation. A Cambridge Structural Database [
10] (CSD) search (the CSD, V 5.36 including updates to November 2014 was searched for structures with 3-D coordinates) indicates that α-CD crystallizes in the channel-packing type in 52% of the total 96 hits, followed by the brick-type in 26% and the herringbone-type in 22% of the structures. In the channel-packing type favored by α-CD, guest molecules are inserted lengthwise into the host’s cavity, forming infinite columns.
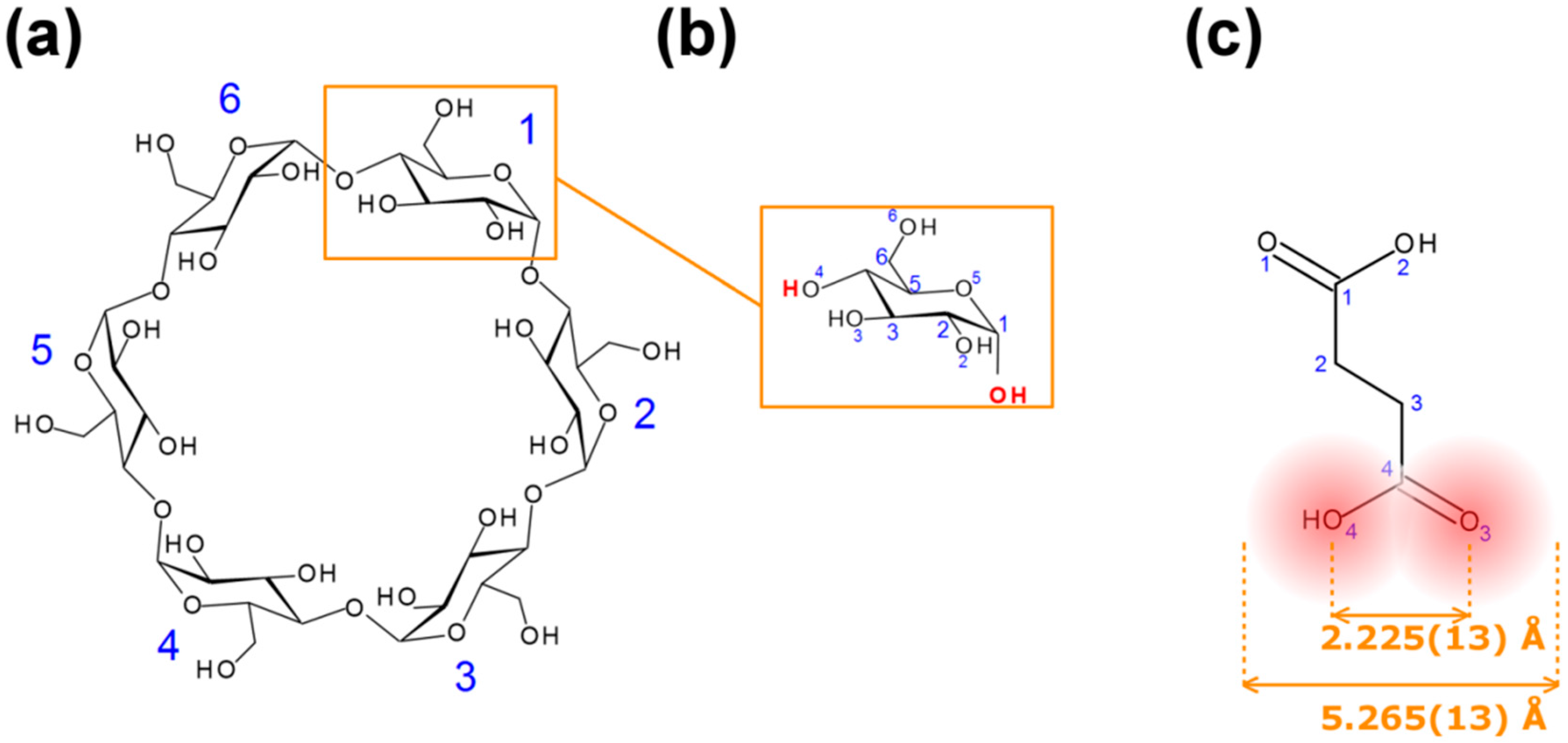
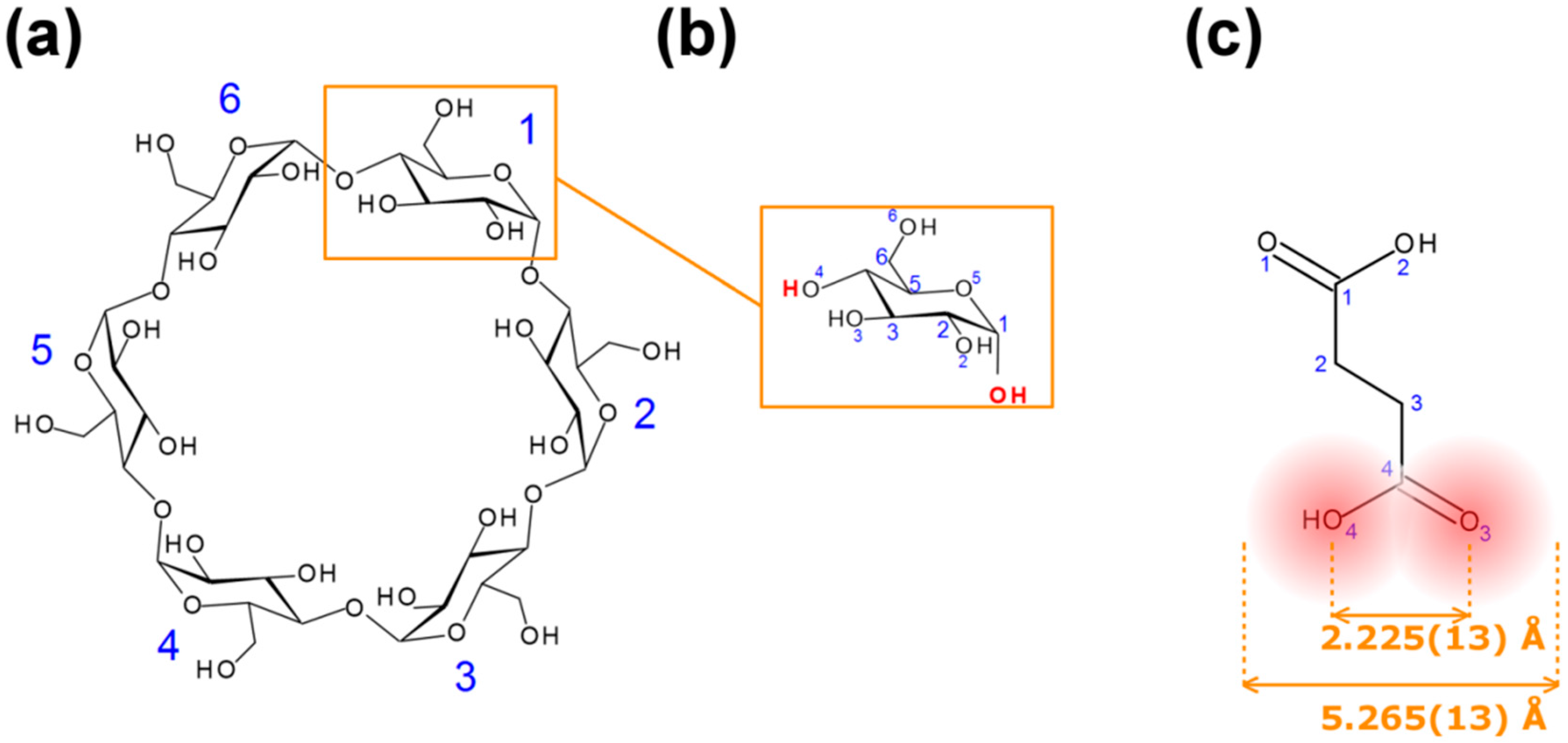
Figure 1.
Diagrams and numbering schemes of: (a) α-cyclodextrins; (b) α-D-glucopyranose; (c) succinic acid (SA): the mean distance O(3)–O(4) was computed from 72 structures of SA in the Cambridge Structural Database (CSD), the width of SA is the (mean distance O(3)–O(4) plus twice the van der Waals radius of an oxygen atom.
Figure 1.
Diagrams and numbering schemes of: (a) α-cyclodextrins; (b) α-D-glucopyranose; (c) succinic acid (SA): the mean distance O(3)–O(4) was computed from 72 structures of SA in the Cambridge Structural Database (CSD), the width of SA is the (mean distance O(3)–O(4) plus twice the van der Waals radius of an oxygen atom.
Succinic acid (SA,
Figure 1), an aliphatic dicarboxylic acid, is essential in aerobic cellular metabolism by intervening in the citric acid cycle, a metabolic pathway for the regeneration of adenosine triphosphate (ATP), which is the main energy source of most cellular functions [
11]. SA is a FDA-Generally Recognized As SAFE (GRAS) substance also used in the pharmaceutical industry for the preparation of succinate ester derivatives of active pharmaceutical ingredients. The structure of a β-CD∙SA inclusion complex (CSD refcode KIJSEC) has been previously obtained while investigating the enhancement of succinic anhydride’s reactivity using β-CD as molecular cages in aqueous solutions [
12]. We found that this complex can easily be obtained with SA instead of succinic anhydride; the large cavity size of β-CD, which is 6.0–6.5 Å in diameter [
13], can easily accommodate SA and the crystal structure of the complex shows several intermolecular interactions between host, guest and solvent molecules [
12]. We hypothesize that with a width (5.265(13) Å, see
Figure 1) commensurable with the cavity diameter of α-CD (4.7–5.3 Å) [
13], SA could in principle form a crystalline complex with α-CD. The literature shows that similar linear compounds form inclusion complexes with α-CD (see for example CSD refcodes BUPDEV [
14], CDKABA [
15] and XIGBOE [
16]).
2. Results and Discussion
Low-temperature crystallization of an equimolar mixture of α-CD with SA in water led to the formation of hexagonal prism shaped crystals. A first polarized microscopy analysis of the crystals through the hexagonal face indicated an absence of light extinction characteristic of uniaxial crystals. This microscopic analysis endorsed the choice of the unit cell from X-ray diffraction, with successful indexing of the reflections using a rhombohedral unit cell. The reflections could also be indexed using a lower symmetry monoclinic unit cell, through the transformation matrix (−⅓ −⅔ −⅔, 1 0 0, −⅓ −⅔ ⅓). The choice of a trigonal crystal system dictates that both host and guest molecules sit on a 3-fold rotation axis going through α-CD cavities. Heavy disorder of the guest, evident from the electron density maps, could be modelled in the higher symmetry space group and this was finally chosen for refinement.
The α-CD∙SA 1:1 inclusion complex crystallizes in space group
R32 with 3 × ⅓ α-CD molecules in the asymmetric unit, here named A, B and C (
Figure 2). Each α-CD sits on the 3-fold rotation axis and encloses one SA molecule disordered over six positions; crystallographically, this is modelled by having two SA molecules per cavity each with ⅙ site occupancy. α-CD C is further disordered over two positions with ⅔ (C) and ⅓ (C′) site occupancies. In addition the unit cell contains
ca. 12 water molecules making the complex a
pseudo dodecahydrate.
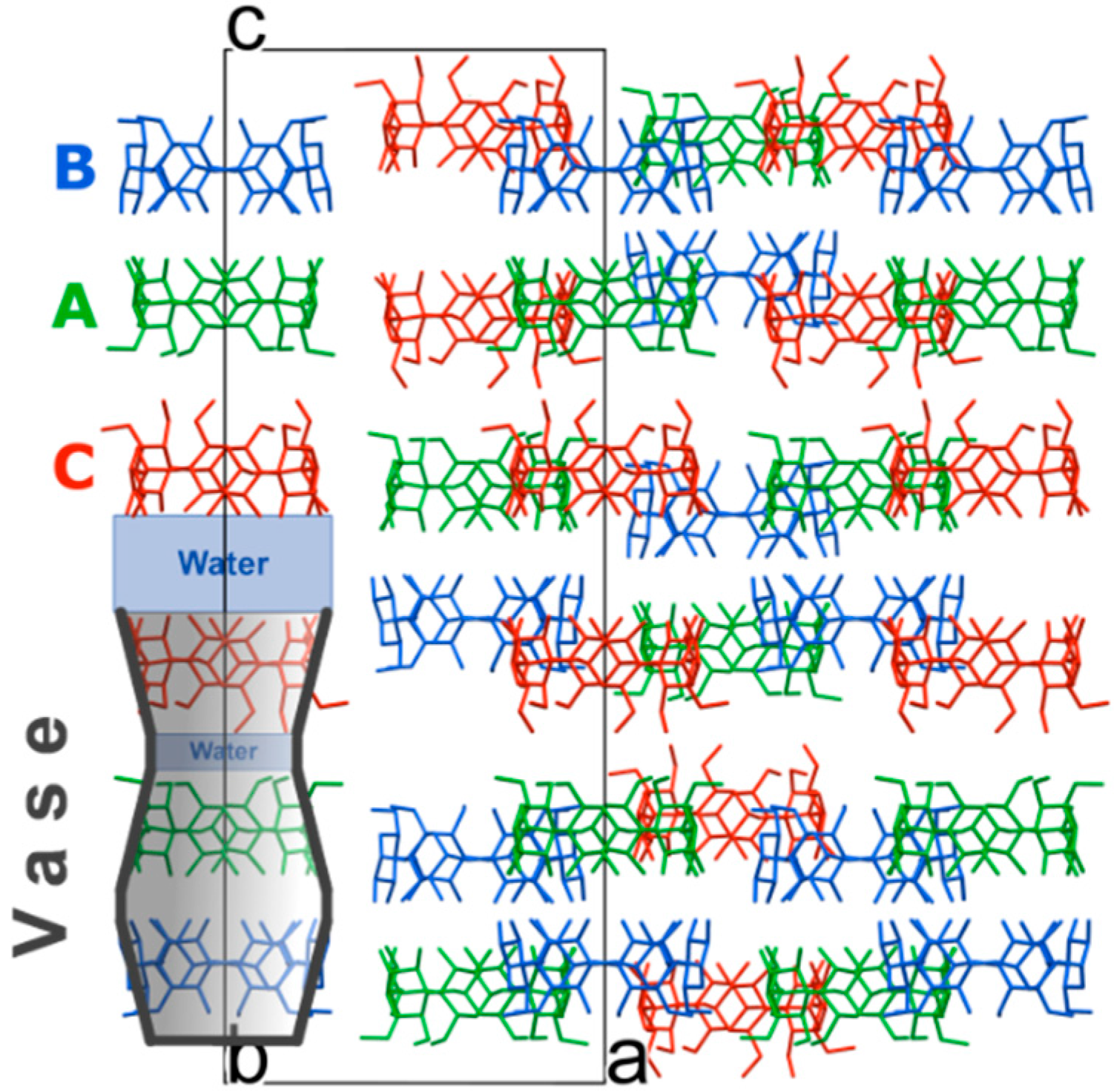
Figure 2.
Crystal packing of α-CD molecules viewed along the b-axis. H atoms, disorder of α-CD C, SA and water molecules have been omitted for clarity. Symmetry-equivalent molecules are color coded.
Figure 2.
Crystal packing of α-CD molecules viewed along the b-axis. H atoms, disorder of α-CD C, SA and water molecules have been omitted for clarity. Symmetry-equivalent molecules are color coded.
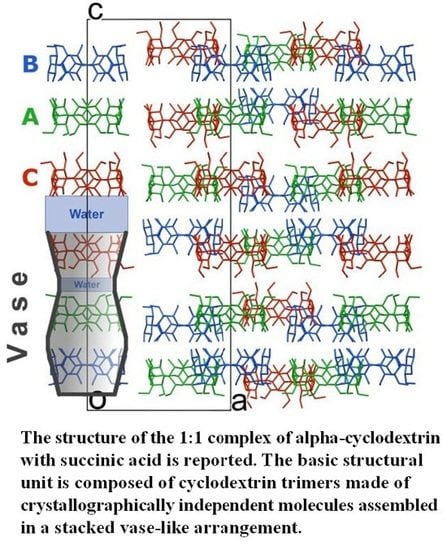
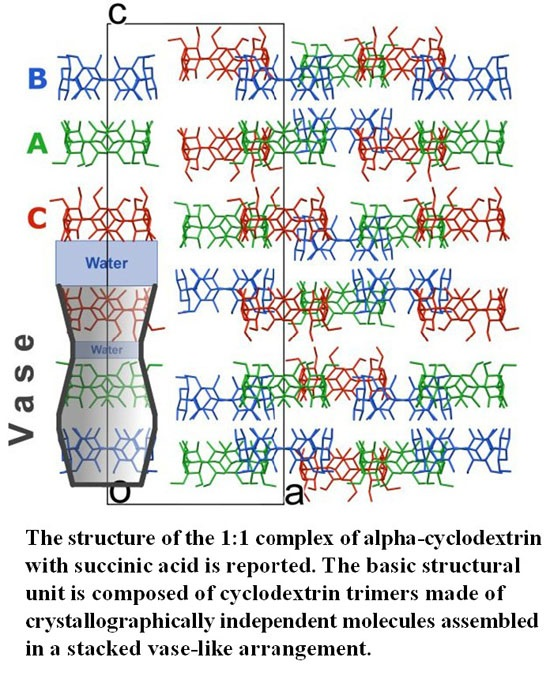
In the solid state α-CD molecules have always been known, to the best of our knowledge, to pack as either distinct entities or dimers [
6,
8]. In this work, a new building block is observed, namely α-CD trimers packed along the
c-axis. Each trimer is made of crystallographically independent molecules assembled in a stacked vase-like cluster (
Figure 2). The stacked trimer motif is not unknown for γ-CD molecules (see for example CSD refcodes FEJFIJ, FEJFOP, NUNRIX, SIBJAO, SIBJES) [
17,
18,
19]; however the concept of vase-like packing has not been previously reported. It has also been reported that β-CD molecules crystallize as trimers (CSD refcodes RIPKIL, OCIGAK) [
20,
21] or tetramers [
22,
23]. The structure of the title compound appears to be very similar to that of α-CD∙hexa-ethylene glycol reported by Harada
et al. (CSD refcode LOJTUZ, no 3D coordinates deposited in the CSD) [
24]; however, this publication neither describe the distinctive trimer arrangement in detail, nor identifies a new packing type. In contrast, in a conference abstract, Caira
et al. have recently reported that the α-CD-lipoic acid system “crystallizes in the trigonal system, space group
R32, with three independent CD molecules in the asymmetric unit and is not isostructural with any known CD complex” [
25]. These observations make the reported packing type rare but not unknown.
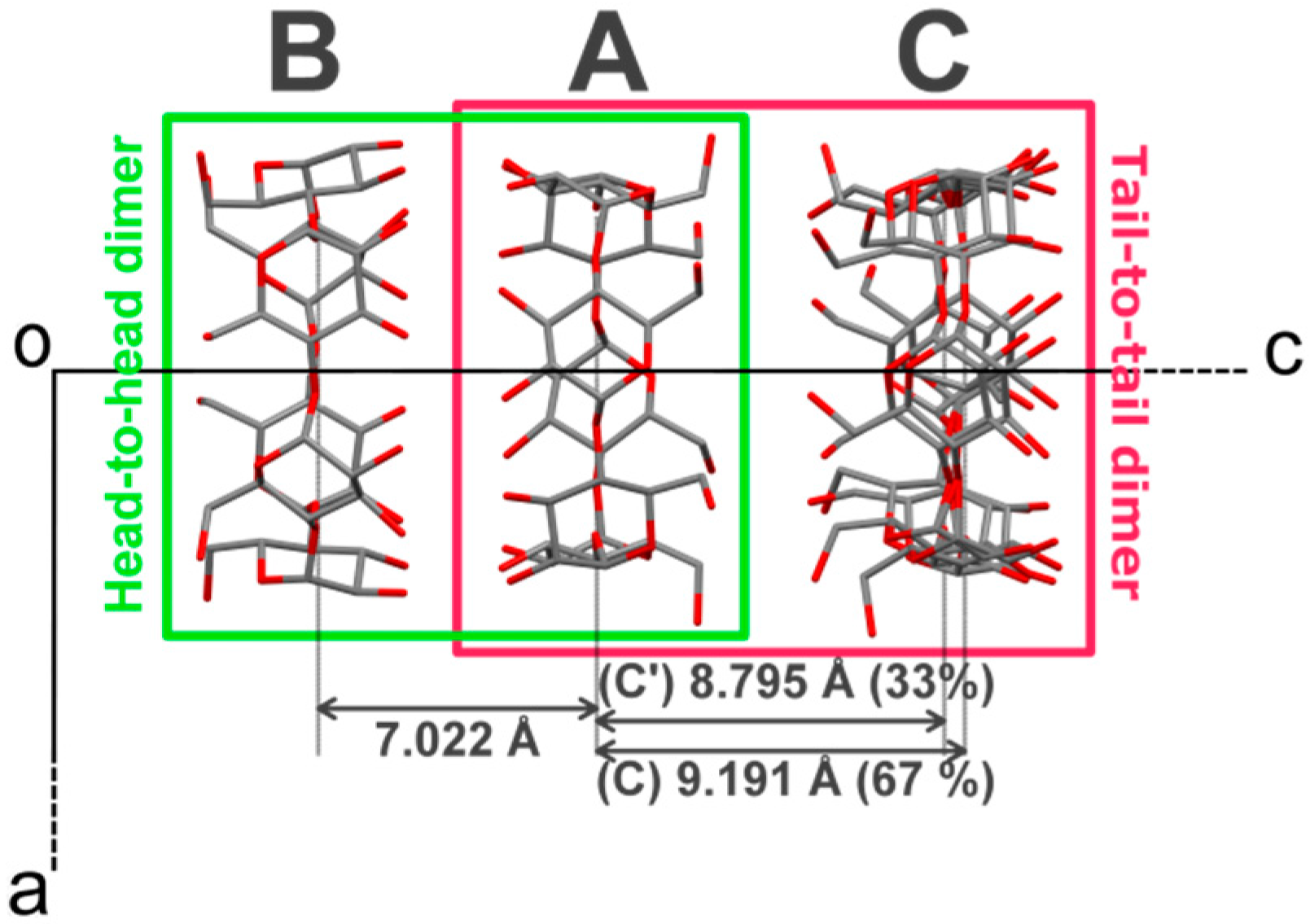
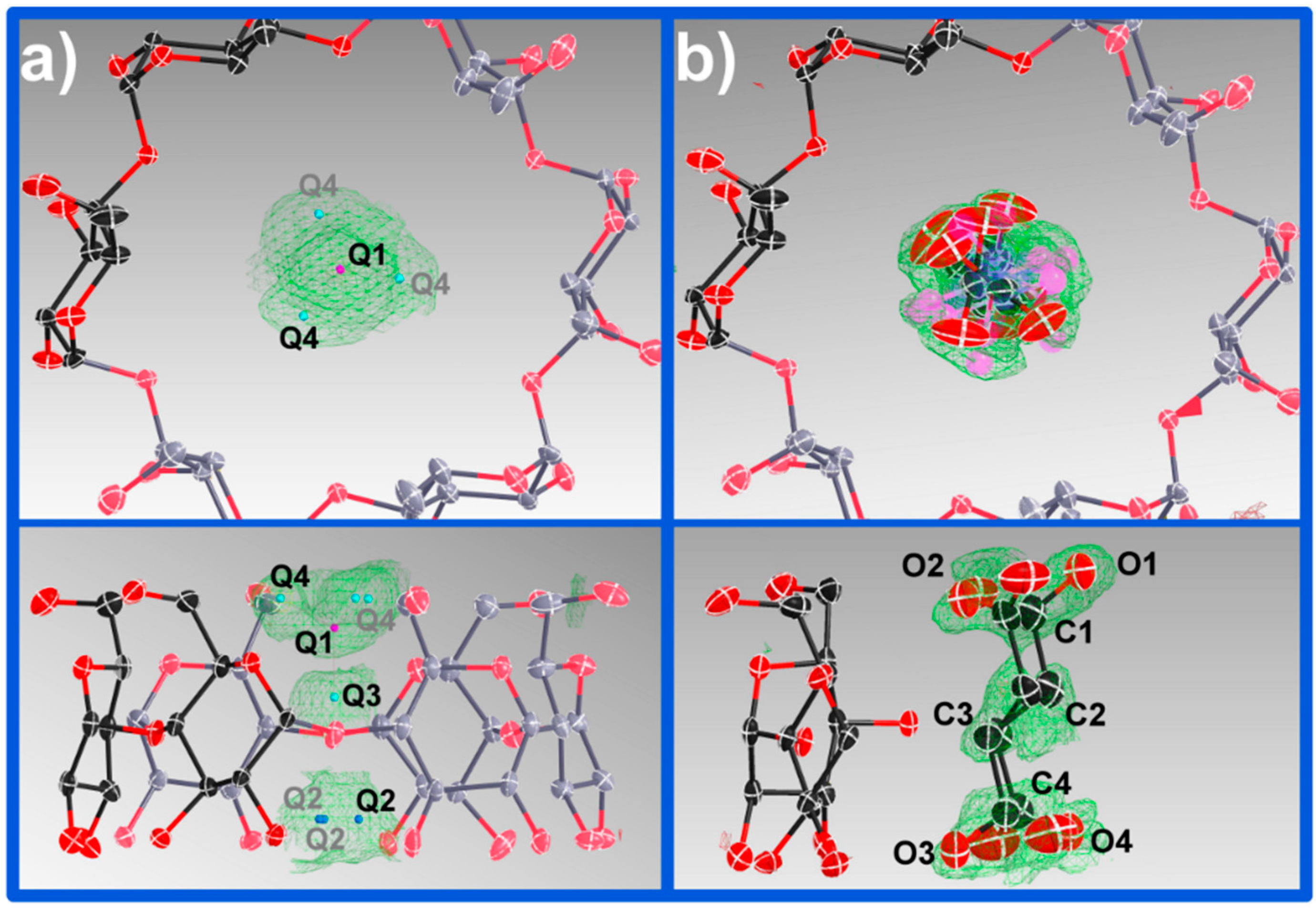
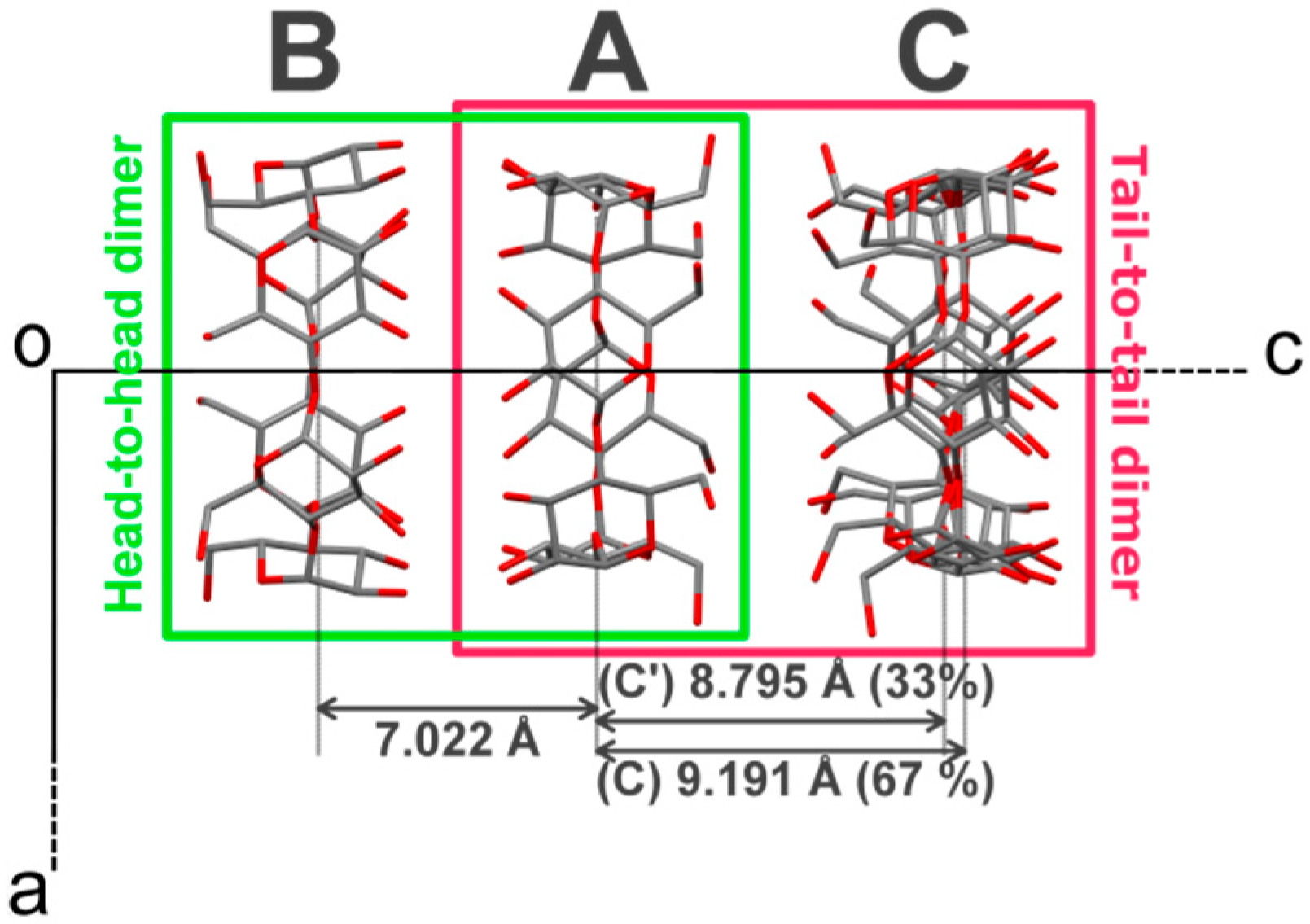
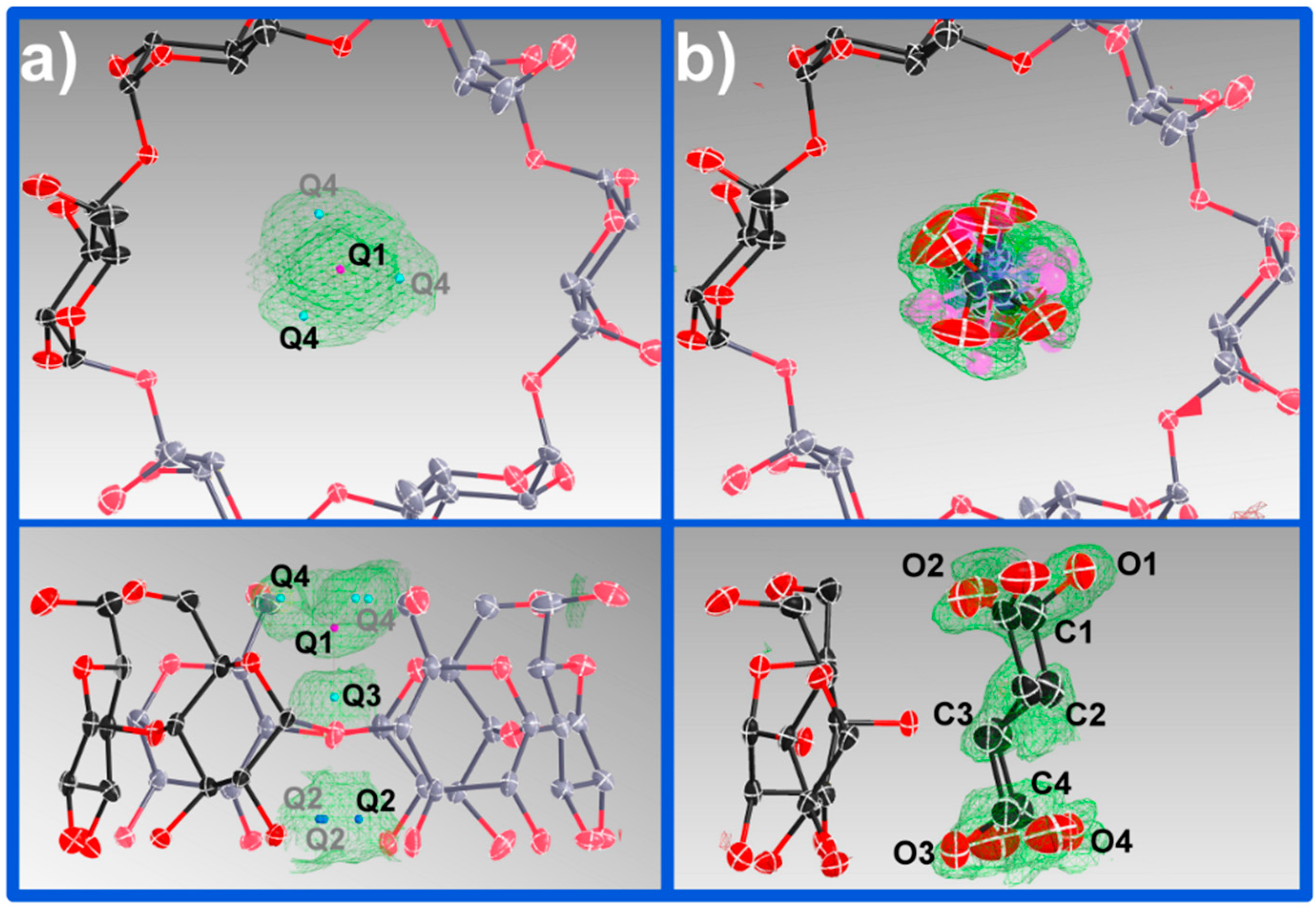
The vase-like cluster in the α-CD∙SA inclusion complex is formed by two sub-dimers: (1) a head-to-head dimer, which is stabilized by H-bonds between secondary hydroxy groups of α-CDs A and B; and (2) a tail-to-tail dimer between α-CDs A and C interconnected through a cluster of water molecules. Analysis of planes formed by glycosidic O-atoms shows that the interplanar distance in the head-to-head dimer is 7.022 Å, compared to a mean distance of 9.060 Å for the tail-to-tail dimer (
Figure 3). Successive vase-like clusters, stacked via two-fold rotation symmetry, are separated by a layer of water molecules forming a complex H-bonded network that holds two vase-like structures together.
The three α-CD molecules forming the vase-like cluster exhibit an almost ideal cylindrical shape and form infinite linear channels extending along the
c-axis of the unit cell in a honeycomb-type arrangement. A honeycomb or
quasi-hexagonal pattern ensures effective close packing [
19,
26], and for CDs it was reported for the first time by Saenger in 1980, while describing the general arrangement of CDs in crystals, as “
hexagonal packing of stacks” [
5]. A careful analysis of the reported channel-type structures of α-CD inclusion complexes, including hydrates, in the CSD shows that honeycomb packing is observed in 27 out of 50 structures (CSD refcodes of these structures are reported in the
Supplementary Information), though this particular structural feature has been rarely reported explicitly. Instead, authors usually report the type of α-CD dimer arrangement, head-to-head or head-to-tail, in channel-type structures.
Figure 3.
Projection of α-CD∙SA structure along the b-axis. The crystallographically-independent CD molecules are named A, B and C, see main text for details. H atoms, SA and water molecules have been omitted for clarity. The distances refer to gap in Å between the least-square planes formed by all O(4) atoms involved in the glycosidic bonds.
Figure 3.
Projection of α-CD∙SA structure along the b-axis. The crystallographically-independent CD molecules are named A, B and C, see main text for details. H atoms, SA and water molecules have been omitted for clarity. The distances refer to gap in Å between the least-square planes formed by all O(4) atoms involved in the glycosidic bonds.
One of the features of CDs is the flexibility of the primary-hydroxy groups. The conformation of the hydroxy group is defined by the value of the O(5)–C(5)–C(6)–O(6) torsion angle (
Figure 1): a preferred (−)-gauche conformation in which the hydroxy group is facing the exterior of the cavity, and a less preferred (+)-gauche conformation where the hydroxy group is facing the inner cavity [
27]. All but one primary hydroxy groups in α-CD exhibit the (−)-gauche conformation and are directed away from the cavity. The rotation of the primary hydroxy group to the less favored (+)-gauche conformation in molecule C can be explained by the formation of the short H-bonds [2.52(4) and 2.79(1) Å] with a disordered water molecule (named O(10)_91 in the structure). A CSD search (V 5.36) shows that (−)-gauche conformers are not unknown for α-CD inclusion complexes and indicate the absence of direct H-bonds between the guest and the host molecules, which could explain the high degree of rotational disorder of the guest observed here. In contrast, in the structure of β-CD∙SA which contains one full guest and host molecules in the asymmetric unit, SA is H-bonded to two primary CD hydroxy groups, which are facing the inner cavity, through one water molecule each. Interestingly, the SA molecule in the β-CD∙SA inclusion complex does not exhibit disorder.
A search in the CSD (V 5.36 including updates to November 2014) based on the C(1)–C(2)–C(3)–C(4) torsion angle of the guest (
Figure 1) shows that SA molecules exhibit two conformations in the solid state, trans and gauche, with an incidence of 90 and 10%, respectively, out of a total of 165 structures. Lisnyak
et al. reported that the trans conformer is energetically more favorable than the gauche one with an energy difference of 31.4 kJ∙mol
−1 [
12]. In β-CD
.SA (CSD refcode KIJSEC [
12]), SA molecules lie almost equatorially in the β-CD cavity and exhibit the gauche conformer. In contrast, the electron density maps of α-CD∙SA point to the trans conformation with SA molecules extended along the
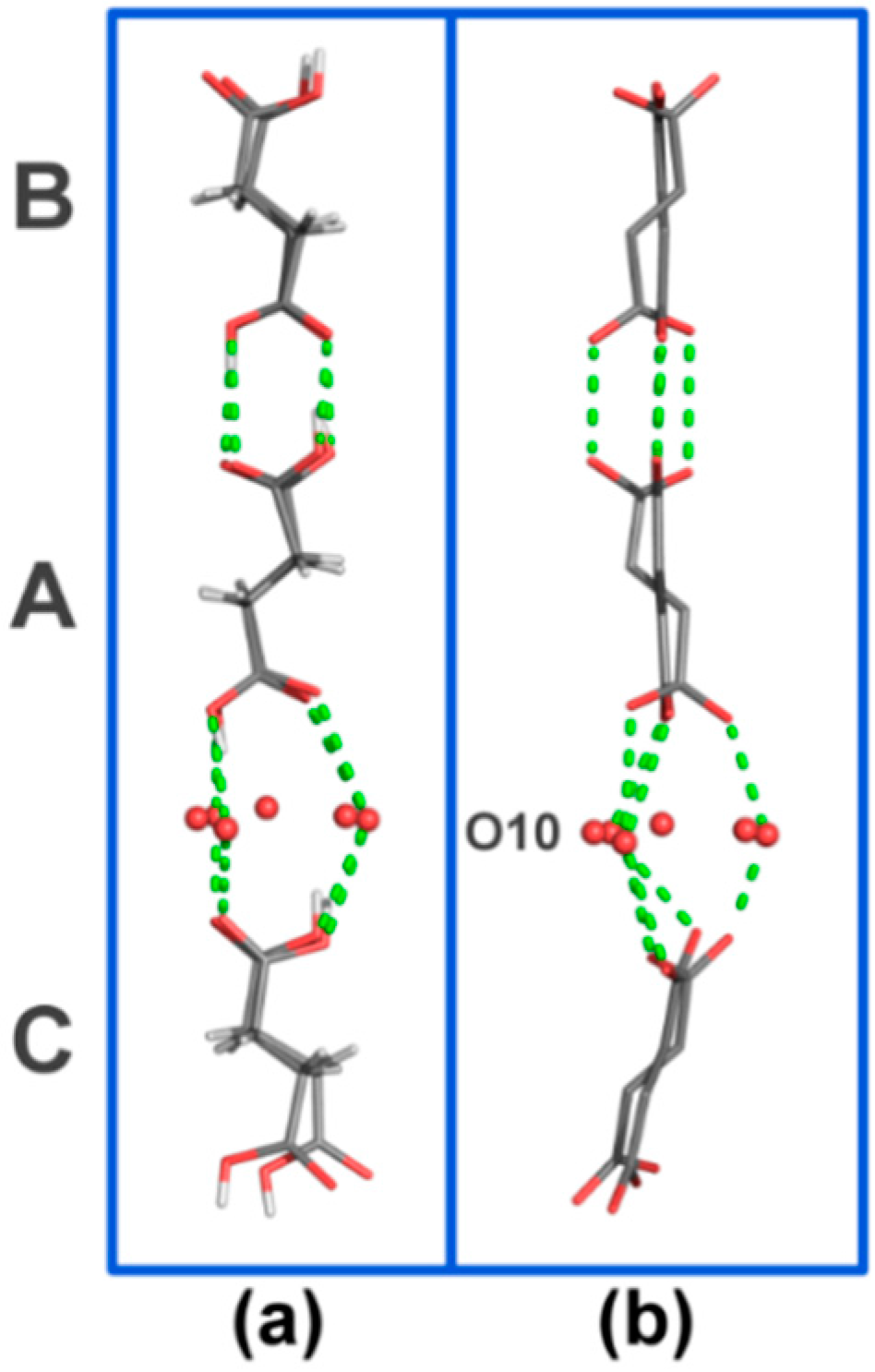
c-axis. The difference in conformation may be directly related to the cavity size differences between α- and β-CD, with an axial inclusion mode being more commensurable with the size of the α-CD cavity. Although as a result of this one might expect a tighter fit of SA inside α-CD, the guest is actually found be disordered. This is in line with the almost ideal cylindrical shape of the host, yielding a more evenly distributed cavity space, and the absence of direct H-bonds between the guest and host molecules. In α-CD∙SA the axial inclusion mode is described by the almost orthogonal angular differences between the planes formed by the glycosidic O-atoms of the ordered CD molecules A and B and to the planes formed by the SA molecules inside the respective cavities (SA planes were calculated using C(1)–C(2)–C(3)–C(4)). For α-CD C, similar calculations show that the same planes are 13 to 18° off from being orthogonal. The different inclusion modes of SA inside α- and β-CD are also associated with different crystal packing: the trans SA molecules in α-CD∙SA form a H-bonded linear chain (
Figure 4) that is associated with the formation of a channel-type packing. On the other hand, in the β-CD
.SA complex gauche SA molecules are “buried” in the cavities and the herringbone-type packing observed in the hydrated β-CD and small-guest molecules structures is conserved. This comparative analysis based on size, shape and functionality of the guest molecule supports the work by Saenger and Steiner [
7].
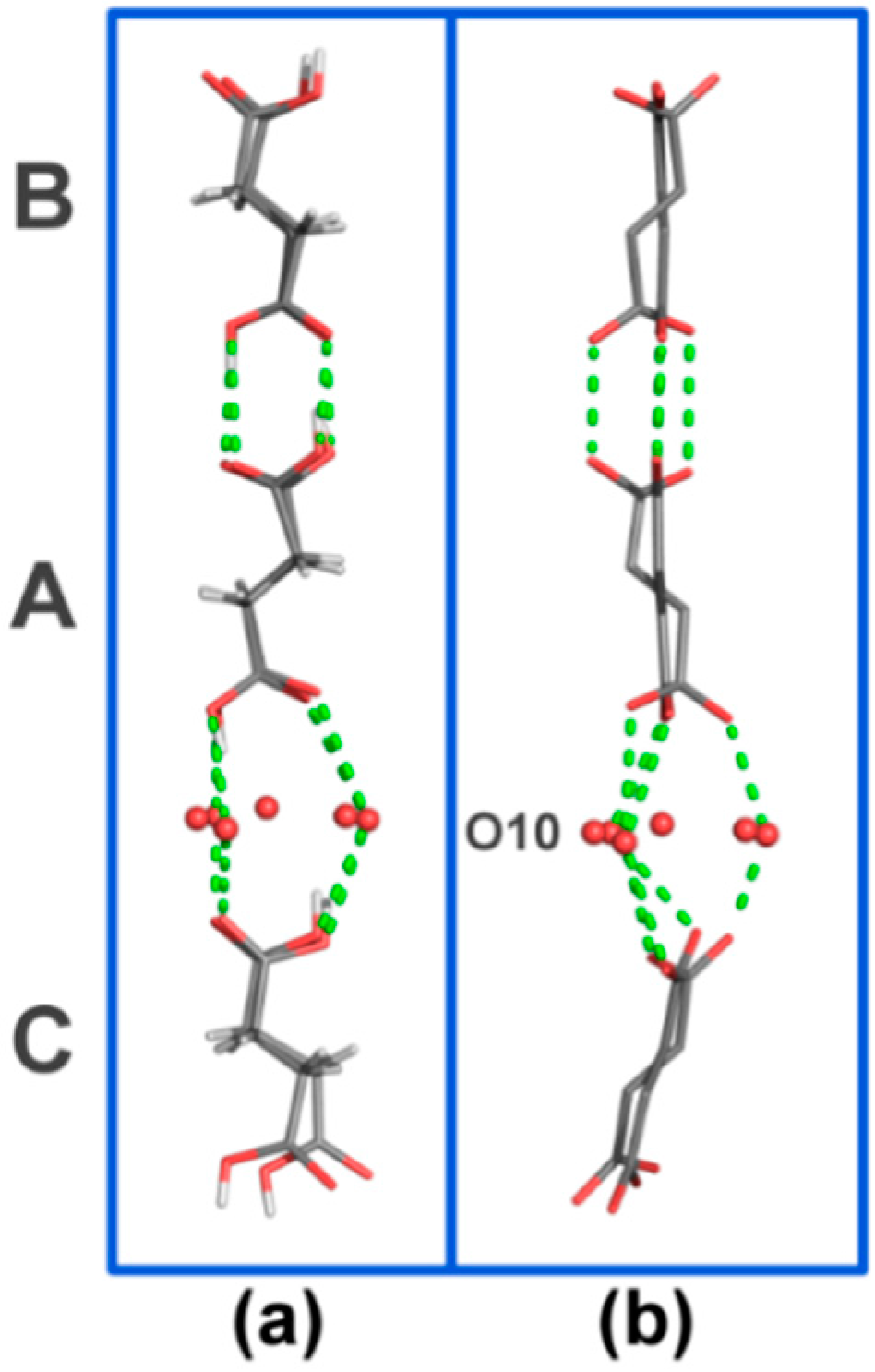
Figure 4.
H-bonded motif formed by SA molecules in the asymmetric unit viewed (a) along the a-axis and (b) along the b-axis. O–O contacts are represented by dashed green lines. H-atoms (in b), α-CD and water molecules have been omitted for clarity. SA molecules are contained in the cavities of the crystallographically-independent CD molecules named A, B and C, see main text for details.
Figure 4.
H-bonded motif formed by SA molecules in the asymmetric unit viewed (a) along the a-axis and (b) along the b-axis. O–O contacts are represented by dashed green lines. H-atoms (in b), α-CD and water molecules have been omitted for clarity. SA molecules are contained in the cavities of the crystallographically-independent CD molecules named A, B and C, see main text for details.
2.1. Insight into Disorder
In an attempt to gain a better insight into the disorder of α-CD∙SA inclusion complex and to investigate whether a temperature-dependent phase transition takes place, we extended our initial work to a multi-temperature investigation covering the 100–270 K temperature range, in an ascending temperature ramp.
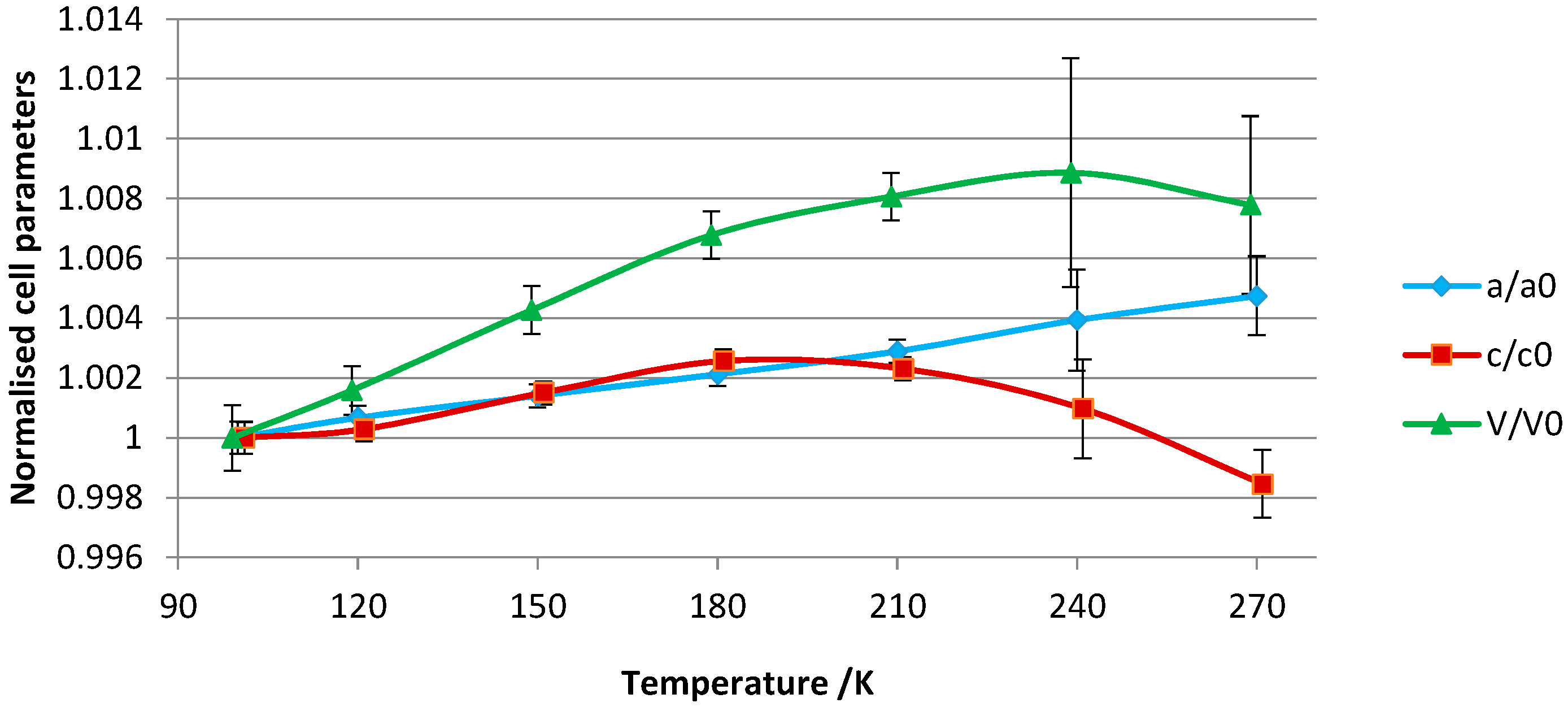
The crystallographic data of all structures, summarized in
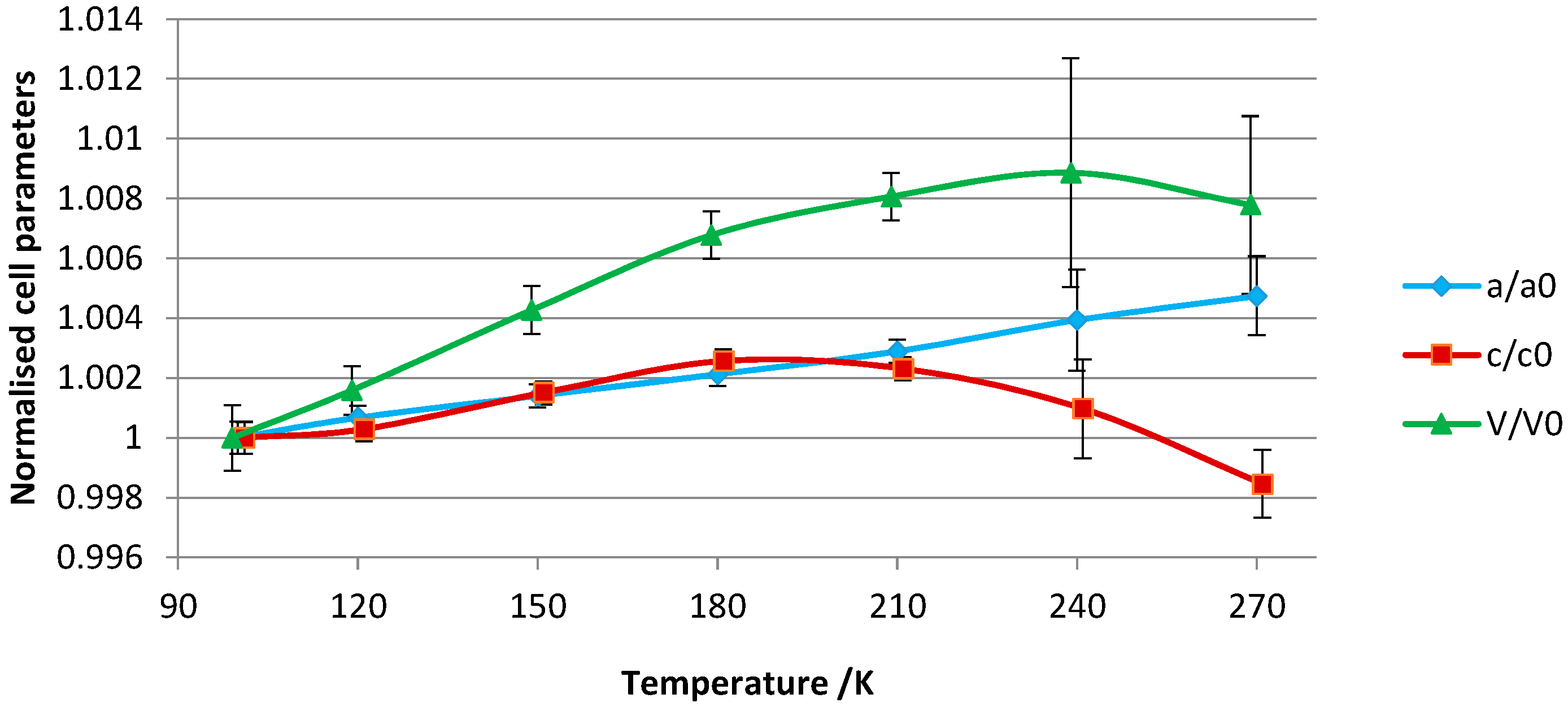
Table S1, show small differences in the lattice constants across the temperature range. The lattice parameters were normalized to the unit cell values of the 100 K crystal structure and are visually represented in
Figure 5. Approximately uniform thermal expansion up to 180 K is observed for both the
c- and
a-axes. For the unit cell volume, the rate of thermal expansion is approximately linear up to this temperature. From 180 to 240 K, the complex overall expands but the
c-axis shows negative thermal expansion. The negative thermal expansion for this axis is marked between 240 and 270 K so that a slight volume contraction is concomitantly observed.
The variations in the unit-cell parameters can be related to the following structural features: (1) lengthening of the
a-axis (
a =
b) as a function of increasing temperature is associated with lengthening of the intermolecular contacts between adjacent α-CD molecules along the same axis; (2) the negative thermal expansion along the
c-axis is associated with a compression of the vase-like cluster (see
Figure S1), mainly arising from compression of the thin water layer that stabilizes the α-CD A–C tail-to-tail dimer (
Figure 2). This thin water layer is formed by one disordered water molecule (named O(10)_91 in the structure), and its symmetry equivalents, H-bonded to the (+)-gauche primary hydroxy group of α-CD molecule C (
Figure 4).
An apparent ordering of α-CD C is observed at 270 K although it should be noted that the structure has large temperature factors. In fact, the low-temperature study points to the presence of dynamic disorder, whereby low temperatures freeze the movement of α-CD C into two distinct positions. Ordering at 270 K is associated with a rearrangement of hydroxy groups: α-CD C has two (+)-gauche primary hydroxy conformers, instead of one observed at low temperature. The new (+)-gauche conformer, pointing towards the inner cavity, is H-bonded to the disordered water molecule O(10)_91 with a distance of 2.52(9) Å. This indicates that there is interplay between movement of α-CD and water molecules towards stabilization of the structure. The multi-temperature experiment did not provide information on the nature of the disorder of the guest molecules, for which it is likely that a combination of dynamic and static disorder is present.
The 100 K structural model was refined based on the atomic coordinates of the 90 K model, and was then used as a starting model for all other temperature points. Modeling of disordered water molecules in a highly hydrated structure is subject to user bias. Temperature factors and site occupancy parameters suffer from correlation and in the absence of independent information, e.g., on water content determined by thermal analysis, the user must make judicious use of refinement tools in order to obtain a reasonable model, which does not necessarily lead to the lowest
R-factor. Comparing Fourier and difference Fourier maps can give an indication on the mean position and site occupancy of the atoms provided a consistent data reduction and refinement strategy for all data points is applied. To minimize model bias and double check the stability of the model, the
SHELXL WIGL command was used as a cross-validation method during the refinement [
28]. Two sets of results were then generated. In the first set, all water molecules were placed and refined according to the difference Fourier electron density maps (results reported in
Table S1). In the second set, all water molecules were omitted during refinement in order to compare the evolution of the maps as a function of increasing temperature. The latter set of structures can be used to emphasize the change in the positions of water molecules and site occupancies by two methods:
- (a)
An analysis with
SHELXLE [
29] allows a qualitative analysis of the changes in the difference density as a function of temperature.
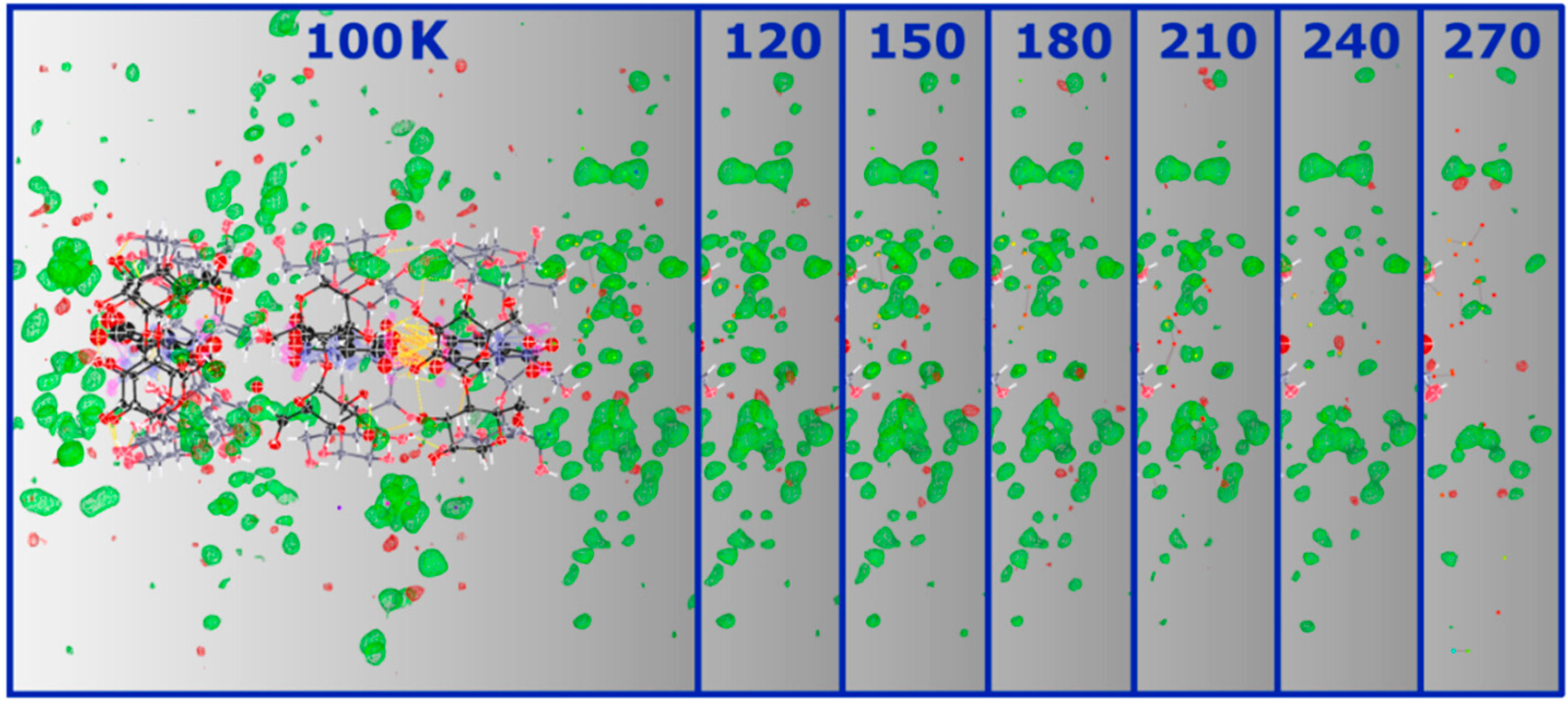
Figure 6 clearly illustrates the loss of this signal from the solvent region as temperature is increased.
- (b)
Void analysis performed with
MERCURY [
30] using a probe radius of 1.2 Å and a grid spacing of 0.2 Å, calculated using the contact surface algorithm, reveals that the volume occupied by the solvent does not change significantly as temperature is increased (
Figure S2).
An analysis, based on the fully refined structures, of the anisotropic displacement parameters (ADP) as function of temperature indicates larger thermal motion at higher temperatures, as expected (see
Figure S3). Analysis of the refined
Uij values of O(1)→O(6) water molecules, which are located in the space between adjacent α-CD columns (
Figure 2), shows comparable expansion of the three diagonal elements
U11,
U22,
U33 of the ADP tensor. These elements are parallel to the reciprocal unit-cell axes
a*,
b* and
c*, respectively, and in the case of a rhombohedral crystal system,
U33 is parallel to the
c-axis. For α-CD atoms, the thermal expansion of
U33 is much more pronounced compared to the two other directions. Hence, while water molecules increase their vibrations in all directions as temperature increases, α-CD molecules have more freedom along the
c-direction.
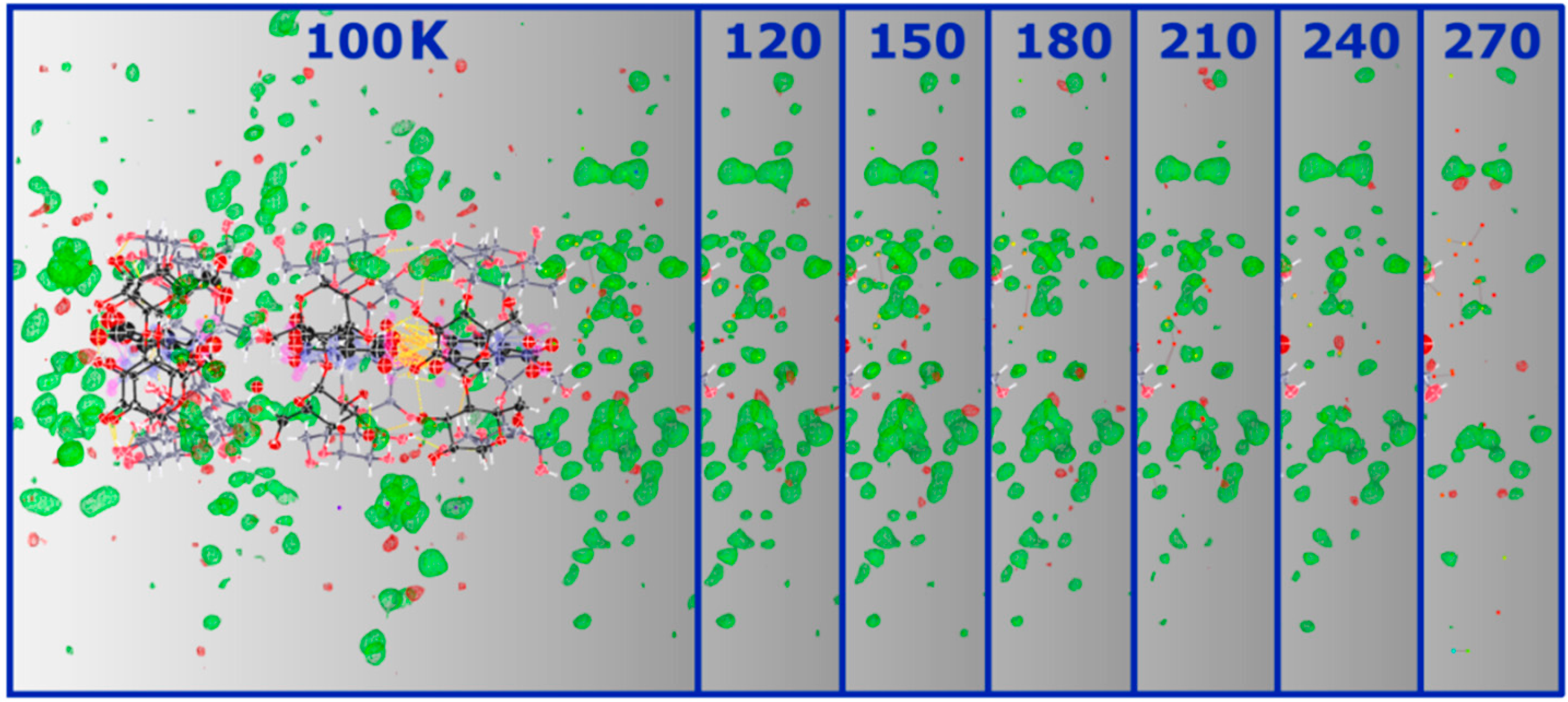
Figure 6.
SHELXLE Fobs-Fcalc maps (at 0.40 e−/Å3) of α-CD∙SA inclusion complex at 100 K and the respective solvent region at the bottom (right-hand side in the picture) of the vase-like cluster at the different temperatures showing the decrease of the signal as a function of increasing temperature. A similar decrease, not shown here, is observed for the other side.
Figure 6.
SHELXLE Fobs-Fcalc maps (at 0.40 e−/Å3) of α-CD∙SA inclusion complex at 100 K and the respective solvent region at the bottom (right-hand side in the picture) of the vase-like cluster at the different temperatures showing the decrease of the signal as a function of increasing temperature. A similar decrease, not shown here, is observed for the other side.
To summarize, the loss of diffraction quality at higher temperatures can be directly ascribed to structural disorder, primarily of the solvent region. It is not unlikely that at even higher temperatures, water molecules vibrate excessively leading to disruption of the crystal packing and structural instability as testified by complete loss of diffraction at 298 K. Given the channel-type packing adopted by the structure, it is conceivable that water molecules find their way out of the structure. Moreover, the multi-temperature study also shows that the guest molecules inside α-CD cavities are freely rotating with no apparent ordering induced by lowering the temperature. This is due to the lack of H-bond interactions between the guest and the host molecules, which, in theory, should help to stabilize the position of the guest molecules. A CSD search reveals that the lack of direct intermolecular contacts between host and guest molecules is not unusual for CD inclusion complexes, and that the guest is indirectly H-bonded to the host via water molecules.
2.2. High-Pressure Crystallization Results
We have investigated complex formation at high pressure up to 0.7 GPa and in the 277–293 K temperature range. Two avenues were explored: first, a diamond-anvil cell (DAC) was loaded with a saturated 1:1 aqueous solution of α-CD and SA and pressure was increased to induce precipitation. No crystals were obtained in this manner. In a second set of experiments, a single crystal of the α-CD∙SA complex crystallized at ambient-pressure conditions was loaded in a DAC together with either its mother liquor or water. Pressure was increased and gradual dissolution of the crystal was observed, similar to what was reported for pure α-CD hydrate form I [
9]. Subsequent to dissolution, no crystallization event was recorded. Upon releasing pressure, there was no microscopic evidence for crystallization of either SA or α-CD alone. While our experiments were not comprehensive, the lack of complex formation at high pressure may be ascribed to concentration, the formation of a particularly stable complex in solution stabilized at high pressure and kinetic effects; it is also conceivable that structural disorder is essential for stability of the α-CD∙SA complex (the entropic contributions of structural disorder in molecular compounds have for instance been discussed in [
31,
32,
33,
34]) and that high pressure, favoring more dense, and in general more ordered states, hinders the
in situ formation of a complex.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}