1. Introduction
Alclometasone dipropionate is a synthetic steroidal prodrug widely used as a topical corticosteroid active pharmaceutical ingredient (API) for the treatment of inflammatory dermatoses such as atopic dermatitis [
1,
2,
3]. As a prodrug, it undergoes metabolic conversion into a pharmacologically active compound after topical administration. First disclosed and patented by Schering Corporation in 1978 [
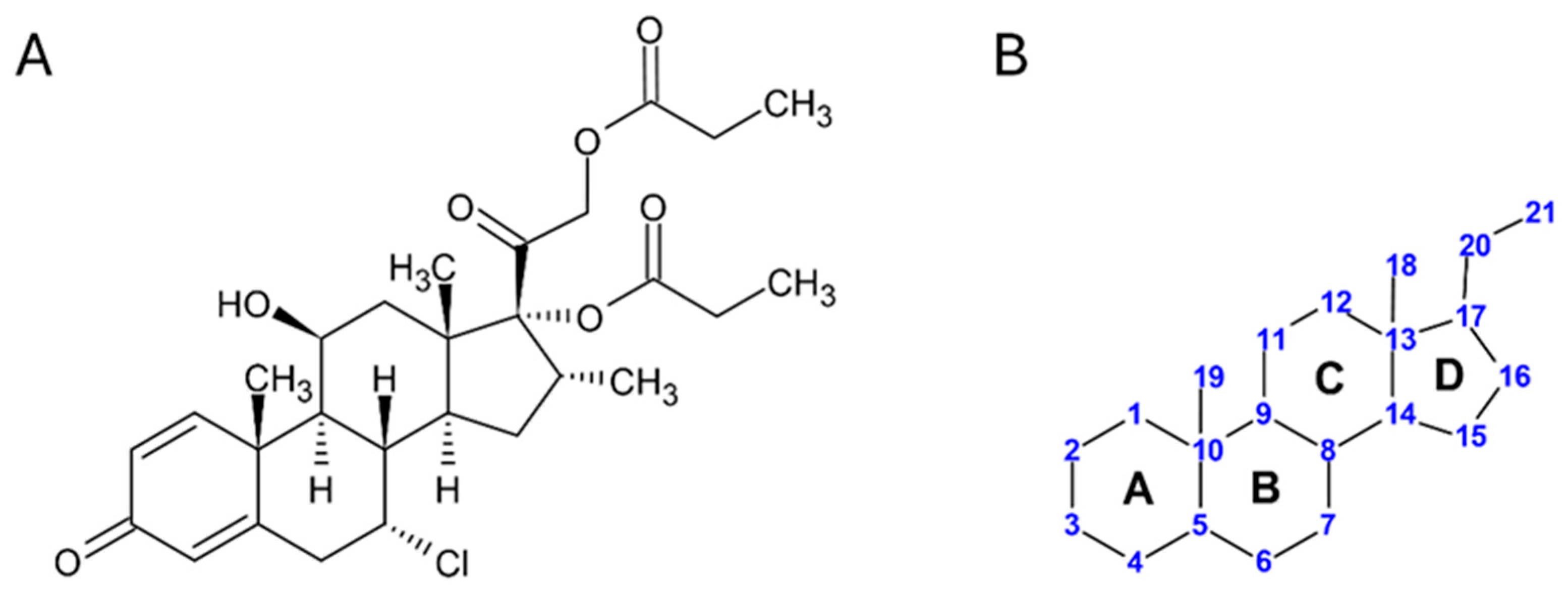
4], the compound was later marketed by GlaxoSmithKline under the brand name “Aclovate,” available as a 0.05% cream or ointment, with several generic versions subsequently launched. Its International Union of Pure and Applied Chemistry (IUPAC) name is [2-[(7R,8S,9S,10R,11S,13S,14S,16R,17R)-7-chloro-11-hydroxy-10,13,16-trimethyl-3-oxo-17-propanoyloxy-7,8,9,11,12,14,15,16-octahydro-6H-cyclopenta[a]phenanthren17-yl]-2-oxoethy] propanoate, with Chemical Abstracts Service (CAS) Registry number 66734-13-2. A two-dimensional molecular representation is shown in
Figure 1A, while
Figure 1B shows the IUPAC-approved ring lettering and carbon numbering for the steroidal pregnane-like skeleton of Alclometasone [
5].
Although Alclometasone dipropionate has been known and clinically used for over four decades, knowledge of its solid-state properties remains limited. In particular, no polymorph-selective crystallization procedures have been described in the literature, and no detailed studies have been published on the crystal form used in commercial drug formulations. This represents a significant knowledge gap, especially given the critical role of polymorphism in defining the pharmaceutical and biological profile of an API. Notably, the current United States Pharmacopeia (USP) monograph for Alclometasone dipropionate does not mention polymorphism.
Polymorphism can significantly influence the key properties of an API, such as solubility, stability, bioavailability, and skin absorption, parameters that are critical for pharmaceutical formulation of the API [
6,
7]. Moreover, a deep understanding of an API’s polymorphic behavior is crucial in Process Chemistry, as we recently highlighted in our study on the Estetrol API [
8]. It is well known that in following a different route of synthesis, as very often happens in the production of API for the generic drug market, different potential impurities can be formed, and it is not always the case that the crystallization method used to isolate the API and the known crystal forms are the right ones to ensure the purging of the process impurities. For this reason, the discovery of new polymorphs of an already existing commercial API is often useful for the improvement of the purification process of the API on an industrial scale [
9].
To date, the only crystallographic study on Alclometasone dipropionate was performed by Kaduk et al. in 2020 [
10], characterizing the USP reference standard using synchrotron powder X-ray diffraction (PXRD) (Beamline 11-BM of the Advanced Photon Source at Argonne National Laboratory, USA).
Their analysis revealed the co-existence of two polymorphs (Form I and Form II), structurally identical except for the conformation of the methyl groups belonging to the propionate chains.
Building on these findings, our research and development (R&D) team initiated a study aimed at optimizing the crystallization process of Alclometasone dipropionate to achieve consistent crystal form, morphology, and particle size distribution, key factors to ensure the robustness of the industrial manufacturing process. PXRD analyses confirmed that the crystallization method, as reported in patents, reproduces the same polymorphic mixture described by Kaduk et al. [
10], thereby reinforcing the link between crystallization procedure and solid-state outcome.
During process modifications, using an acetone/water solvent system, we unexpectedly crystallized Alclometasone dipropionate as pure polymorph Form II, and we also isolated the API in a previously unreported polymorphic form. This new form, herein referred to as Form III, displayed a unique PXRD pattern, distinct thermal behavior, and a characteristic rod-like morphology under Scanning Electron Microscopy (SEM).
In this study, we report the crystal structure elucidation and physicochemical characterization of the novel Form III of Alclometasone dipropionate. Comprehensive structural and thermal analyses were performed using PXRD, Fourier Transform Infrared (FT-IR) spectroscopy, melting-point determination, Differential Scanning Calorimetry (DSC), Thermogravimetric Analysis (TGA/DTG), optical microscopy, and SEM. In addition, we present the first report of a polymorph-selective crystallization method for Alclometasone dipropionate Form II, providing a reproducible route to obtain a structurally distinct and thermally stable form. Together, these findings contribute new insights into the polymorphic landscape of this API, with potential implications for manufacturing development and process control.
3. Results and Discussion
Alclometasone dipropionate was synthesized following previously reported procedures, as detailed in the 1978 patent [
4], with experimental modifications recently introduced by our R&D team [
11]. The crystallization process was initially based on the method described in the originator’s patent, which employed a ternary solvent mixture of methanol, acetone, and isopropyl ether [
4]. Two more recent patent applications also reported the isolation of Alclometasone dipropionate using the same solvent system, specifying a 2:1:3 solvent ratio [
19,
20]. However, the use of three-solvent mixtures is uncommon in industrial API isolation, primarily due to the cost of using multiple organic solvents and the potential risk for uncontrolled variations in solvent composition during crystallization. Such variations can negatively impact the solid-state properties of the resulting product, including crystallization yield and the consistency of particle size distribution.
In light of these limitations, we tried to purify the material coming from the chemical synthesis using a more traditional solvent/anti-solvent crystallization system, capable of minimizing the risk of losses in yield associated with the residual solubility of the compound in the crystallization mother liquor. In this context, we developed two crystallization methods based on the use of acetone and water. One of these procedures led to the selective isolation of Alclometasone dipropionate in pure Form II, which had not been previously obtained as a single polymorphic phase.
The second procedure, under slightly modified conditions, serendipitously resulted in the formation of a previously unreported polymorph of Alclometasone dipropionate, which we have designated as Form III, as it represents the third polymorph identified to date for this compound.
The experimental conditions for both crystallization processes are described in
Section 2.2.1 and allowed for the isolation of pure Form II and Form III in very high yield on gram scale.
Due to the lack of suitable single crystals, the characterization of both polymorphs was performed on crystalline powder samples. Specifically, both forms were subjected to comprehensive solid-state analysis characterization using melting-point determination, DSC, FT-IR, TGA/DTG, optical microscopy, and PXRD, while the SEM analyses were carried out exclusively on Form III. The results from all these techniques confirmed the distinct nature of the two crystalline forms and provided a basis for further investigation into their physicochemical and thermodynamic properties.
3.1. Optical Microscopy and Scanning Electron Microscopy (SEM)
Optical microscopy images of the Alclometasone dipropionate Form II and Form III are reported in
Figure 2A,B, respectively. The Form II is represented by well-defined prisms of an almost similar particle size. Crystals of Form III showed smaller particle size, whose morphology was difficult to accurately attribute solely by optical microscopy. For this reason, a SEM analysis has been performed, revealing the presence of rod-like particles, as shown in
Figure 2C,D. From a pharmacological and physicochemical perspective, the morphology of the crystals can influence the drug’s dissolution rate, solubility, and, consequently, its bioavailability. This difference in morphology between the polymorphs may have important implications, potentially for the future development of new finished dose forms of the API. These aspects are of considerable interest and present promising avenues for further investigation.
3.2. Crystallographic Study of Alclometasone Dipropionate: Powder Diffraction Characterization
The PXRD data were collected and analyzed to solve the crystal structure of the novel polymorph, Form III, of Alclometasone dipropionate. This newly identified polymorphic form represents a significant addition to the existing crystallographic knowledge of the compound, which, until now, was limited to only two known forms (Form I and Form II). The discovery of Form III expands the current polymorphic landscape of Alclometasone dipropionate, providing a more complete and nuanced understanding of its solid-state behavior and physico-chemical properties.
Figure 3A presents a comparison between the experimental PXRD patterns of Form I, Form II and Form III. Form I and Form II correspond to the crystalline phase previously identified in the polymorphic mixture characterized by Kaduk et al. [
10], and both crystallize in an orthorhombic symmetry with one molecule in the asymmetric unit. In contrast, Form III exhibits a markedly different PXRD profile characterized by a triclinic unit cell, two molecules in the asymmetric unit, and a unique set of reflections not observed in Form I and Form II. This confirms the distinct nature and phase purity of the newly identified polymorph. The results are further supported by complementary solid-state analytical techniques, discussed in the following sections.
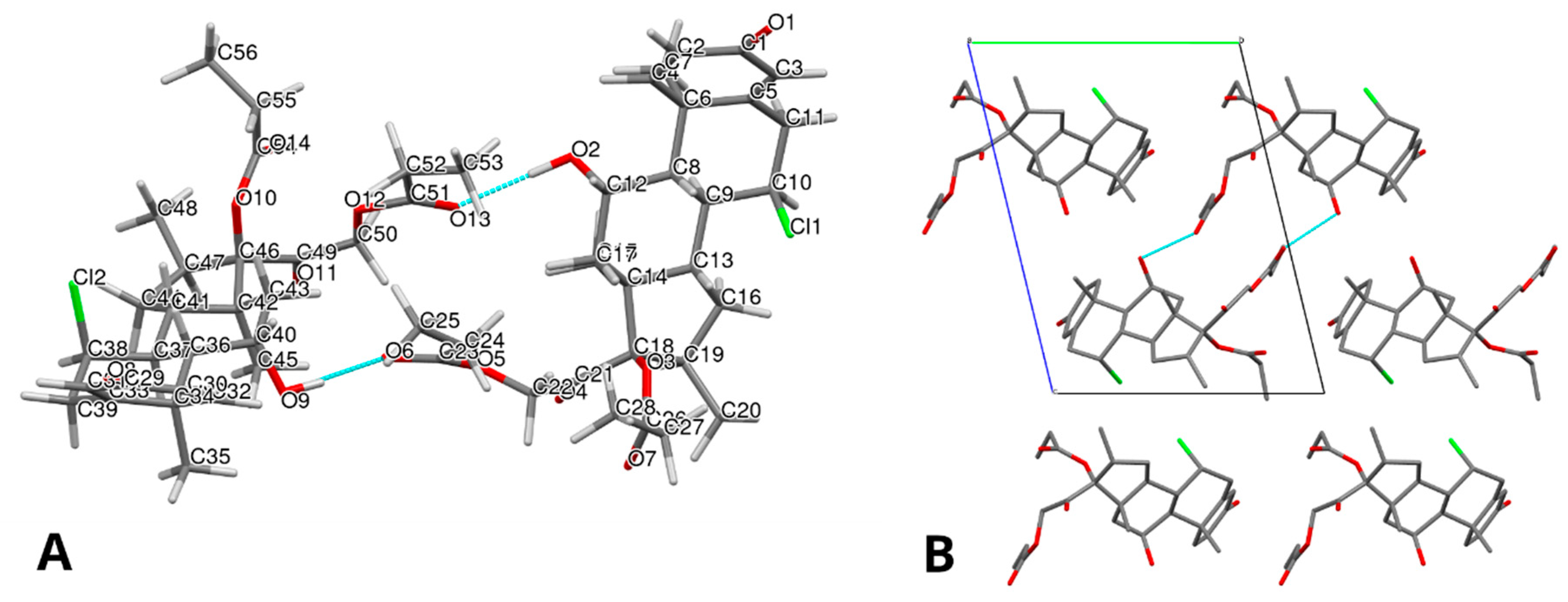
Regarding the structural analysis, a complete crystal structure solution was successfully achieved for Form III. The main PXRD parameters and crystallographic data are summarized in
Table 1. The refined molecular structure, obtained through Rietveld refinement, is shown in
Figure 4A,B, providing insight into the molecular arrangement and packing of the new form.
The corresponding Rietveld plot (
Figure 3B) confirms the good agreement between the experimental and calculated PXRD patterns derived from the solved crystal structure.
To further support the structural assignment,
Figure 3C presents the calculated PXRD patterns of Form I, Form II, and Form III derived from the corresponding solved crystal structures. This comparison highlights the structural similarities between Form I and Form II, and underscores the distinct differences in Form III relative to both. These findings are consistent with the performed complementary solid-state analyses.
The crystal packing of Form III is mainly stabilized by two O–H···O hydrogen bonds: O2–H25···O13 and O9–H62···O6. The hydrogen bonds in Alclometasone dipropionate Form III are listed in
Table 2. These interactions, involving hydroxyl groups as donors and carbonyl oxygen atoms of an ester group as acceptors, generate a centrosymmetric R
22(22) ring motif that links two crystallographically independent molecules, designated as molecule A and molecule B, into hydrogen-bonded dimers. These dimers are further interconnected through a network of weaker C–H···O and C–H···Cl interactions, forming a robust three-dimensional supramolecular framework. These crystallographic observations are further supported by spectroscopic evidence, where the carbonyl band shifts and the analysis of graph-set motifs are fully consistent with the presence of the described O–H···O=C hydrogen bonds.
The crystal structure contains two conformers of the same molecule in the asymmetric unit (Z’ = 2).
Figure 5 shows the superposition of the two molecules, depicted in red and blue, respectively, and highlights appreciable conformational differences in the side chains of the propanoate and 2-oxoethyl propanoate groups. The root-mean-square deviation (RMSD) between the two conformers is 0.5419 Å.
In comparison, Forms I and II are structurally very similar to each other (as described by Kaduk et al. [
10], each with one molecule in the asymmetric unit and nearly identical molecular conformations and packing arrangements. Both forms are stabilized by a single O–H···O hydrogen bond between a hydroxyl and a ketone group, linking pairs of molecules along the c-axis, and supported by C–H···O and C–H···Cl interactions. Their hydrogen-bonding topologies and interaction geometries are largely conserved with only minor differences in terminal methyl orientations and torsional angles.
In contrast, the comparison with Forms I and II reveals pronounced differences in the conformations of both the rings and the side chains, with RMSD values exceeding 1.5 Å.
These structural differences may have implications for the physical properties and stability of this new polymorphic form, and they provide a solid foundation for further studies on polymorph control, formulation strategies, and solid-state behavior of Alclometasone dipropionate.
3.3. Thermal Analyses
In this study, melting-point determination, DSC and TGA were employed to investigate the thermal properties and stability of Form II and Form III of Alclometasone dipropionate. Additionally, DTG was used to provide a more detailed understanding of the sample’s mass loss behavior, offering insight into the thermal degradation mechanisms of each form.
Regarding the melting-point determination, the instrument did not provide a specific outcome, likely due to a particular behavior observed through the instrument camera for both the crystal forms: in fact, in the range of about 190–194 °C for Form II, and 226–230 °C for Form III, samples started to develop gas bubbles, with the simultaneous liquefaction of the solid mass.
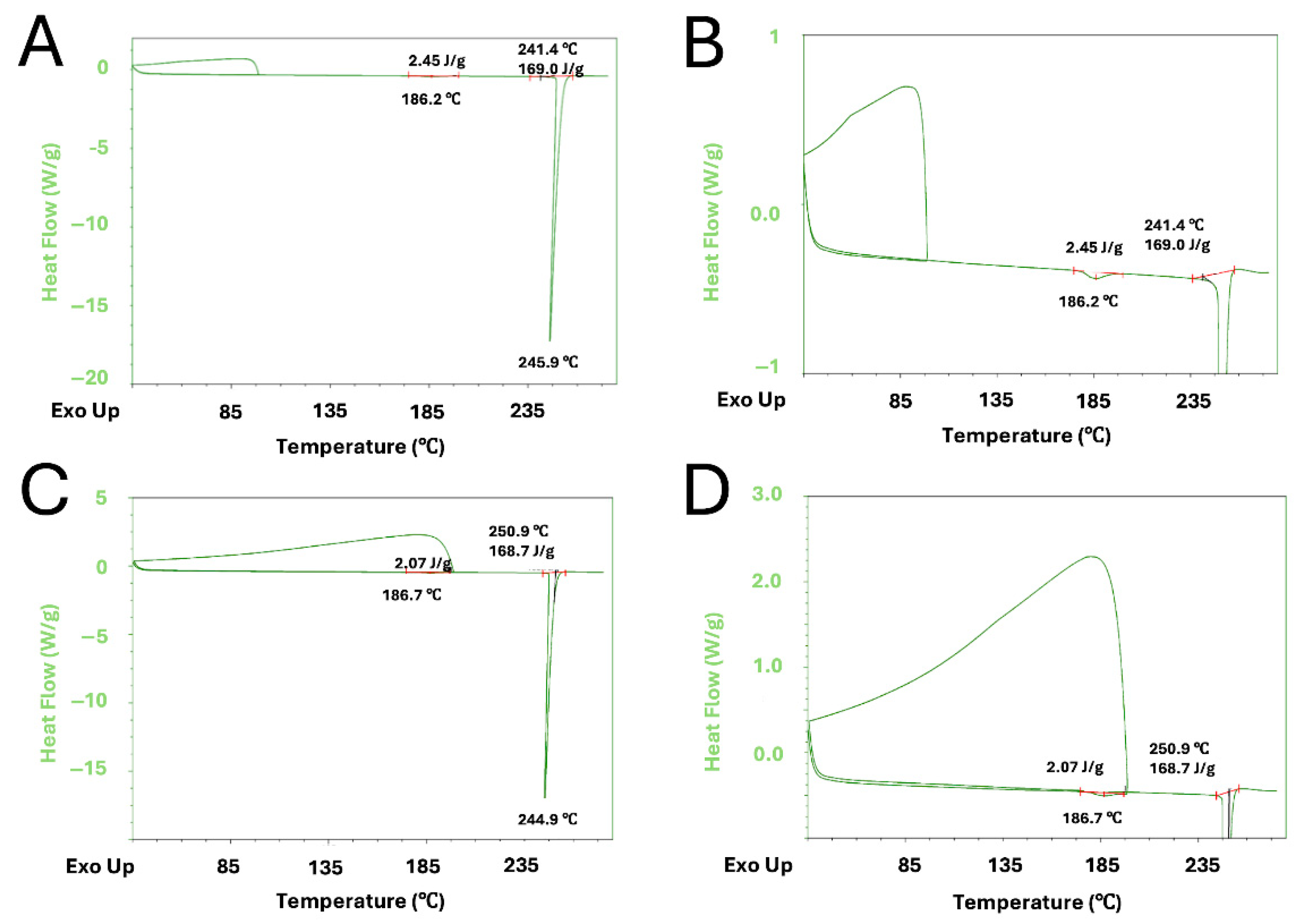
The DSC profiles of Form II and Form III are shown in
Figure 6A,B, respectively.
The thermal analysis reveals significant structural and energetic differences between the two crystalline forms. The DSC thermogram of Form II displays a single sharp endothermic peak around 223.7 °C, almost 30 °C above the gas-evolution/melting event observed in the determination of the melting point. In contrast, the DSC curve of Form III exhibits three distinct thermal events: a small endothermic peak at ~96.0 °C; a second endothermic event at ~188.5 °C, and a major endothermic peak at ~243.0 °C, 13 °C higher than the event that happened in the melting-point experiment.
In order to investigate the nature of the two minor endothermic events of Form III, two additional DSC experiments were conducted. In the first experiment, the sample was heated to 100 °C, in correspondence of the end value of the first minor endothermic event, then cooled and again subjected to a new DSC heating cycle, while in the second experiment the first heating was prolonged to 200 °C, where the second event ended. Thermograms are shown in
Figure 7A–D.
In the first experiment, the heating of Form III to 100 °C did not indicate any thermic event, and the repetition of the complete heating cycle after cooling did not provide any change if compared to the original DSC thermogram (
Figure 7A,B).
On the other hand, in the second experiment, in which the crystal Form III was first heated at 200 °C, the endothermic peak at ~185 °C was no longer observed during the second heating performed after cooling, suggesting the irreversibility of such a thermic event. However, no change in the temperature range of the main endothermic event at ~245 °C was observed.
The corresponding TGA/DTG profiles are shown in
Figure 8A,B.
No considerable weight loss has been observed below ~200 °C, meaning a certain chemical stability up to this temperature for both forms. This behavior finally excludes any involved degradation for both the minor endothermic peaks detected in DSC of Form III. In particular, the peak at ~96.0 °C could be likely related to some traces of residual solvent, not observed in the new DSC runs probably due to spontaneous loss of solvent molecules during the compound storage, considering that the additional experiments were performed several months after the original DSC experiment (
Figure 6B). Regarding the endothermic event detected at ~185 °C, an irreversible solid–solid transition could be supposed, likely causing the conversion of crystal Form III into a different crystal form, known or unknown.
Above 200 °C, both TGA profiles reveal multi-step degradation behaviors, with a clear difference observed in the onset of the first major decomposition step. This step occurs at approximately 200 °C and ends at ~235 °C for Form II, while it begins at around 220 °C and ends at ~250 °C for Form III, covering a temperature range that includes both the events observed in melting-point experiments and the major endothermic peak detected by DSC. The gas evolution observed during the melting-point determination could be proof of a decomposition event, most likely due to the dehydrochlorination of the molecule, with the development of gaseous hydrogen chloride (calculated weight loss = 7%), providing the corresponding 6,7-unsaturated derivative as the main product of degradation [
22]. Anyway, in the whole first decomposition step detected by TGA, both compounds show a comparable mass loss of approximately 10%, indicating the presence of an additional reaction of decomposition besides the loss of hydrogen chloride. In particular, considering the molecular structure of Alclometasone dipropionate, the missing 3.0% of weight loss could be attributed to the dehydration of the secondary hydroxyl group in position 11 (calculated weight loss = 3.5%).
Considering all the above data collected by the different thermal experiments, as well as the potential presence of a degradative multi-step pathway in the first decomposition event shown in TGA, the absence of a corresponding detectable peak in the DSC thermograms in most of the duration of this whole degradative step could be attributable to a situation of apparent flat baseline, probably derived from the simultaneous presence of both exothermic (likely attributable to reactions of decomposition) and endothermic events (likely attributable to the melting and removal of the volatile decomposition products, such as hydrogen chloride and water) having a comparable heating exchange. Therefore, the detection of the main endothermic peak observed in DSC at ~224 °C and ~243 °C for Form II and III, respectively, could be related not to a new specific transformation, but could be just derived from the reduction in the intensity of the exothermic events, with subsequent preponderance of the endothermic ones.
In addition to the first decomposition step, both crystal forms also showed a similar TGA/DTG pattern in the subsequent degradation steps.
3.4. FT-IR Spectroscopy
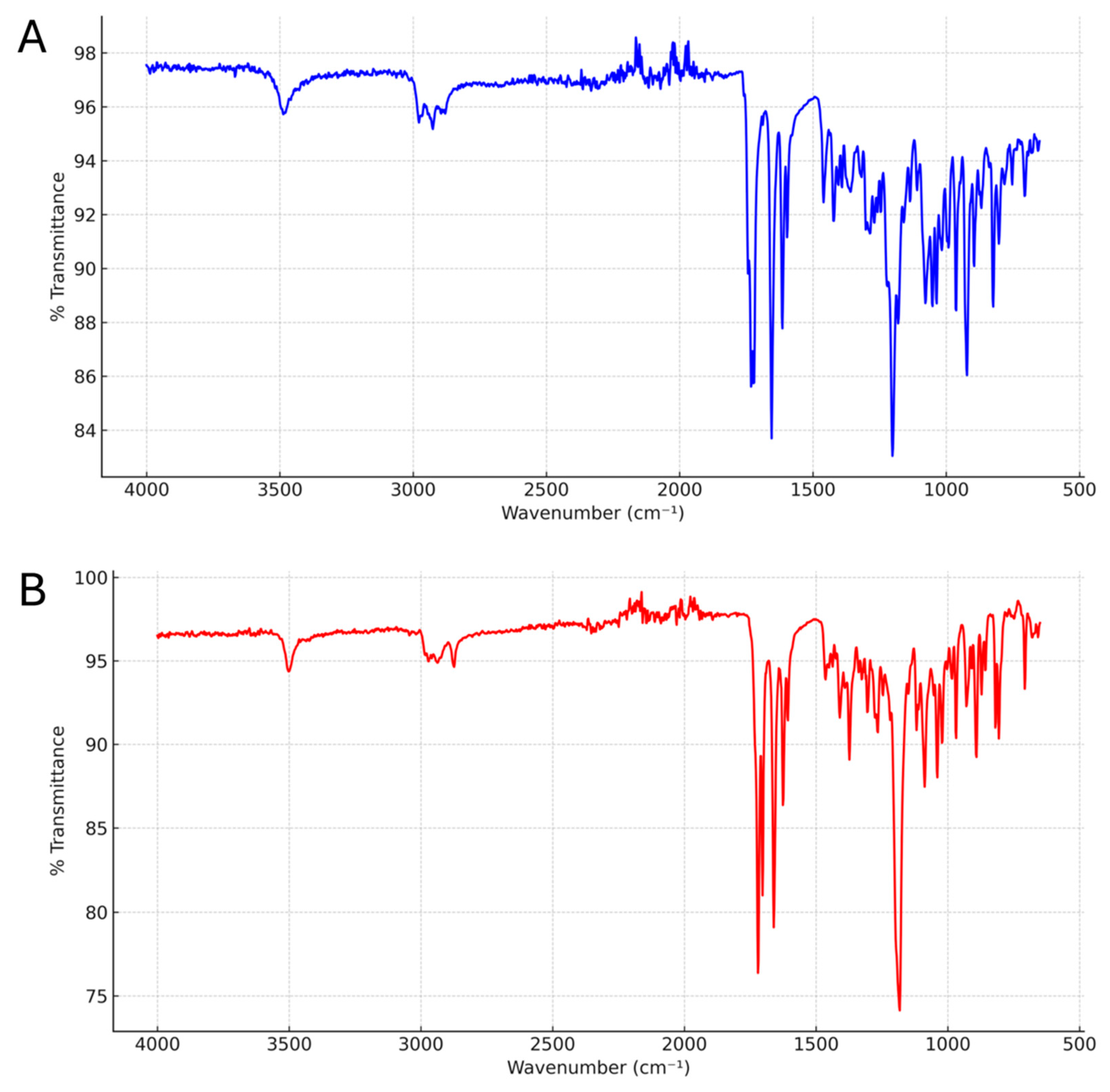
The FT-IR spectra of the Alclometasone dipropionate Form II and Form III are shown in
Figure 9A,B, respectively. Given that the two crystals share the same chemical structure, both spectra exhibit the same fundamental functional groups and largely overlap, with only minor variations in relative peak intensities within the 650–1100 cm
−1 region.
Compared to Form II, Form III shows substantial shifts in the stretching frequencies of both ketone and ester carbonyls, as well as the C–O stretching vibrations of the esters. Specifically, the spectrum of Form II displays three distinct peaks at 1743, 1732, and 1722 cm−1, attributed to the ester carbonyl groups of the two propionate chains and to the unconjugated ketone. In contrast, Form III presents a single, broader peak at 1720 cm−1, and an additional peak at 1703 cm−1, suggesting reduced differentiation between the C=O stretching modes, likely due to altered intermolecular interactions.
These spectral differences are consistent with the presence of intermolecular hydrogen bonding in Form III. The crystal structure analysis confirms the formation of two O–H···O=C hydrogen bonds (specifically O2–H25···O13 and O9–H62···O6), as discussed in detail in
Section 3.2.
The observed redshift of up to ~40 cm
−1 for the carbonyl stretching bands in Form III, compared to Form II, is in line with medium-strength hydrogen bonding as categorized by Jeffrey [
23,
24]. According to this classification, such shifts correspond to interaction energies in the range of approximately 15–25 kJ/mol. The broadening of these bands further supports the involvement of ester carbonyl oxygen atoms in hydrogen bond formation.
Moreover, graph-set analysis of the hydrogen bonding motifs in Form III reveals the presence of both C(4) chains, formed by linear repetition of O–H···O interactions along a crystallographic axis, and a R
22(22) ring motif, involving two molecules connected through two hydrogen bonds forming a closed supramolecular loop of 22 atoms (11 from each molecule, including hydrogen atoms). These graph-set motifs are assigned according to the formalism developed by Etter and Bernstein et al. [
25,
26]. The resulting hydrogen-bond network contributes to the structural cohesion and enhanced thermal stability of Form III. The conjugated ketone (linked to C=C double bonds in the steroidal A ring) is detected at 1650 cm
−1 in Form II and 1660 cm
−1 in Form III.
A similar shift is observed in the C–O stretching region, with characteristic peaks appearing at 1200 cm
−1 in Form II and at 1180 cm
−1 in Form III. Additionally, C=C stretching vibrations involving the carbon atoms in positions 1–2 and 4–5 are present in both samples. In Form III, these peaks are located at 1625 and 1606 cm
−1, while in Form II, they appear at slightly lower frequencies: 1614 and 1597 cm
−1 [
27,
28].
These FT-IR results, taken together with PXRD and thermal analysis data, provide a comprehensive understanding of the structural and intermolecular differences between the polymorphs, further supporting the uniqueness of Form III.
4. Conclusions
In this study, the crystallization method for the isolation of pure Form II of Alclometasone dipropionate was described, and a new polymorph, designated as Form III, was identified and characterized. The optimization of the crystallization method facilitated the isolation of this novel crystalline form, which differs from the previously known polymorphs (Form I and Form II) in terms of its structural and thermal properties.
PXRD analysis clearly distinguishes Form III from the known forms. FT-IR spectroscopy revealed variations in vibrational signals, suggesting a different conformational arrangement. Additionally, melting-point, DSC, and TGA/DTG analyses demonstrated differences between the polymorph Forms II and III, and lastly, SEM showed rod-shaped crystals for Form III, further differentiating the two forms.
This study contributes to a deeper understanding of the solid-state properties of Alclometasone dipropionate, which could significantly impact both the industrial chemical manufacturing of the drug substance and the future pharmaceutical development of the new Form III of the API. In particular, the distinct particle shape of Form III may offer advantages in terms of formulation processability.
Even though we successfully discovered a new crystalline form of Alclometasone dipropionate, the in-depth exploration of its solid state is still ongoing, with the aim of finding new polymorphs and expanding the possibility of choosing a form of crystallization which will enable the most efficient industrial purification of crude Alclometasone dipropionate manufactured according to our own route of synthesis.







 ,
, 






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}