

Structural Analysis of PlyKp104, a Novel Phage Endoysin

Abstract
1. Introduction
2. Materials and Methods
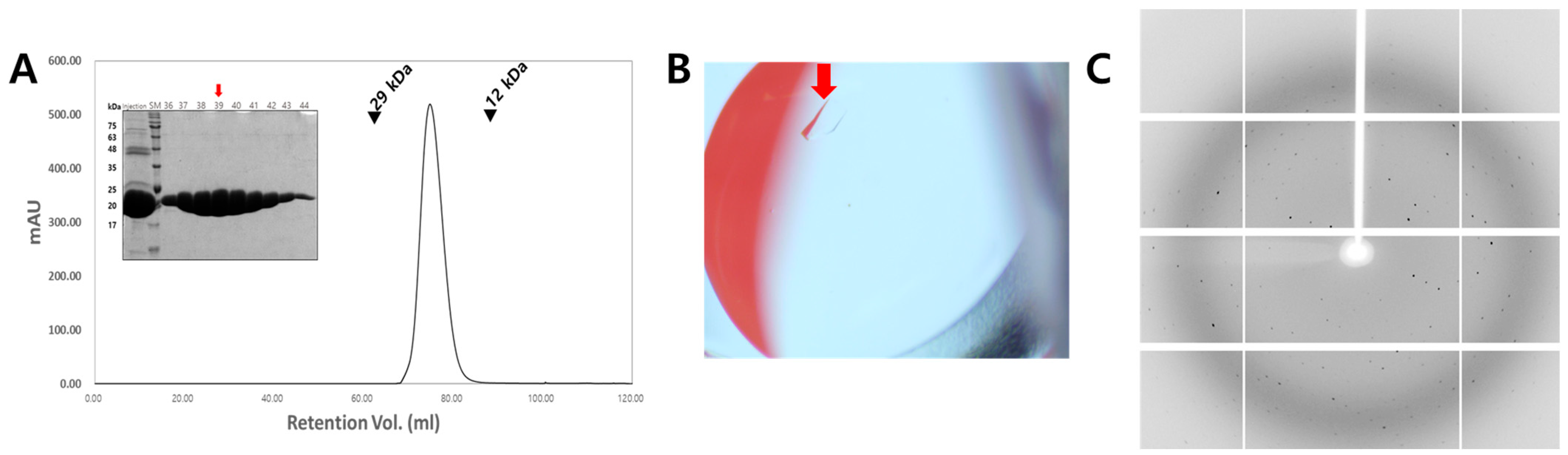
2.1. Cloning, Protein Expression, and Purification
2.2. Crystallization
2.3. X-Ray Diffraction Data Collection
2.4. Structure Determination
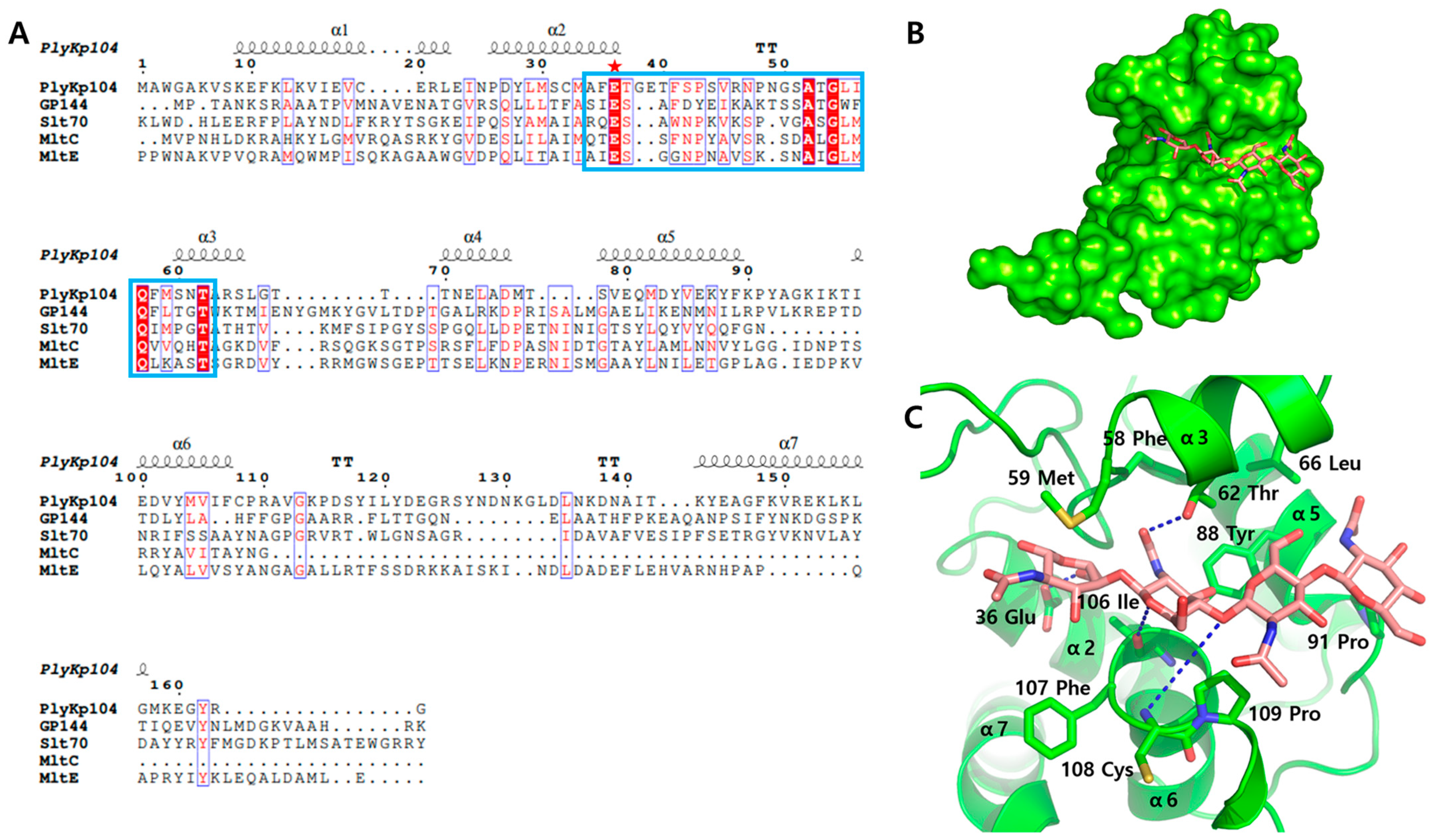
2.5. Bioinformatics and Structure Analysis
3. Results
3.1. Structure Determination
3.2. Crystal Structure of PlyKp104
3.3. Active Site Structure of PlyKp104
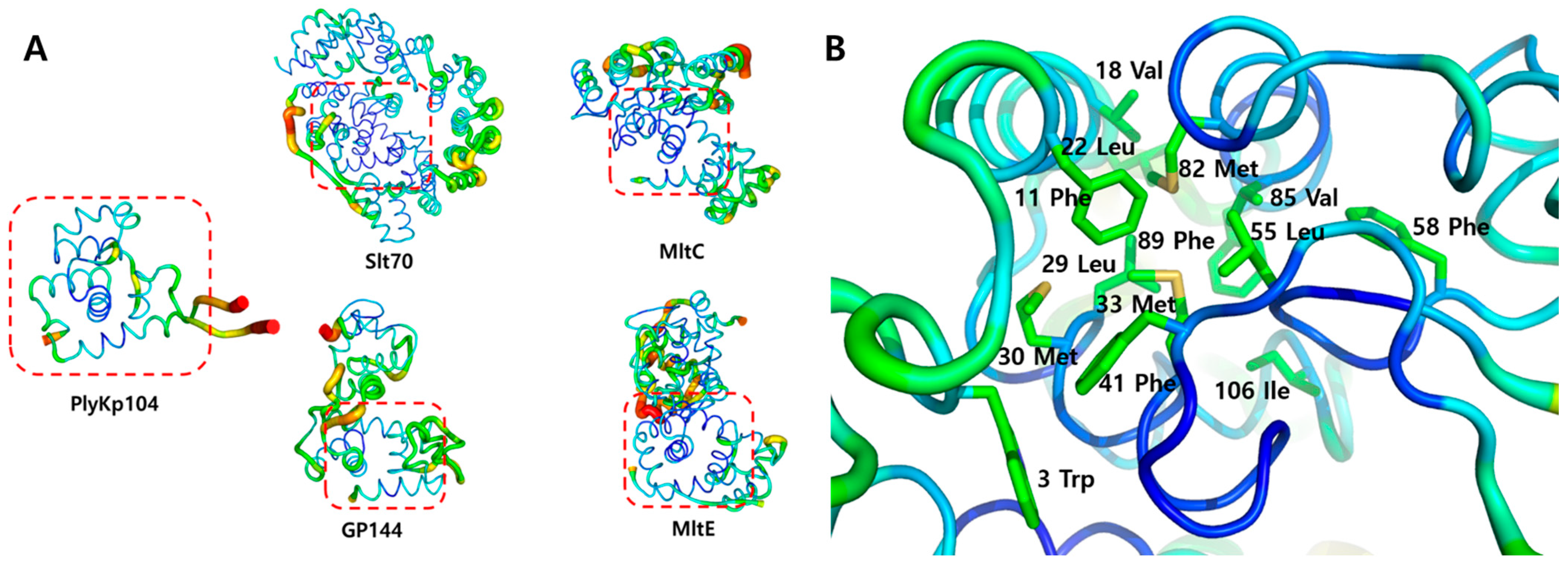
3.4. Structural Stability Analysis of PlyKp104
3.5. Potential Lipid-Binding Site of PlyKp104
4. Discussion
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Breijyeh, Z.; Jubeh, B.; Karaman, R. Resistance of Gram-Negative Bacteria to Current Antibacterial Agents and Approaches to Resolve It. Molecules 2020, 25, 1340. [Google Scholar] [CrossRef] [PubMed]
- Wright, G.D. Q&A: Antibiotic resistance: Where does it come from and what can we do about it? BMC Biol. 2010, 8, 123. [Google Scholar]
- Baran, A.; Kwiatkowska, A.; Potocki, L. Antibiotics and bacterial resistance—A short story of an endless arms race. Int. J. Mol. Sci. 2023, 24, 5777. [Google Scholar] [CrossRef] [PubMed]
- Uddin, T.M.; Chakraborty, A.J.; Khusro, A.; Zidan, B.R.M.; Mitra, S.; Emran, T.B.; Dhama, K.; Ripon, M.K.H.; Gajdács, M.; Sahibzada, M.U.K. Antibiotic resistance in microbes: History, mechanisms, therapeutic strategies and future prospects. J. Infect. Public Health 2021, 14, 1750–1766. [Google Scholar] [CrossRef]
- Baharoglu, Z.; Garriss, G.; Mazel, D. Multiple pathways of genome plasticity leading to development of antibiotic resistance. Antibiotics 2013, 2, 288–315. [Google Scholar] [CrossRef]
- Pulingam, T.; Parumasivam, T.; Gazzali, A.M.; Sulaiman, A.M.; Chee, J.Y.; Lakshmanan, M.; Chin, C.F.; Sudesh, K. Antimicrobial resistance: Prevalence, economic burden, mechanisms of resistance and strategies to overcome. Eur. J. Pharm. Sci. 2022, 170, 106103. [Google Scholar] [CrossRef]
- Salam, M.A.; Al-Amin, M.Y.; Salam, M.T.; Pawar, J.S.; Akhter, N.; Rabaan, A.A.; Alqumber, M.A.A. Antimicrobial Resistance: A Growing Serious Threat for Global Public Health. Healthcare 2023, 11, 1946. [Google Scholar] [CrossRef]
- Murugaiyan, J.; Kumar, P.A.; Rao, G.S.; Iskandar, K.; Hawser, S.; Hays, J.P.; Mohsen, Y.; Adukkadukkam, S.; Awuah, W.A.; Jose, R.A.M.; et al. Progress in Alternative Strategies to Combat Antimicrobial Resistance: Focus on Antibiotics. Antibiotics 2022, 11, 200. [Google Scholar] [CrossRef]
- Gupta, R.; Sharma, S. Role of alternatives to antibiotics in mitigating the antimicrobial resistance crisis. Indian J. Med. Res. 2022, 156, 464–477. [Google Scholar] [CrossRef]
- Schmelcher, M.; Donovan, D.M.; Loessner, M.J. Bacteriophage endolysins as novel antimicrobials. Future Microbiol. 2012, 7, 1147–1171. [Google Scholar] [CrossRef]
- Lu, Y.; Wang, Y.; Wang, J.; Zhao, Y.; Zhong, Q.; Li, G.; Fu, Z.; Lu, S. Phage endolysin LysP108 showed promising antibacterial potential against methicillin-resistant Staphylococcus aureus. Front. Cell. Infect. Microbiol. 2021, 11, 668430. [Google Scholar] [CrossRef] [PubMed]
- Sisson, H.M.; Jackson, S.A.; Fagerlund, R.D.; Warring, S.L.; Fineran, P.C. Gram-negative endolysins: Overcoming the outer membrane obstacle. Curr. Opin. Microbiol. 2024, 78, 102433. [Google Scholar] [CrossRef]
- Vasina, D.V.; Antonova, N.P.; Gushchin, V.A.; Aleshkin, A.V.; Fursov, M.V.; Fursova, A.D.; Gancheva, P.G.; Grigoriev, I.V.; Grinkevich, P.; Kondratev, A.V.; et al. Development of novel antimicrobials with engineered endolysin LysECD7-SMAP to combat Gram-negative bacterial infections. J. Biomed. Sci. 2024, 31, 75. [Google Scholar] [CrossRef]
- Cernooka, E.; Rumnieks, J.; Zrelovs, N.; Tars, K.; Kazaks, A. Diversity of the lysozyme fold: Structure of the catalytic domain from an unusual endolysin encoded by phage Enc34. Sci. Rep. 2022, 12, 5005. [Google Scholar] [CrossRef]
- Euler Chad, W.; Raz, A.; Hernandez, A.; Serrano, A.; Xu, S.; Andersson, M.; Zou, G.; Zhang, Y.; Fischetti Vincent, A.; Li, J. PlyKp104, a Novel Phage Lysin for the Treatment of Klebsiella pneumoniae, Pseudomonas aeruginosa, and Other Gram-Negative ESKAPE Pathogens. Antimicrob. Agents Chemother. 2023, 67, e01519-22. [Google Scholar] [CrossRef]
- Park, S.-Y.; Ha, S.-C.; Kim, Y.-G. The protein crystallography beamlines at the pohang light source II. Biodesign 2017, 5, 30–34. [Google Scholar]
- Otwinowski, Z.; Minor, W. Processing of X-ray diffraction data collected in oscillation mode. In Methods Enzymol; Academic Press: Cambridge, MA, USA, 1997; Volume 276, pp. 307–326. [Google Scholar]
- Winter, G.; Waterman, D.G.; Parkhurst, J.M.; Brewster, A.S.; Gildea, R.J.; Gerstel, M.; Fuentes-Montero, L.; Vollmar, M.; Michels-Clark, T.; Young, I.D. DIALS: Implementation and evaluation of a new integration package. Biol. Crystallogr. 2018, 74, 85–97. [Google Scholar] [CrossRef]
- Liebschner, D.; Afonine, P.V.; Baker, M.L.; Bunkóczi, G.; Chen, V.B.; Croll, T.I.; Hintze, B.; Hung, L.-W.; Jain, S.; McCoy, A.J. Macromolecular structure determination using X-rays, neutrons and electrons: Recent developments in Phenix. Biol. Crystallogr. 2019, 75, 861–877. [Google Scholar] [CrossRef]
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Žídek, A.; Potapenko, A.; et al. Highly accurate protein structure prediction with AlphaFold. Nature 2021, 596, 583–589. [Google Scholar] [CrossRef]
- Emsley, P.; Lohkamp, B.; Scott, W.G.; Cowtan, K. Features and development of Coot. Biol. Crystallogr. 2010, 66, 486–501. [Google Scholar] [CrossRef]
- Holm, L.; Laiho, A.; Törönen, P.; Salgado, M. DALI shines a light on remote homologs: One hundred discoveries. Protein Sci. 2023, 32, e4519. [Google Scholar] [CrossRef] [PubMed]
- Sievers, F.; Higgins, D.G. Clustal Omega for making accurate alignments of many protein sequences. Protein Sci. 2018, 27, 135–145. [Google Scholar] [CrossRef] [PubMed]
- Robert, X.; Gouet, P. Deciphering key features in protein structures with the new ENDscript server. Nucleic Acids Res. 2014, 42, W320–W324. [Google Scholar] [CrossRef] [PubMed]
- Fokine, A.; Miroshnikov, K.A.; Shneider, M.M.; Mesyanzhinov, V.V.; Rossmann, M.G. Structure of the bacteriophage φKZ lytic transglycosylase gp144. J. Biol. Chem. 2008, 283, 7242–7250. [Google Scholar] [CrossRef]
- Dik, D.A.; Marous, D.R.; Fisher, J.F.; Mobashery, S. Lytic transglycosylases: Concinnity in concision of the bacterial cell wall. Crit. Rev. Biochem. Mol. Biol. 2017, 52, 503–542. [Google Scholar] [CrossRef]
- Vermassen, A.; Leroy, S.; Talon, R.; Provot, C.; Popowska, M.; Desvaux, M. Cell Wall Hydrolases in Bacteria: Insight on the Diversity of Cell Wall Amidases, Glycosidases and Peptidases Toward Peptidoglycan. Front. Microbiol. 2019, 10, 331. [Google Scholar] [CrossRef]
- Alcorlo, M.; Martínez-Caballero, S.; Molina, R.; Hermoso, J.A. Carbohydrate recognition and lysis by bacterial peptidoglycan hydrolases. Curr. Opin. Struct. Biol. 2017, 44, 87–100. [Google Scholar] [CrossRef]
- Sun, Z.; Liu, Q.; Qu, G.; Feng, Y.; Reetz, M.T. Utility of B-Factors in Protein Science: Interpreting Rigidity, Flexibility, and Internal Motion and Engineering Thermostability. Chem. Rev. 2019, 119, 1626–1665. [Google Scholar] [CrossRef]
- Broendum, S.S.; Buckle, A.M.; McGowan, S. Catalytic diversity and cell wall binding repeats in the phage-encoded endolysins. Mol. Microbiol. 2018, 110, 879–896. [Google Scholar] [CrossRef]
- Artola-Recolons, C.; Lee, M.; Bernardo-García, N.; Blázquez, B.; Hesek, D.; Bartual, S.; Mahasenan, K.; Lastochkin, E.; Pi, H.; Boggess, B. Structure and cell wall cleavage by modular lytic transglycosylase MltC of Escherichia coli. ACS Chem. Biol. 2014, 9, 2058–2066. [Google Scholar] [CrossRef]
- Artola-Recolons, C.; Carrasco-López, C.; Llarrull, L.I.; Kumarasiri, M.; Lastochkin, E.; Martínez de Ilarduya, I.; Meindl, K.; Usón, I.; Mobashery, S.; Hermoso, J.A. High-Resolution Crystal Structure of MltE, an Outer Membrane-Anchored Endolytic Peptidoglycan Lytic Transglycosylase from Escherichia coli. Biochemistry 2011, 50, 2384–2386. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameter | |
| X-ray source | 7A beamline, PLS-II |
| Wavelength (Å) | 0.9793 |
| Space group | C2221 |
| Cell dimension | |
| a, b, c (Å) | 67.73, 89.58, 67.74 |
| α, β, γ (°) | 90, 90, 90 |
| Resolution (Å) | 50.00–1.85 (1.88–1.85) |
| Unique reflections | 18,434 (710) |
| Completeness (%) | 98.7 (77.8) |
| Redundancy | 11.8 (6.5) |
| I/σ | 22.3 (1.3) |
| Rmerge | 0.123 (0.694) |
| Rmeas | 0.129 (0.747) |
| CC1/2 | 0.996 (0.867) |
| CC* | 0.999 (0.964) |
| Refinement | |
| Resolution (Å) | 30.29–1.85 |
| Rwork a | 0.19 (0.26) |
| Rfree b | 0.22 (0.30) |
| R.m.s. deviations | |
| Bonds (Å) | 0.007 |
| Angles (°) | 0.877 |
| B factors (Å2) | |
| Protein | 27.02 |
| Ramachandran plot | |
| Favored (%) | 98.70 |
| Allowed (%) | 1.30 |
| Disallowed (%) | 0.00 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Choi, J.-M. Structural Analysis of PlyKp104, a Novel Phage Endoysin. Crystals 2025, 15, 448. https://doi.org/10.3390/cryst15050448
Choi J-M. Structural Analysis of PlyKp104, a Novel Phage Endoysin. Crystals. 2025; 15(5):448. https://doi.org/10.3390/cryst15050448
Chicago/Turabian StyleChoi, Jung-Min. 2025. "Structural Analysis of PlyKp104, a Novel Phage Endoysin" Crystals 15, no. 5: 448. https://doi.org/10.3390/cryst15050448
APA StyleChoi, J.-M. (2025). Structural Analysis of PlyKp104, a Novel Phage Endoysin. Crystals, 15(5), 448. https://doi.org/10.3390/cryst15050448