

Structural Study of Thermostable Ginsenoside β-Glucosidase from Caldicellulosiruptor bescii

Abstract
1. Introduction
2. Materials and Methods
2.1. Cloning, Protein Expression, and Purification
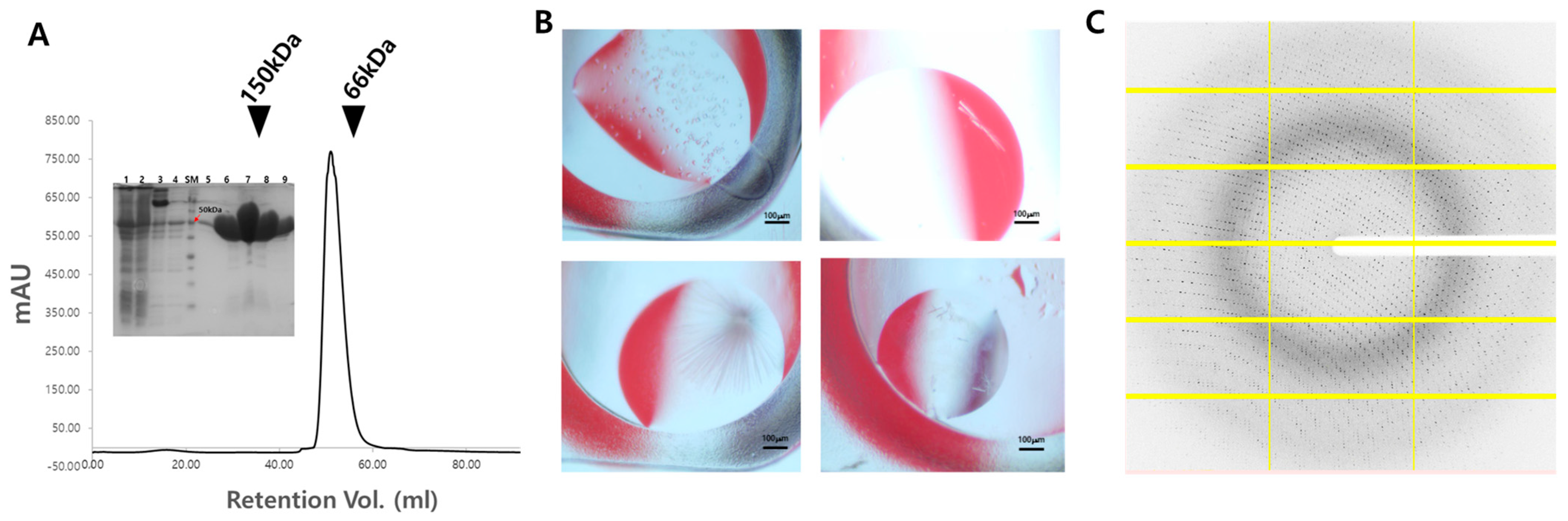
2.2. Crystallization
2.3. X-Ray Diffraction Data Collection
2.4. Structure Determination
2.5. Bioinformatics and Structure Analysis
3. Results
3.1. Structure Determination of CbBGL
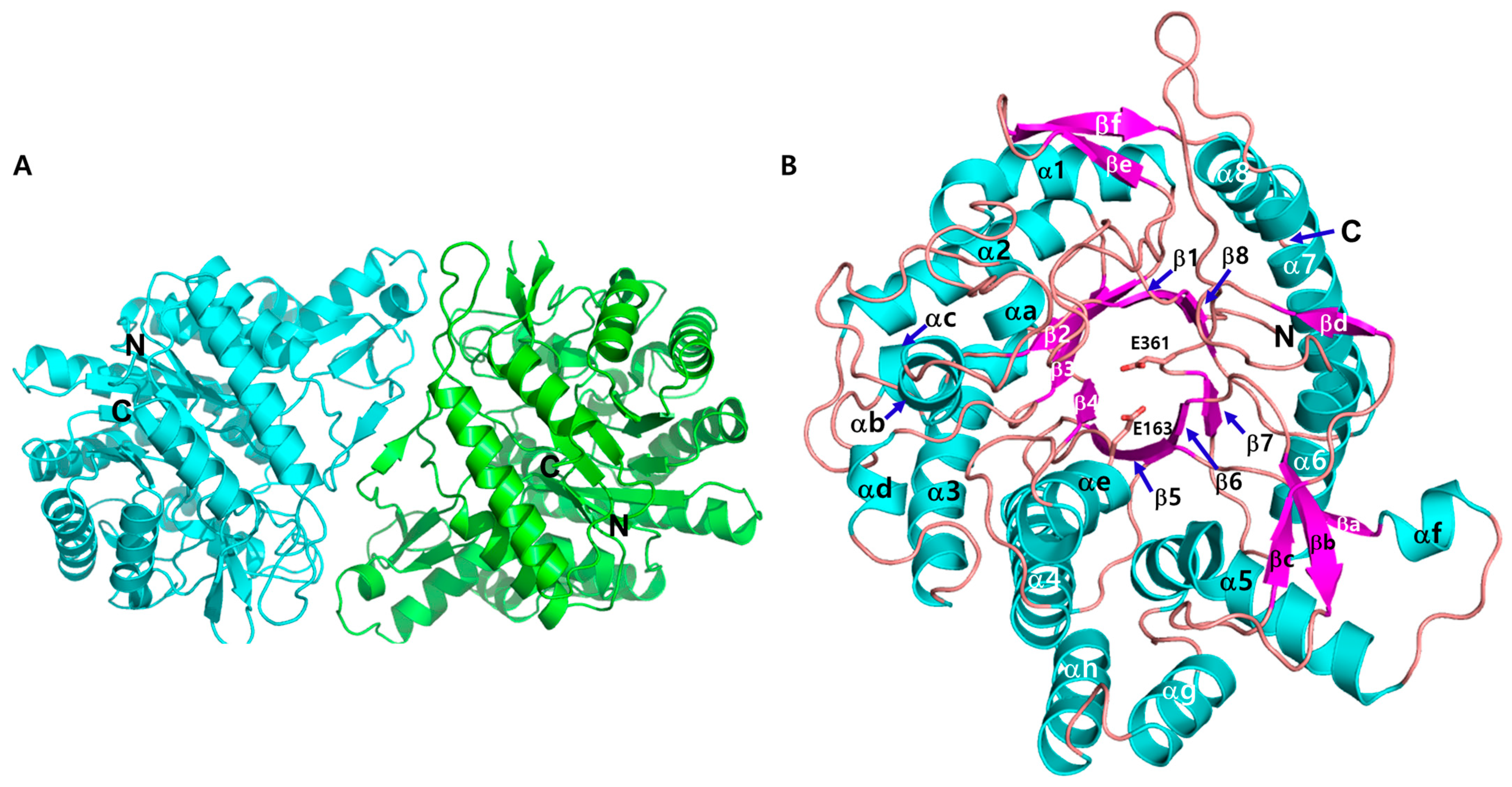
3.2. Crystal Structure of CbBGL
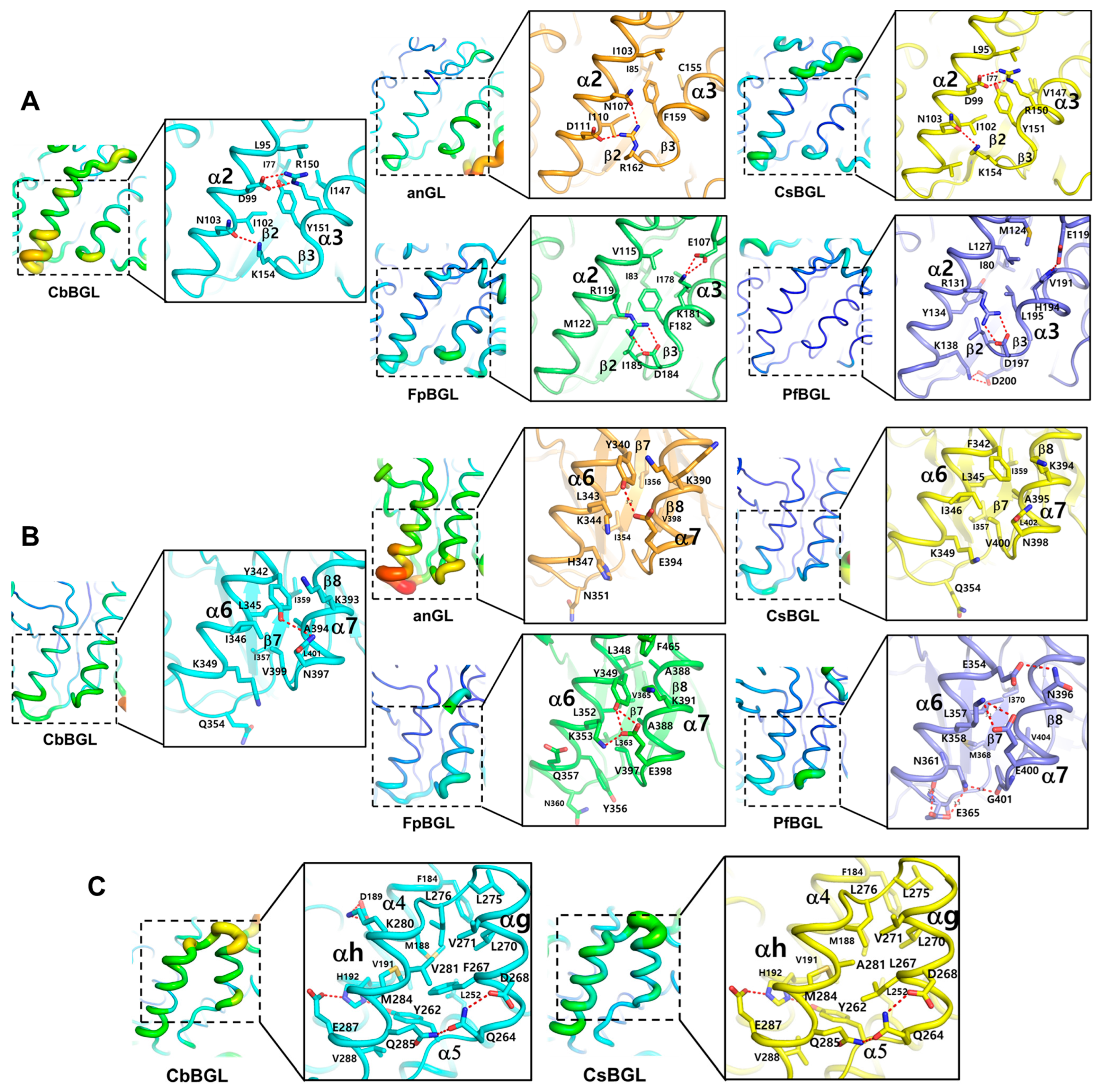
3.3. Comparative Structural Investigation of CbBGL Thermostability
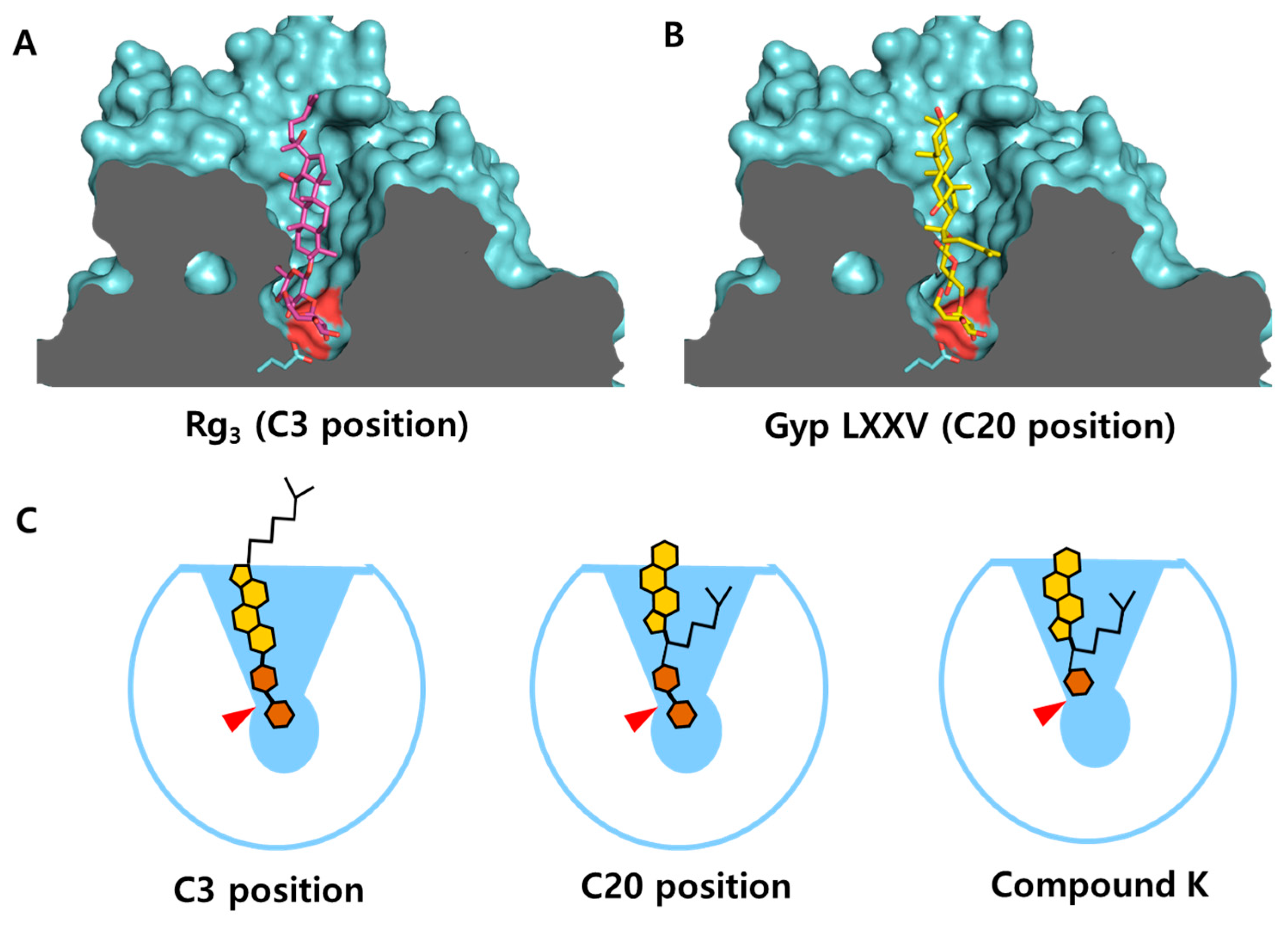
3.4. Structural Characterization of the Substrate Binding Cleft of CbBGL
4. Discussion
Supplementary Materials
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Jia, L.; Zhao, Y. Current evaluation of the millennium phytomedicine–ginseng (I): Etymology, pharmacognosy, phytochemistry, market and regulations. Curr. Med. Chem. 2009, 16, 2475–2484. [Google Scholar] [CrossRef] [PubMed]
- Lü, J.M.; Yao, Q.; Chen, C. Ginseng compounds: An update on their molecular mechanisms and medical applications. Curr. Vasc. Pharmacol. 2009, 7, 293–302. [Google Scholar] [CrossRef] [PubMed]
- Newman, D.J.; Cragg, G.M. Natural products as sources of new drugs over the 30 years from 1981 to 2010. J. Nat. Prod. 2012, 75, 311–335. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.R.; Leung, C.Y.; Ho, H.M.; Chai, S.; Yau, L.F.; Zhao, Z.Z.; Jiang, Z.H. Quantitative comparison of ginsenosides and polyacetylenes in wild and cultivated American ginseng. Chem. Biodivers. 2010, 7, 975–983. [Google Scholar] [CrossRef]
- Chen, W.; Balan, P.; Popovich, D.G. Analysis of Ginsenoside Content (Panax ginseng) from Different Regions. Molecules 2019, 24, 3491. [Google Scholar] [CrossRef]
- Park, S.J.; Choi, J.M.; Kyeong, H.H.; Kim, S.G.; Kim, H.S. Rational design of a β-glycosidase with high regiospecificity for triterpenoid tailoring. ChemBioChem 2015, 16, 854–860. [Google Scholar] [CrossRef]
- Zhu, H.; Zhang, R.; Huang, Z.; Zhou, J. Progress in the Conversion of Ginsenoside Rb1 into Minor Ginsenosides Using β-Glucosidases. Foods 2023, 12, 397. [Google Scholar] [CrossRef]
- Chen, R.J.; Chung, T.Y.; Li, F.Y.; Lin, N.H.; Tzen, J.T. Effect of sugar positions in ginsenosides and their inhibitory potency on Na+/K+-ATPase activity. Acta Pharmacol. Sin. 2009, 30, 61–69. [Google Scholar] [CrossRef]
- Wang, W.; Zhang, X.; Qin, J.J.; Voruganti, S.; Nag, S.A.; Wang, M.H.; Wang, H.; Zhang, R. Natural product ginsenoside 25-OCH3-PPD inhibits breast cancer growth and metastasis through down-regulating MDM2. PLoS ONE 2012, 7, e41586. [Google Scholar] [CrossRef]
- Wu, Y.L.; Wan, Y.; Jin, X.J.; OuYang, B.Q.; Bai, T.; Zhao, Y.Q.; Nan, J.X. 25-OCH3-PPD induces the apoptosis of activated t-HSC/Cl-6 cells via c-FLIP-mediated NF-kappaB activation. Chem. Biol. Interact. 2011, 194, 106–112. [Google Scholar] [CrossRef]
- Xu, Q.F.; Fang, X.L.; Chen, D.F. Pharmacokinetics and bioavailability of ginsenoside Rb1 and Rg1 from Panax notoginseng in rats. J. Ethnopharmacol. 2003, 84, 187–192. [Google Scholar] [CrossRef]
- Cheng, L.Q.; Kim, M.K.; Lee, J.W.; Lee, Y.J.; Yang, D.C. Conversion of major ginsenoside Rb1 to ginsenoside F2 by Caulobacter leidyia. Biotechnol. Lett. 2006, 28, 1121–1127. [Google Scholar] [CrossRef]
- Weymouth-Wilson, A.C. The role of carbohydrates in biologically active natural products. Nat. Prod. Rep. 1997, 14, 99–110. [Google Scholar] [CrossRef]
- Chang, Y.H.; Ng, P.K. Effects of extrusion process variables on extractable ginsenosides in wheat-ginseng extrudates. J. Agric. Food Chem. 2009, 57, 2356–2362. [Google Scholar] [CrossRef] [PubMed]
- Han, B.H.; Park, M.H.; Han, Y.N.; Woo, L.K.; Sankawa, U.; Yahara, S.; Tanaka, O. Degradation of ginseng saponins under mild acidic conditions. Planta Med. 1982, 44, 146–149. [Google Scholar] [CrossRef] [PubMed]
- Li, W.N.; Fan, D.D. Biocatalytic strategies for the production of ginsenosides using glycosidase: Current state and perspectives. Appl. Microbiol. Biotechnol. 2020, 104, 3807–3823. [Google Scholar] [CrossRef]
- Li, W.; Jiang, Y.; Liu, Y.; Li, C.; Fan, D. Biocatalytic strategies in producing ginsenoside by glycosidase-a review. Sheng Wu Gong Cheng Xue Bao 2019, 35, 1590–1606. [Google Scholar] [PubMed]
- Park, C.S.; Yoo, M.H.; Noh, K.H.; Oh, D.K. Biotransformation of ginsenosides by hydrolyzing the sugar moieties of ginsenosides using microbial glycosidases. Appl. Microbiol. Biotechnol. 2010, 87, 9–19. [Google Scholar] [CrossRef]
- Cui, C.H.; Kim, J.K.; Kim, S.C.; Im, W.T. Characterization of a Ginsenoside-Transforming beta-glucosidase from Paenibacillus mucilaginosus and Its Application for Enhanced Production of Minor Ginsenoside F2. PLoS ONE 2014, 9, e85727. [Google Scholar]
- Kim, J.K.; Cui, C.H.; Liu, Q.; Yoon, M.H.; Kim, S.C.; Im, W.T. Mass production of the ginsenoside Rg3(S) through the combinative use of two glycoside hydrolases. Food Chem. 2013, 141, 1369–1377. [Google Scholar] [CrossRef]
- Kim, M.-J.; Upadhyaya, J.; Yoon, M.-S.; Ryu, N.S.; Song, Y.E.; Park, H.-W.; Kim, Y.-H.; Kim, M.-K. Highly regioselective biotransformation of ginsenoside Rb2 into compound Y and compound K by β-glycosidase purified from Armillaria mellea mycelia. J. Ginseng Res. 2018, 42, 504–511. [Google Scholar] [CrossRef]
- Shin, K.C.; Kim, T.H.; Choi, J.H.; Oh, D.K. Complete Biotransformation of Protopanaxadiol-Type Ginsenosides to 20-O-β-Glucopyranosyl-20(S)-protopanaxadiol Using a Novel and Thermostable β-Glucosidase. J. Agric. Food Chem. 2018, 66, 2822–2829. [Google Scholar] [CrossRef]
- Noh, K.-H.; Son, J.-W.; Kim, H.-J.; Oh, D.-K. Ginsenoside Compound K Production from Ginseng Root Extract by a Thermostable β-Glycosidase from Sulfolobus solfataricus. Biosci. Biotechnol. Biochem. 2009, 73, 316–321. [Google Scholar] [CrossRef]
- Yang, L.; Zou, H.; Gao, Y.; Luo, J.; Xie, X.; Meng, W.; Zhou, H.; Tan, Z. Insights into gastrointestinal microbiota-generated ginsenoside metabolites and their bioactivities. Drug Metab. Rev. 2020, 52, 125–138. [Google Scholar] [CrossRef] [PubMed]
- Li, X.Y.; Sun, L.W.; Zhao, D.Q. Current Status and Problem-Solving Strategies for Ginseng Industry. Chin. J. Integr. Med. 2019, 25, 883–886. [Google Scholar] [CrossRef]
- Park, S.-Y.; Ha, S.-C.; Kim, Y.-G. The protein crystallography beamlines at the pohang light source II. Biodesign 2017, 5, 30–34. [Google Scholar]
- Minor, W.; Cymborowski, M.; Otwinowski, Z.; Chruszcz, M. HKL-3000: The integration of data reduction and structure solution--from diffraction images to an initial model in minutes. Acta Crystallogr. Sect. D Biol. Crystallogr. 2006, 62 Pt 8, 859–866. [Google Scholar] [CrossRef] [PubMed]
- Liebschner, D.; Afonine, P.V.; Baker, M.L.; Bunkóczi, G.; Chen, V.B.; Croll, T.I.; Hintze, B.; Hung, L.-W.; Jain, S.; McCoy, A.J. Macromolecular structure determination using X-rays, neutrons and electrons: Recent developments in Phenix. Biol. Crystallogr. 2019, 75, 861–877. [Google Scholar] [CrossRef] [PubMed]
- Gamiz-Arco, G.; Gutierrez-Rus, L.I.; Risso, V.A.; Ibarra-Molero, B.; Hoshino, Y.; Petrovic, D.; Justicia, J.; Cuerva, J.M.; Romero-Rivera, A.; Seelig, B.; et al. Heme-binding enables allosteric modulation in an ancient TIM-barrel glycosidase. Nat. Commun. 2021, 12, 380. [Google Scholar] [CrossRef]
- Emsley, P.; Lohkamp, B.; Scott, W.G.; Cowtan, K. Features and development of Coot. Biol. Crystallogr. 2010, 66, 486–501. [Google Scholar] [CrossRef]
- Sievers, F.; Higgins, D.G. Clustal Omega for making accurate alignments of many protein sequences. Protein Sci. 2018, 27, 135–145. [Google Scholar] [CrossRef]
- Robert, X.; Gouet, P. Deciphering key features in protein structures with the new ENDscript server. Nucleic Acids Res. 2014, 42, W320–W324. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.R.; Makhatadze, G.I. ProteinVolume: Calculating molecular van der Waals and void volumes in proteins. BMC Bioinform. 2015, 16, 101. [Google Scholar] [CrossRef] [PubMed]
- Ferruz, N.; Schmidt, S.; Höcker, B. ProteinTools: A toolkit to analyze protein structures. Nucleic Acids Res. 2021, 49, W559–W566. [Google Scholar] [CrossRef] [PubMed]
- Heinig, M.; Frishman, D. STRIDE: A web server for secondary structure assignment from known atomic coordinates of proteins. Nucleic Acids Res. 2004, 32, W500–W502. [Google Scholar] [CrossRef]
- Koes, D.R.; Baumgartner, M.P.; Camacho, C.J. Lessons learned in empirical scoring with smina from the CSAR 2011 benchmarking exercise. J. Chem. Inf. Model. 2013, 53, 1893–1904. [Google Scholar] [CrossRef]
- Karplus, P.A.; Diederichs, K. Assessing and maximizing data quality in macromolecular crystallography. Curr. Opin. Struct. Biol. 2015, 34, 60–68. [Google Scholar] [CrossRef]
- Henrissat, B. A classification of glycosyl hydrolases based on amino acid sequence similarities. Biochem. J. 1991, 280 Pt 2, 309–316. [Google Scholar] [CrossRef]
- Sotiropoulou, A.I.; Hatzinikolaou, D.G.; Chrysina, E.D. Structural studies of β-glucosidase from the thermophilic bacterium Caldicellulosiruptor saccharolyticus. Biol. Crystallogr. 2024, 80, 733–743. [Google Scholar] [CrossRef]
- Oh, H.-J.; Shin, K.-C.; Oh, D.-K. Production of ginsenosides Rg1 and Rh1 by hydrolyzing the outer glycoside at the C-6 position in protopanaxatriol-type ginsenosides using β-glucosidase from Pyrococcus furiosus. Biotechnol. Lett. 2014, 36, 113–119. [Google Scholar] [CrossRef]
- Zhou, K.; Zhang, Y.; Xu, M.; Zhou, Y.; Sun, A.; Zhou, H.; Han, Y.; Zhao, D.; Yu, S. A GH1 β-glucosidase from the Fervidobacterium pennivorans DSM9078 showed extraordinary thermostability and distinctive ability in the efficient transformation of ginsenosides. Bioorg. Chem. 2025, 154, 108049. [Google Scholar] [CrossRef] [PubMed]
- Nakabayashi, M.; Kataoka, M.; Mishima, Y.; Maeno, Y.; Ishikawa, K. Structural analysis of beta-glucosidase mutants derived from a hyperthermophilic tetrameric structure. Acta Crystallogr. Sect. D Biol. Crystallogr. 2014, 70 Pt 3, 877–888. [Google Scholar] [CrossRef]
- Linse, S.; Thulin, E.; Nilsson, H.; Stigler, J. Benefits and constrains of covalency: The role of loop length in protein stability and ligand binding. Sci. Rep. 2020, 10, 20108. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, A.; Sumreen, A.; Bibi, A.; Batool, K. In silico approach to elucidate factors associated with GH1 β-Glucosidase thermostability. J. Pure Appl. Microbiol. 2019, 13, 1953–1968. [Google Scholar] [CrossRef]
- Grigas, A.T.; Liu, Z.; Logan, J.A.; Shattuck, M.D.; O’Hern, C.S. Protein Folding as a Jamming Transition. PRX Life 2025, 3, 013018. [Google Scholar] [CrossRef]
- Sun, Z.; Liu, Q.; Qu, G.; Feng, Y.; Reetz, M.T. Utility of B-factors in Protein Science: Interpreting Rigidity, Flexibility, and Internal Motion and Engineering Thermostability. Chem. Rev. 2019, 119, 1626–1665. [Google Scholar] [CrossRef]
- Hung, C.-L.; Kuo, Y.-H.; Lee, S.W.; Chiang, Y.-W. Protein Stability Depends Critically on the Surface Hydrogen-Bonding Network: A Case Study of Bid Protein. J. Phys. Chem. B 2021, 125, 8373–8382. [Google Scholar] [CrossRef]
- Xu, D.; Tsai, C.J.; Nussinov, R. Hydrogen bonds and salt bridges across protein-protein interfaces. Protein Eng. 1997, 10, 999–1012. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Data Collection | CbBGL |
|---|---|
| X-ray source | 5C beamline, PLS-II |
| Wavelength (Å) | 1.0 |
| Space group | P212121 |
| Cell dimension | |
| a, b, c (Å) | 74.60, 96.87, 143.01 |
| α, β, γ (◦) | 90, 90, 90 |
| Resolution (Å) | 50.0–1.90 (1.93–1.90) |
| Unique reflections | 142,431 (1466) |
| Completeness (%) | 95.6 (35.0) |
| Redundancy | 11.4 (7.7) |
| I/σ | 22.05 (5.0) |
| Rmerge | 0.095 (0.270) |
| Rmeas | 0.099 (0.287) |
| CC1/2 | 0.996 (0.946) |
| CC * | 0.999 (0.986) |
| Refinement | |
| Resolution (Å) | 37.11–1.90 |
| Rwork a | 0.1537 |
| Rfree b | 0.1807 |
| R.m.s. deviations | |
| Bonds (Å) | 0.008 |
| Angles (◦) | 0.972 |
| B factors (Å2) | |
| Protein | 20.10 |
| Ramachandran plot | |
| Favored (%) | 97.56 |
| Allowed (%) | 2.33 |
| Disallowed (%) | 0.11 |
| β-Glucosidases | Optimum Activity Temperature (°C) | Packing Density of Protein Core | Area of Hydrophobic Clusters (Å2) * | No. of Mobile Loops ** | No. of Salt Bridges *** | No. of H-Bond Networks |
|---|---|---|---|---|---|---|
| anGL | 65 | 0.734 | 5405.55 | 6 | 16 | 26 |
| CsBGL | 65 | 0.737 | 7412.17 | 8 | 19 | 35 |
| CbBGL | 80 | 0.735 | 7710.93 | 7 | 20 | 39 |
| FpBGL | 98.8 **** | 0.729 | 4426.40 | 1 | 26 | 39 |
| PfBGL | 109.5 **** | 0.738 | 2741.56 | 4 | 19 | 30 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Choi, J.-M. Structural Study of Thermostable Ginsenoside β-Glucosidase from Caldicellulosiruptor bescii. Crystals 2025, 15, 447. https://doi.org/10.3390/cryst15050447
Choi J-M. Structural Study of Thermostable Ginsenoside β-Glucosidase from Caldicellulosiruptor bescii. Crystals. 2025; 15(5):447. https://doi.org/10.3390/cryst15050447
Chicago/Turabian StyleChoi, Jung-Min. 2025. "Structural Study of Thermostable Ginsenoside β-Glucosidase from Caldicellulosiruptor bescii" Crystals 15, no. 5: 447. https://doi.org/10.3390/cryst15050447
APA StyleChoi, J.-M. (2025). Structural Study of Thermostable Ginsenoside β-Glucosidase from Caldicellulosiruptor bescii. Crystals, 15(5), 447. https://doi.org/10.3390/cryst15050447