
Fine Tuning of Hierarchical Zeolite Beta Acid Sites Strength


Abstract
:1. Introduction
2. Materials and Methods
2.1. Zeolite Beta Synthesis and Cations Insertion
2.2. Methods of Sample Characterization
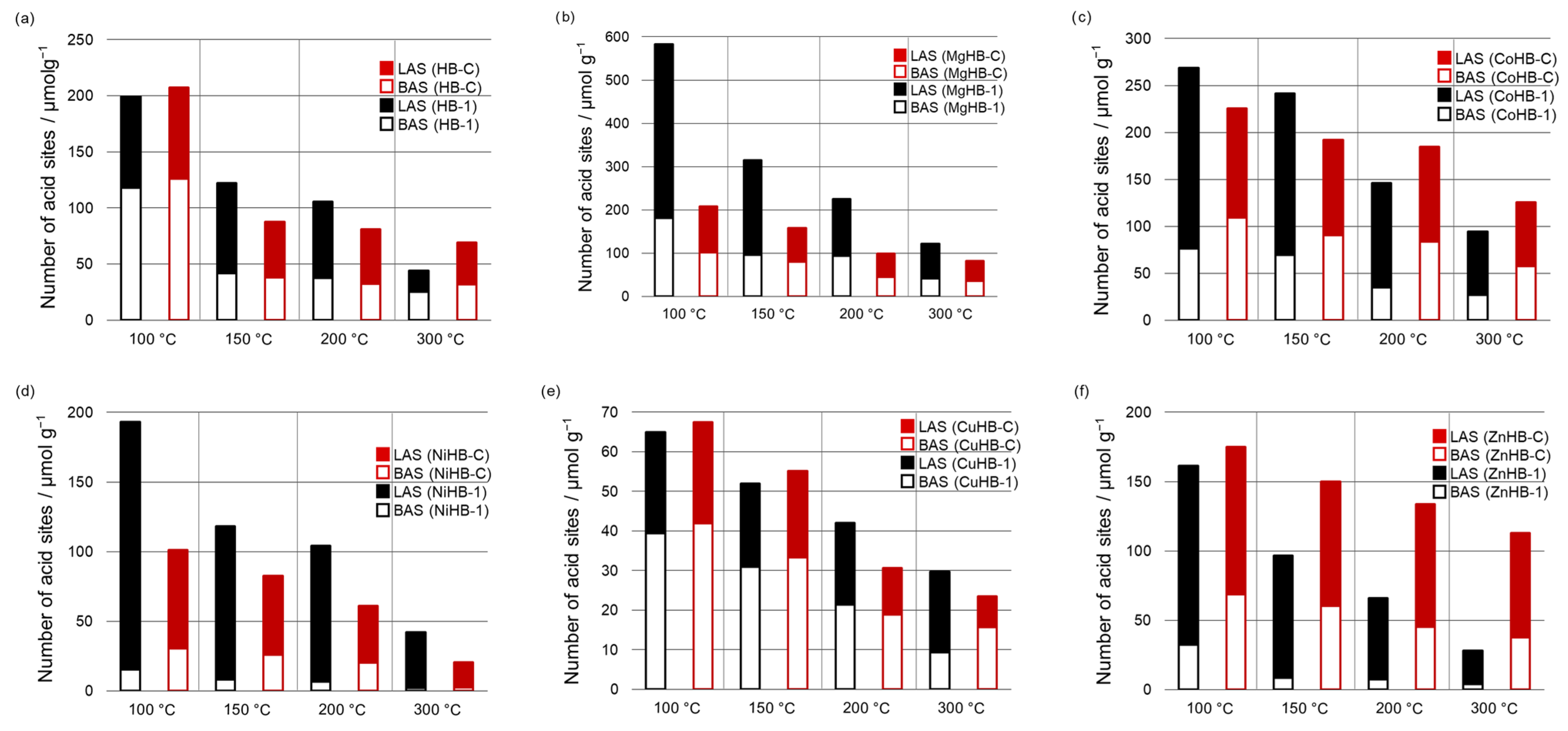
3. Results and discussion
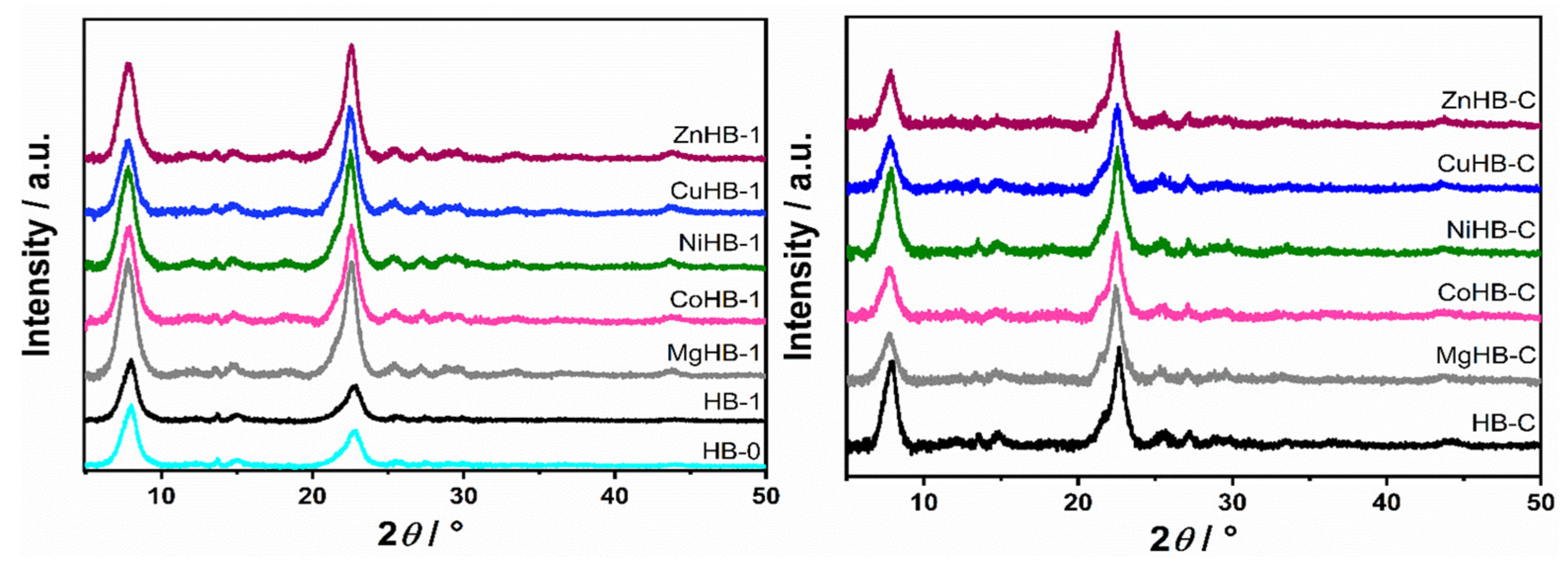
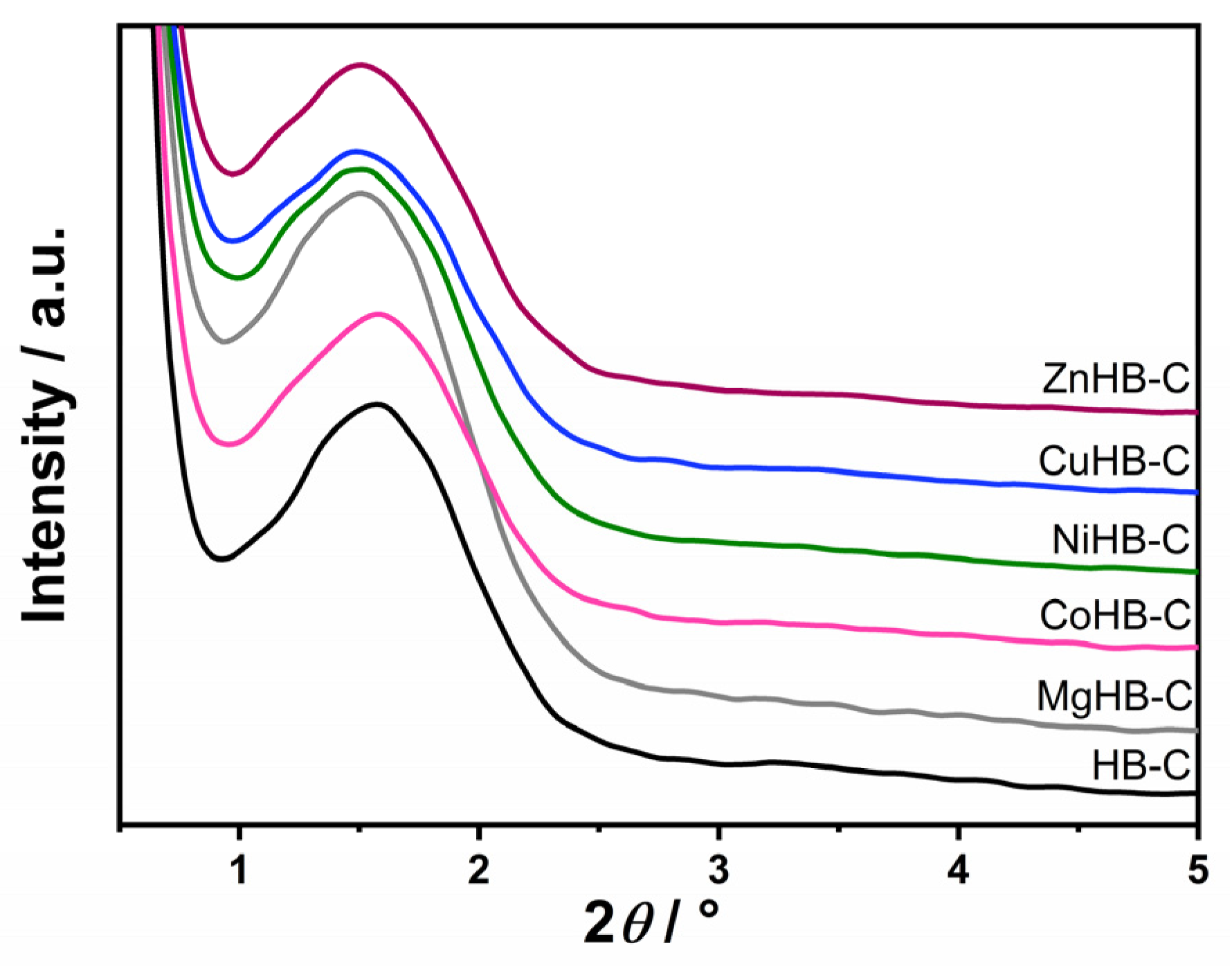
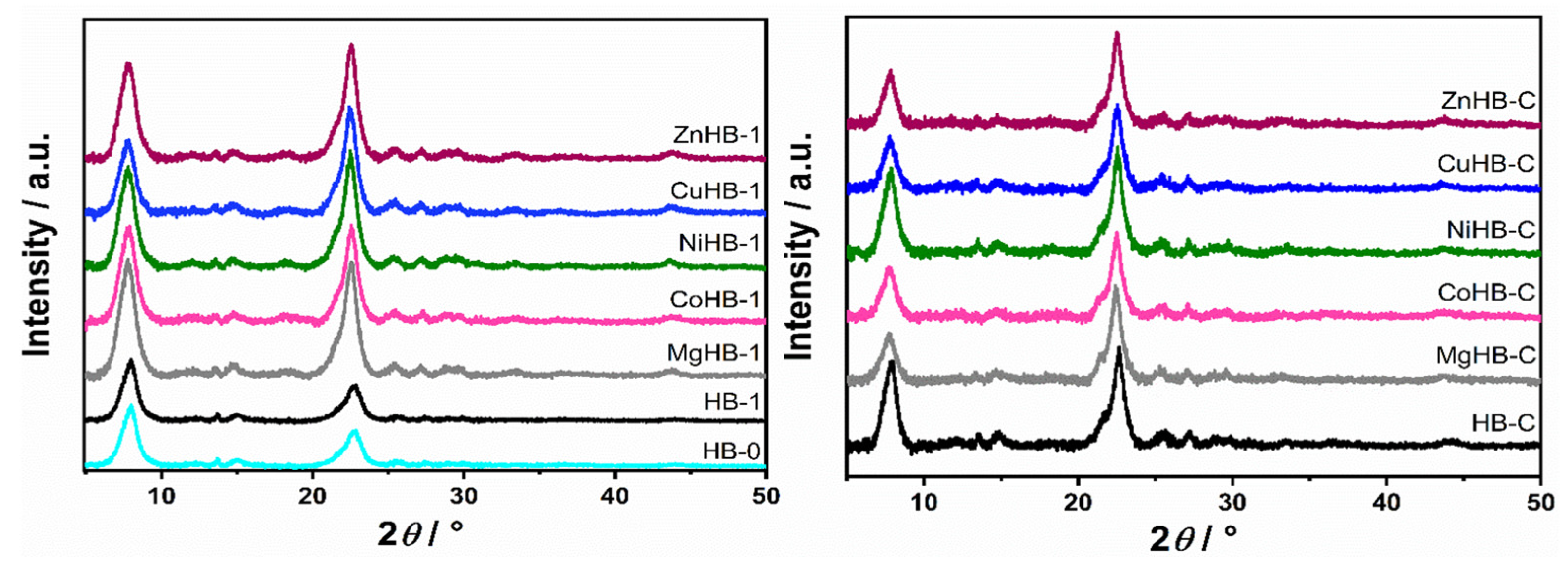
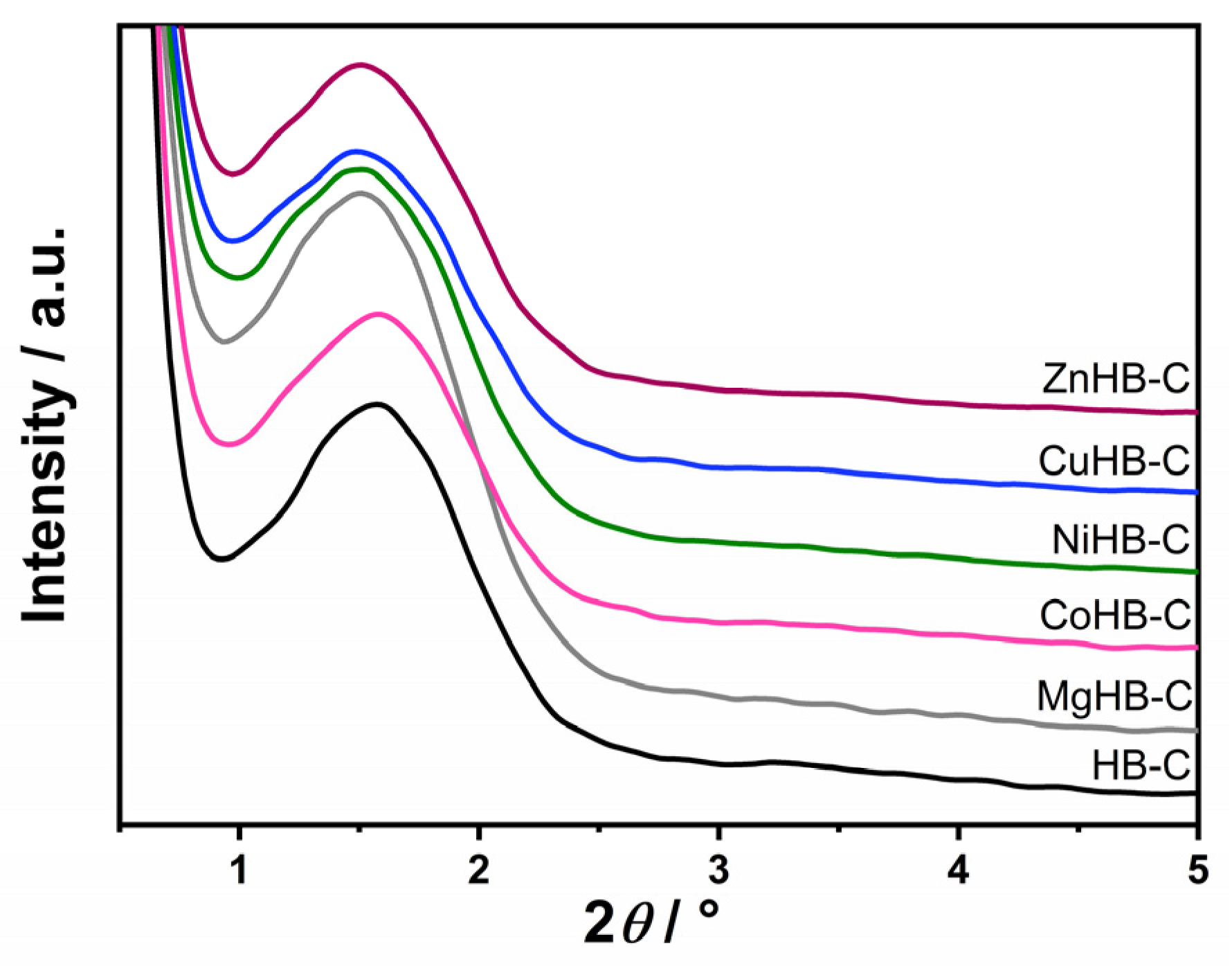
3.1. Powder X-ray Diffraction (PXRD)
3.2. Scanning Electron Microscopy (SEM)
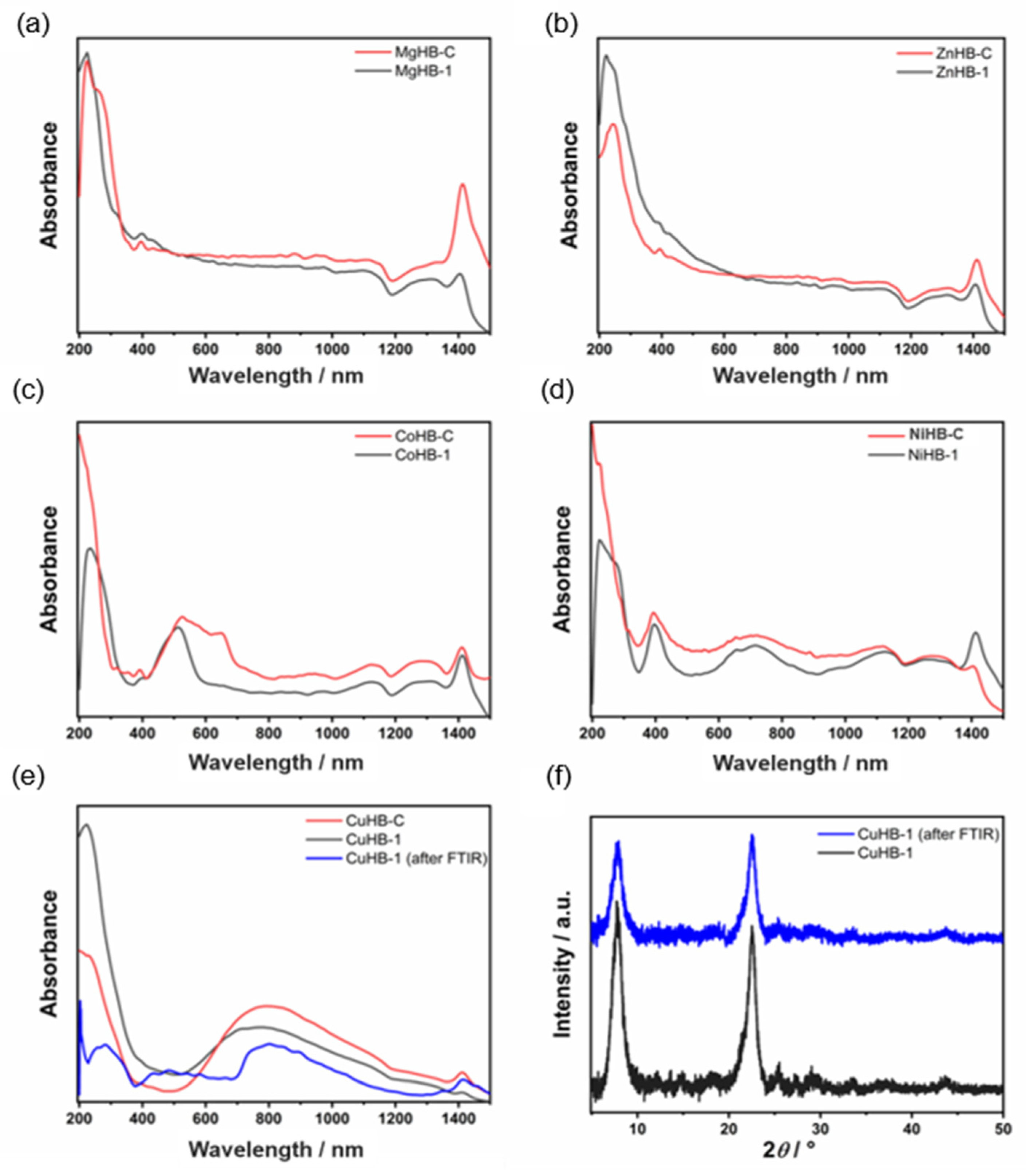
3.3. UV Vis Spectroscopy
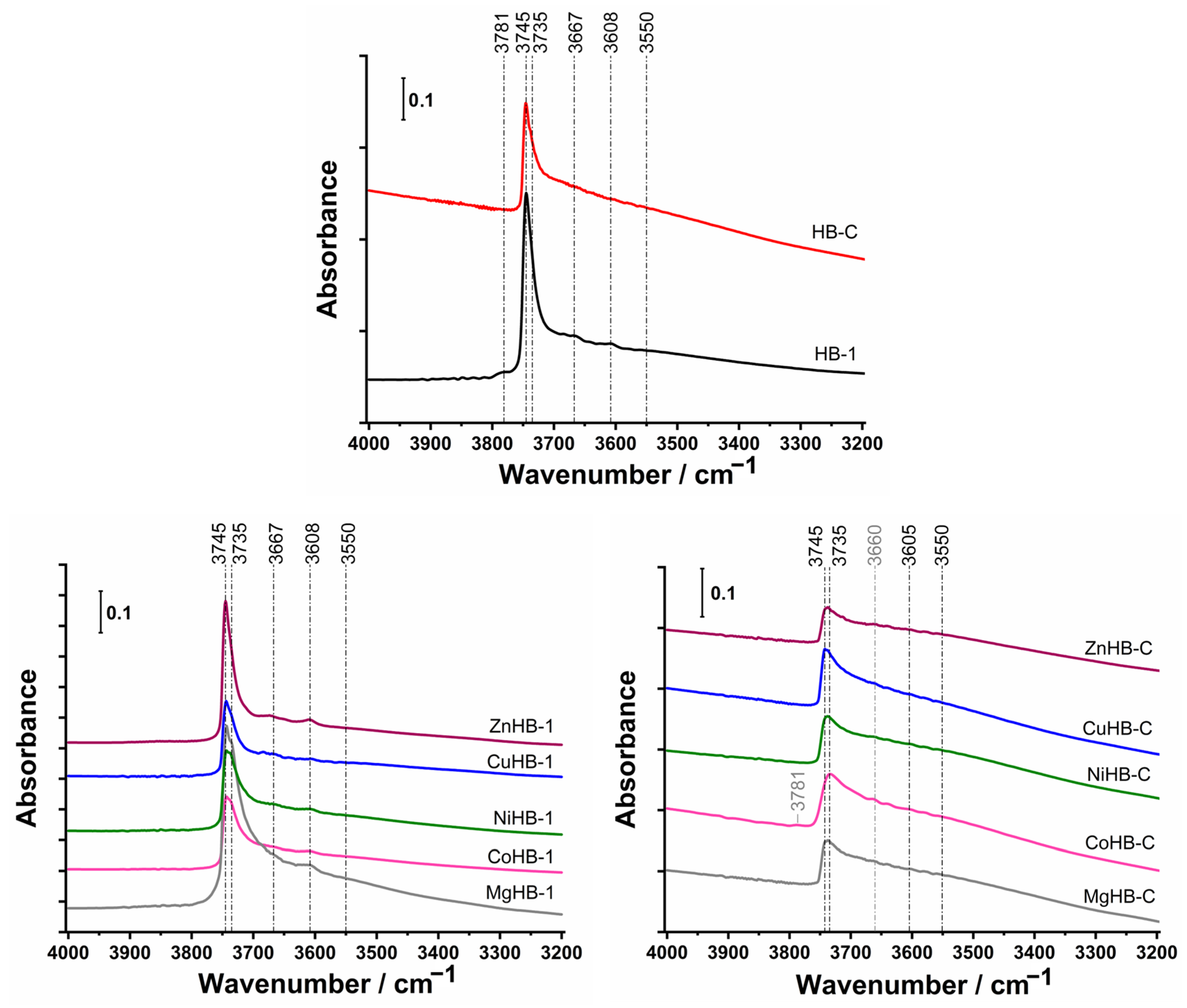
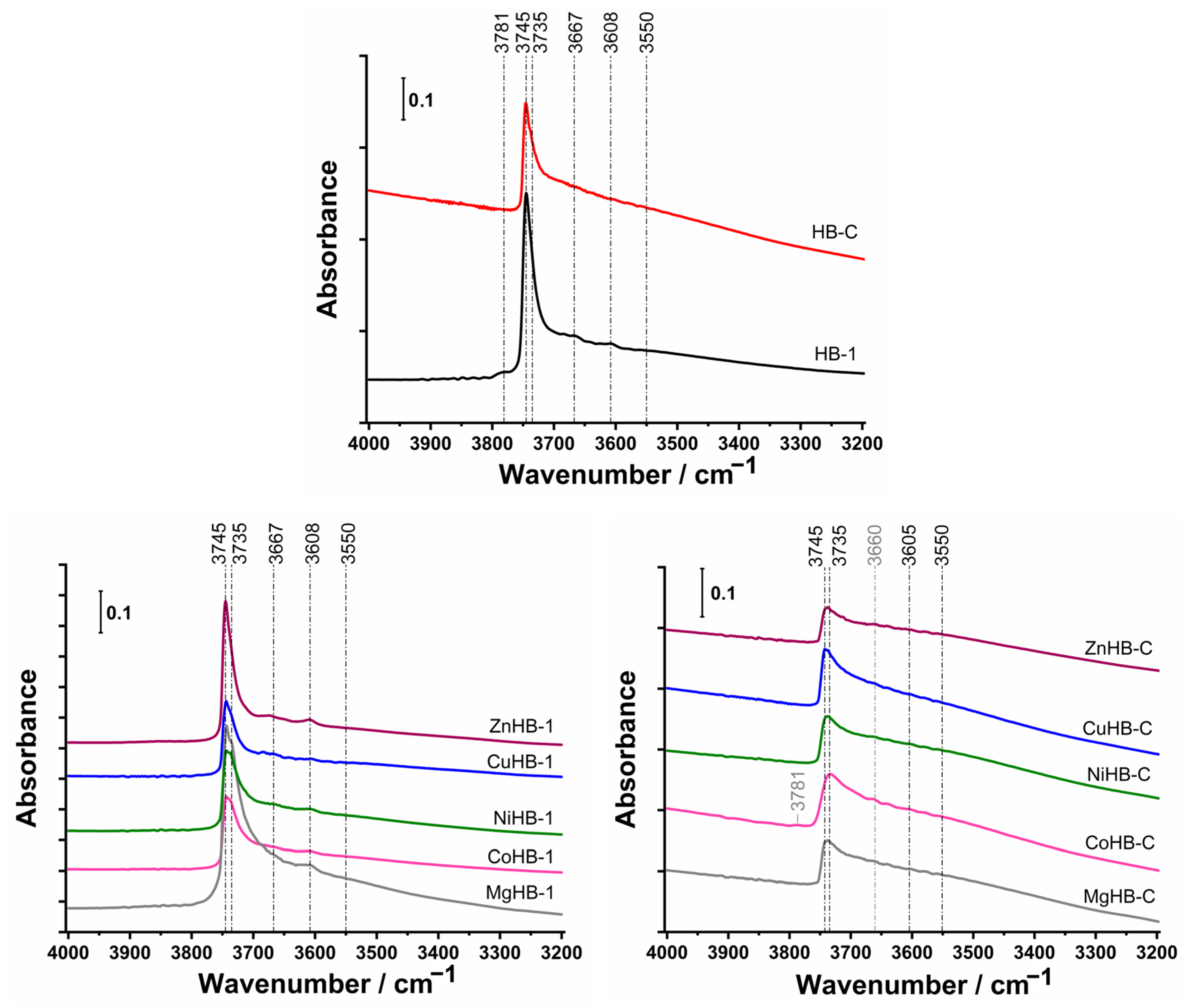
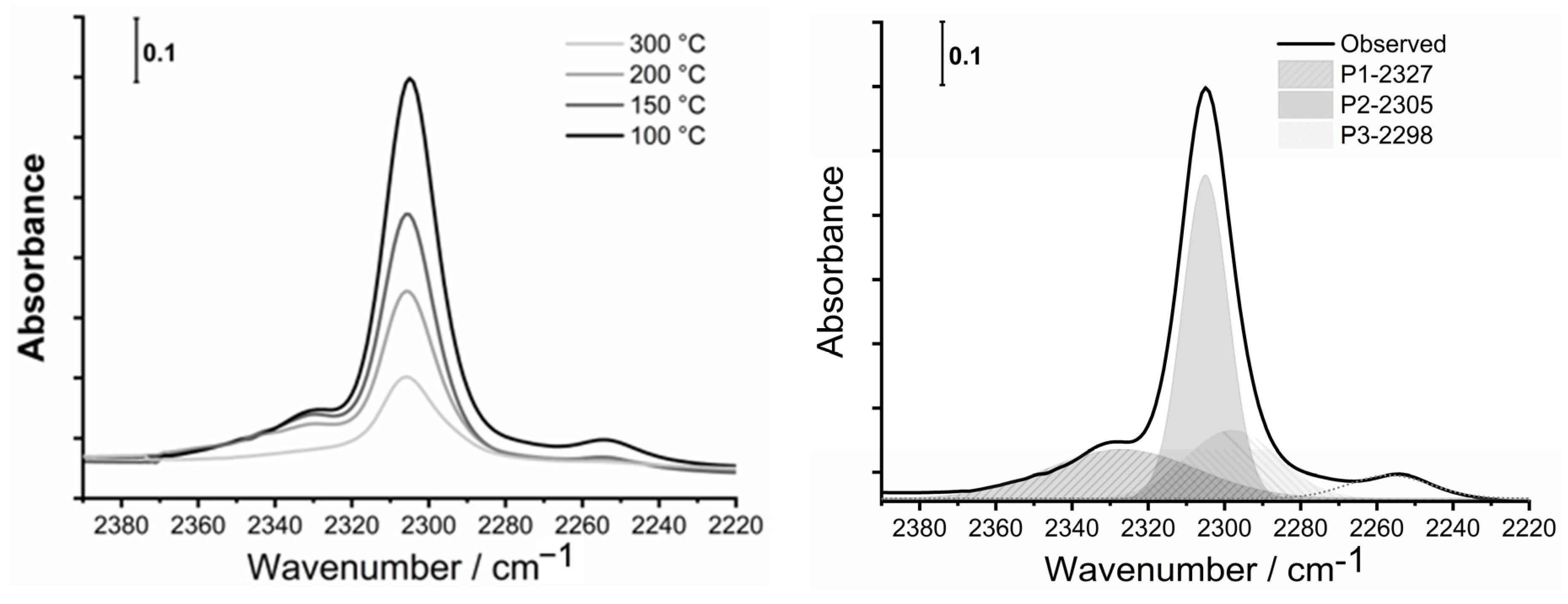
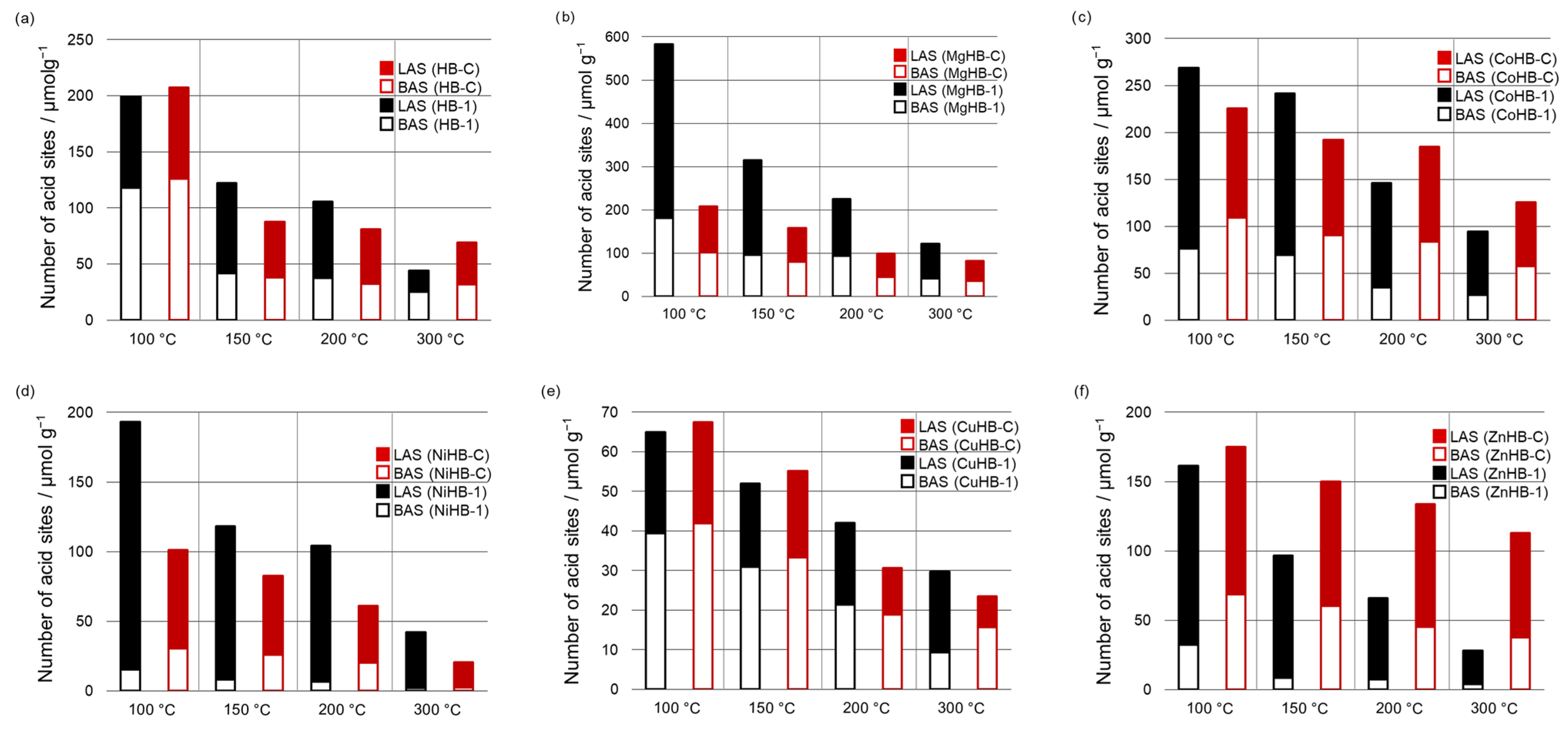
3.4. FTIR Spectroscopy
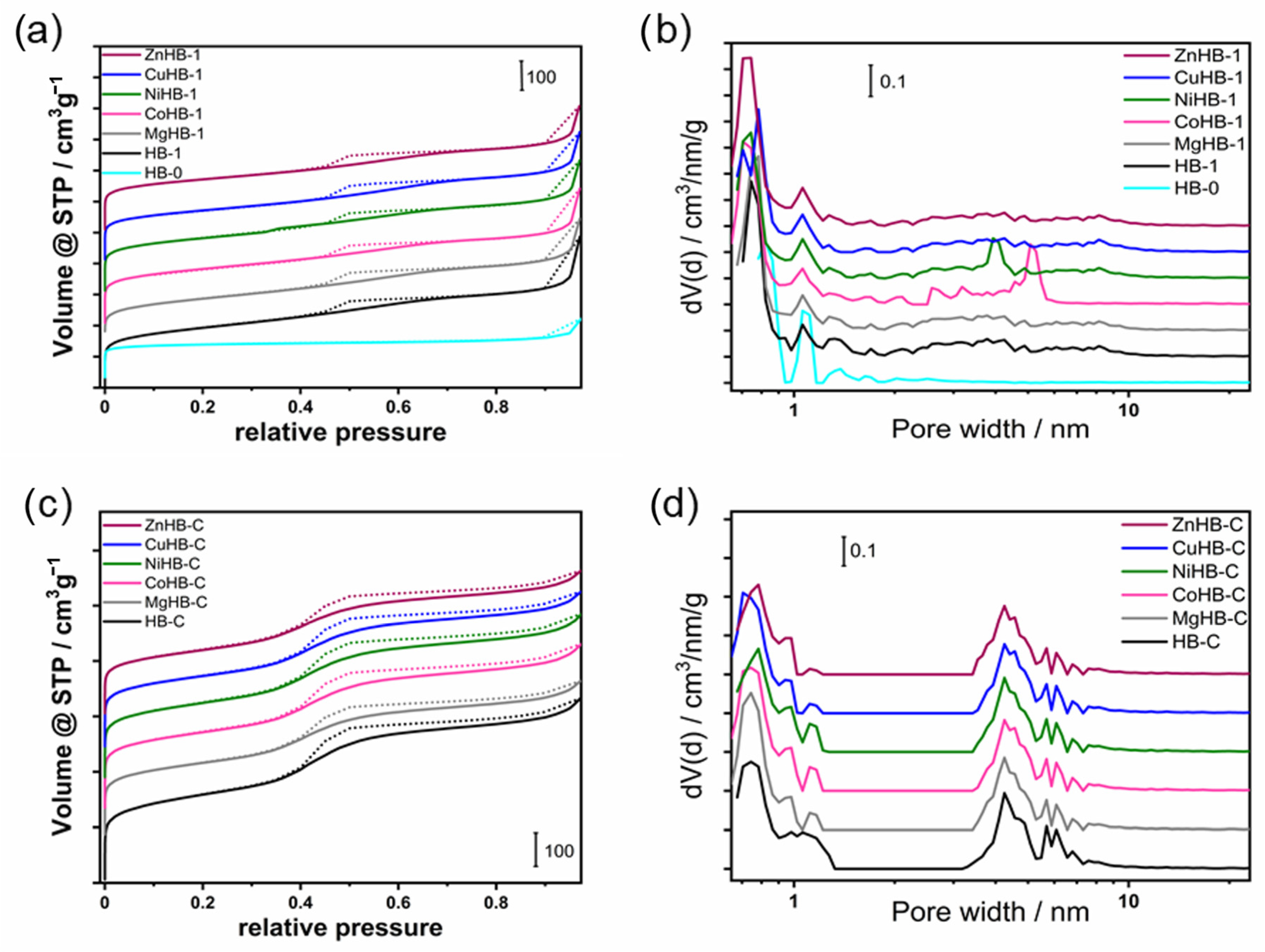
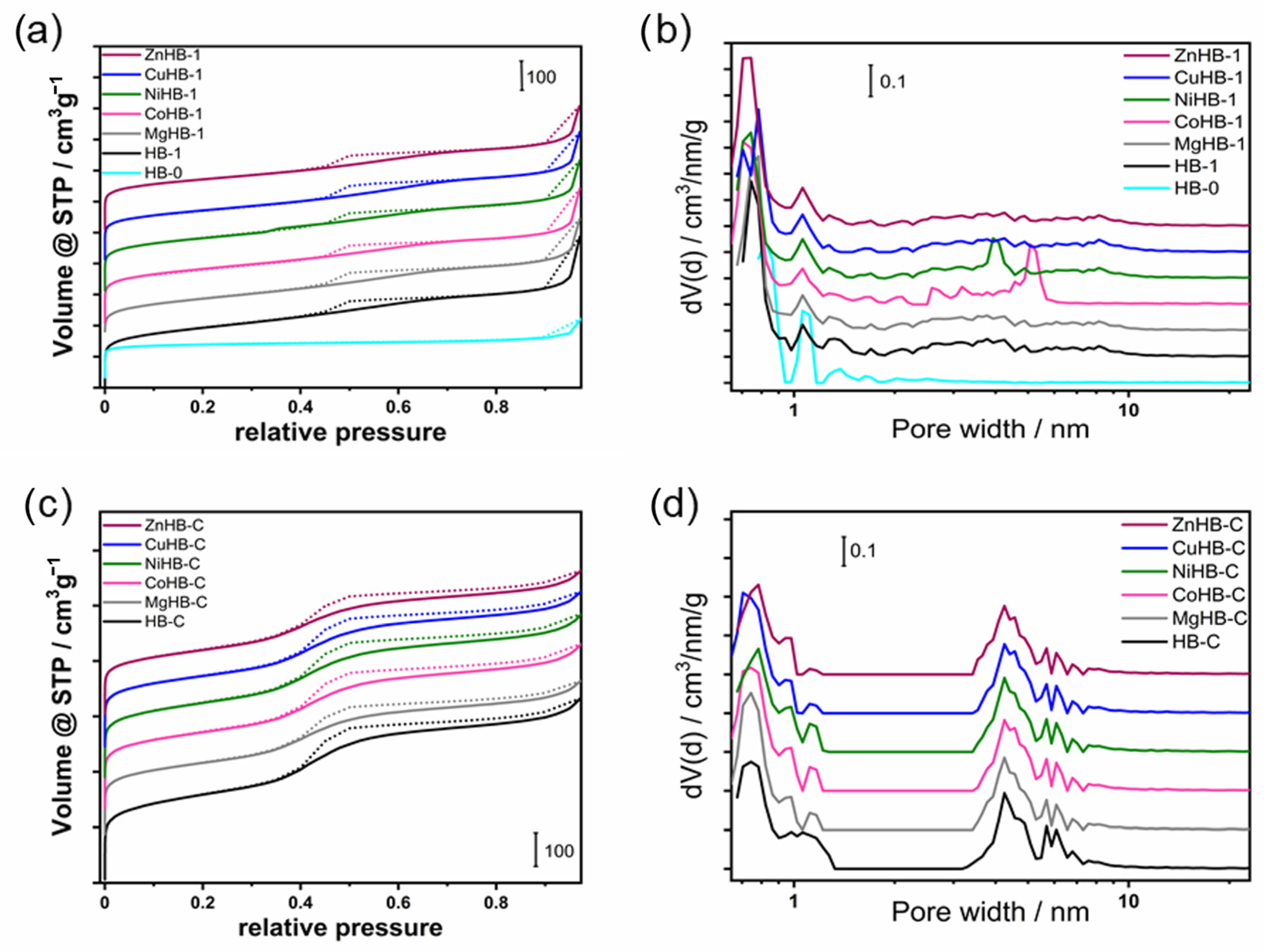
3.5. Textural Properties—Porosimetry (N2 Adsorption/Desorption)
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Wang, J.-J.; Chuang, Y.-Y.; Hsu, H.-Y.; Tsai, T.-C. Toward industrial catalysis of zeolite for linear alkylbenzene synthesis: A mini review. Catal. Today 2017, 298, 109–116. [Google Scholar] [CrossRef]
- Armor, J.N. A history of industrial catalysis. Catal. Today 2011, 163, 3–9. [Google Scholar] [CrossRef]
- Abate, S.; Barbera, K.; Centi, G.; Lanzafame, P.; Perathoner, S. Disruptive catalysis by zeolites. Catal. Sci. Technol. 2016, 6, 2485–2501. [Google Scholar] [CrossRef]
- Hong, L.; Zang, J.; Li, B.; Liu, G.; Wang, Y.; Wu, L. Research Progress on the Synthesis of Nanosized and Hierarchical Beta Zeolites. Inorganics 2023, 11, 214. [Google Scholar] [CrossRef]
- Valtchev, V.; Tosheva, L. Porous Nanosized Particles: Preparation, Properties, and Applications. Chem. Rev. 2013, 113, 6734–6760. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Wang, C.; Zhang, L.; Dai, W.; Chu, Y.; Xu, J.; Wu, G.; Gao, M.; Liu, W.; Xu, Z.; et al. Stabilizing the framework of SAPO-34 zeolite toward long-term methanol-to-olefins conversion. Nat. Commun. 2021, 12, 4661. [Google Scholar] [CrossRef]
- Dalena, F.; Puškarić, A.; Landripet, I.; Jelić, T.A.; Bosnar, S.; Medak, G.; Marino, A.; Giordano, G.; Bronić, J.; Migliori, M. Combining hierarchization and Mg2+ ions insertion in ZSM-5: Acidity-modulation effect on MTO reaction. Mol. Catal. 2023, 545, 113181. [Google Scholar] [CrossRef]
- Bordiga, S.; Lamberti, C.; Bonino, F.; Travert, A.; Thibault-Starzyk, F. Probing zeolites by vibrational spectroscopies. Chem. Soc. Rev. 2015, 44, 7262–7341. [Google Scholar] [CrossRef]
- Hegde, S.; Abdullah, R.; Bhat; Ratnasamy, P. FTi.r. spectroscopic study of gallium beta. Zeolites 1992, 15, 951–956. [Google Scholar] [CrossRef]
- Kefirov, R.; Penkova, A.; Hadjiivanov, K.; Dzwigaj, S.; Che, M. Stabilization of Cu+ ions in BEA zeolite: Study by FTIR spectroscopy of adsorbed CO and TPR. Microporous Mesoporous Mater. 2008, 116, 180–187. [Google Scholar] [CrossRef]
- Anquetil, R.; Saussey, J.; Lavalley, J.-C. Confinement effect on the interaction of hydroxy groups contained in the side pockets of H-mordenite with nitriles; a FT-IR study. Phys. Chem. Chem. Phys. 1999, 1, 555–560. [Google Scholar] [CrossRef]
- Pelmenschikov, A.G.; van Santen, R.A.; Janchen, J.; Meijer, E. Acetonitrile-d3 as a probe of Lewis and Broensted acidity of zeolites. J. Phys. Chem. 1993, 97, 11071–11074. [Google Scholar] [CrossRef]
- Kotrel, S.; Lunsford, J.H.; Knözinger, H. Characterizing Zeolite Acidity by Spectroscopic and Catalytic Means: A Comparison. J. Phys. Chem. B 2001, 105, 3917–3921. [Google Scholar] [CrossRef]
- Góra-Marek, K.; Datka, J.; Dzwigaj, S.; Che, M. Influence of V Content on the Nature and Strength of Acidic Sites in VSiβ Zeolite Evidenced by IR Spectroscopy. J. Phys. Chem. B 2006, 110, 6763–6767. [Google Scholar] [CrossRef] [PubMed]
- Bisio, C.; Massiani, P.; Fajerwerg, K.; Sordelli, L.; Stievano, L.; Silva, E.; Coluccia, S.; Martra, G. Identification of cationic and oxidic caesium species in basic Cs-overloaded BEA zeolites. Microporous Mesoporous Mater. 2006, 90, 175–187. [Google Scholar] [CrossRef]
- Zhang, W.; Ming, W.; Hu, S.; Qin, B.; Ma, J.; Li, R. A Feasible One-Step Synthesis of Hierarchical Zeolite Beta with Uniform Nanocrystals via CTAB. Materials 2018, 12, 651. [Google Scholar] [CrossRef]
- Verboekend, D.; Pérez-Ramírez, J. Design of hierarchical zeolite catalysts by desilication. Catal. Sci. Technol. 2011, 1, 879–890. [Google Scholar] [CrossRef]
- De Baerdemaeker, T.; Yilmaz, B.; Müller, U.; Feyen, M.; Xiao, F.-S.; Zhang, W.; Tatsumi, T.; Gies, H.; Bao, X.; De Vos, D. Catalytic applications of OSDA-free Beta zeolite. J. Catal. 2013, 308, 73–81. [Google Scholar] [CrossRef]
- Chaida-Chenni, F.Z.; Belhadj, F.; Casas, M.S.G.; Márquez-Álvarez, C.; Hamacha, R.; Bengueddach, A.; Pérez-Pariente, J. Synthesis of mesoporous-zeolite materials using Beta zeolite nanoparticles as precursors and their catalytic performance in m-xylene isomerization and disproportionation. Appl. Catal. A Gen. 2018, 568, 148–156. [Google Scholar] [CrossRef]
- Wang, Y.; Yokoi, T.; Namba, S.; Tatsumi, T. Effects of Dealumination and Desilication of Beta Zeolite on Catalytic Performance in n-Hexane Cracking. Catalysts 2016, 6, 8. [Google Scholar] [CrossRef]
- Hao, W.; Zhang, W.; Guo, Z.; Ma, J.; Li, R. Mesoporous Beta Zeolite Catalysts for Benzylation of Naphthalene: Effect of Pore Structure and Acidity. Catalysts 2018, 8, 504. [Google Scholar] [CrossRef]
- Miao, S.; Sun, S.; Lei, Z.; Sun, Y.; Zhao, C.; Zhan, J.; Zhang, W.; Jia, M. Micron-Sized Hierarchical Beta Zeolites Templated by Mesoscale Cationic Polymers as Robust Catalysts for Acylation of Anisole with Acetic Anhydride. Catalysts 2023, 13, 1517. [Google Scholar] [CrossRef]
- Zhao, R.; Zhao, Z.; Li, S.; Parvulescu, A.; Müller, U.; Zhang, W. Excellent Performances of Dealuminated H-Beta Zeolites from Organotemplate-Free Synthesis in Conversion of Biomass-derived 2,5-Dimethylfuran to Renewable p-Xylene. ChemSusChem 2018, 11, 3803–3811. [Google Scholar] [CrossRef] [PubMed]
- Rutkowska, M.; Piwowarska, Z.; Micek, E.; Chmielarz, L. Hierarchical Fe-, Cu-and Co-Beta zeolites obtained by mesotemplate-free method. Part I: Synthesis and catalytic activity in N2O decomposition. Microporous Mesoporous Mater. 2015, 209, 54–65. [Google Scholar] [CrossRef]
- Xu, L.; Shi, C.; Zhang, Z.; Gies, H.; Xiao, F.-S.; De Vos, D.; Yokoi, T.; Bao, X.; Feyen, M.; Maurer, S.; et al. Enhancement of low-temperature activity over Cu-exchanged zeolite beta from organotemplate-free synthesis for the selective catalytic reduction of NOx with NH3 in exhaust gas streams. Microporous Mesoporous Mater. 2014, 200, 304–310. [Google Scholar] [CrossRef]
- Cruz-Cabeza, A.J.; Esquivel, D.; Jiménez-Sanchidrián, C.; Romero-Salguero, F.J. Metal-Exchanged β Zeolites as Catalysts for the Conversion of Acetone to Hydrocarbons. Materials 2012, 5, 121–134. [Google Scholar] [CrossRef] [PubMed]
- Esquivel, D.; Cruz-Cabeza, A.J.; Jiménez-Sanchidrián, C.; Romero-Salguero, F.J. Transition metal exchanged β zeolites: Characterization of the metal state and catalytic application in the methanol conversion to hydrocarbons. Microporous Mesoporous Mater. 2013, 179, 30–39. [Google Scholar] [CrossRef]
- Atoguchi, T.; Kanougi, T. Phenol oxidation over alkaline earth metal ion exchange beta zeolite in the presence of ketone. J. Mol. Catal. A Chem. 2004, 222, 253–257. [Google Scholar] [CrossRef]
- Essid, S.; Ayari, F.; Bulánek, R.; Vaculík, J.; Mhamdi, M.; Delahay, G.; Ghorbel, A. Improvement of the conventional preparation methods in Co/BEA zeolites: Characterization and ethane ammoxidation. Solid State Sci. 2019, 93, 13–23. [Google Scholar] [CrossRef]
- Mintova, S.; Valtchev, V.; Onfroy, T.; Marichal, C.; Knözinger, H.; Bein, T. Variation of the Si/Al ratio in nanosized zeolite Beta crystals. Microporous Mesoporous Mater. 2006, 90, 237–245. [Google Scholar] [CrossRef]
- Wichterlová, B.; Tvarůžková, Z.; Sobalík, Z.; Sarv, P. Determination and properties of acid sites in H-ferrierite: A comparison of ferrierite and MFI structures. Microporous Mesoporous Mater. 1998, 24, 223–233. [Google Scholar] [CrossRef]
- Sadek, R.; Chalupka-Spiewak, K.; Krafft, J.-M.; Millot, Y.; Valentin, L.; Casale, S.; Gurgul, J.; Dzwigaj, S. The Synthesis of Different Series of Cobalt BEA Zeolite Catalysts by Post-Synthesis Methods and Their Characterization. Catalysts 2022, 12, 1644. [Google Scholar] [CrossRef]
- Medak, G.; Puškarić, A.; Bronić, J. The Influence of Inserted Metal Ions on Acid Strength of OH Groups in Faujasite. Crystals 2023, 13, 332. [Google Scholar] [CrossRef]
- Sobuś, N.; Michorczyk, B.; Piotrowski, M.; Kuterasiński, Ł.; Chlebda, D.K.; Łojewska, J.; Jędrzejczyk, R.J.; Jodłowski, P.; Kuśtrowski, P.; Czekaj, I. Design of Co, Cu and Fe–BEA Zeolite Catalysts for Selective Conversion of Lactic Acid into Acrylic Acid. Catal. Lett. 2019, 149, 3349–3360. [Google Scholar] [CrossRef]
- Śrębowata, A.; Baran, R.; Łomot, D.; Lisovytskiy, D.; Onfroy, T.; Dzwigaj, S. Remarkable effect of postsynthesis preparation procedures on catalytic properties of Ni-loaded BEA zeolites in hydrodechlorination of 1,2-dichloroethane. Appl. Catal. B Environ. 2014, 147, 208–220. [Google Scholar] [CrossRef]
- Baran, R.; Averseng, F.; Wierzbicki, D.; Chalupka, K.; Krafft, J.-M.; Grzybek, T.; Dzwigaj, S. Effect of postsynthesis preparation procedure on the state of copper in CuBEA zeolites and its catalytic properties in SCR of NO with NH3. Appl. Catal. A Gen. 2016, 523, 332–342. [Google Scholar] [CrossRef]
- Wang, H.; Xu, R.; Jin, Y.; Zhang, R. Zeolite structure effects on Cu active center, SCR performance and stability of Cu-zeolite catalysts. Catal. Today 2019, 327, 295–307. [Google Scholar] [CrossRef]
- Rutkowska, M.; Díaz, U.; Palomares, A.E.; Chmielarz, L. Cu and Fe modified derivatives of 2D MWW-type zeolites (MCM-22, ITQ-2 and MCM-36) as new catalysts for DeNOx process. Appl. Catal. B Environ. 2015, 168–169, 531–539. [Google Scholar] [CrossRef]
- Groothaert, M.H.; Smeets, P.J.; Sels, B.F.; Jacobs, P.A.; Schoonheydt, R.A. Selective Oxidation of Methane by the Bis(μ-oxo)dicopper Core Stabilized on ZSM-5 and Mordenite Zeolites. J. Am. Chem. Soc. 2005, 127, 1394–1395. [Google Scholar] [CrossRef]
- Smeets, P.J.; Groothaert, M.H.; Schoonheydt, R.A. Cu based zeolites: A UV–vis study of the active site in the selective methane oxidation at low temperatures. Catal. Today 2005, 110, 303–309. [Google Scholar] [CrossRef]
- Dzwigaj, S.; Massiani, P.; Davidson, A.; Che, M. Role of silanol groups in the incorporation of V in β zeolite. J. Mol. Catal. A Chem. 2000, 155, 169–182. [Google Scholar] [CrossRef]
- Maache, M.; Janin, A.; Lavalley, J.; Joly, J.; Benazzi, E. Acidity of zeolites Beta dealuminated by acid leaching: An FTi.r. study using different probe molecules (pyridine, carbon monoxide). Zeolites 1993, 13, 419–426. [Google Scholar] [CrossRef]
- Janin, A.; Maache, M.; Raatz, F.; Lavalley, J.C.; Joly, J.F.; Szydlowski, N. FTIR study of the silanol groups in dealuminated HY zeolites: Nature of the extraframework debris. Zeolites 1991, 11, 391–396. [Google Scholar] [CrossRef]
- Chen, J.; Thomas, J.M.; Sankar, G. IR spectroscopic study of CD3CN adsorbed on ALPO-18 molecular sieve and the solid acid catalysts SAPO-18 and MeAPO-18. J. Chem. Soc. Faraday Trans. 1994, 90, 3455–3459. [Google Scholar] [CrossRef]
- Areán, C.O.; Platero, E.E.; Mentruit, M.P.; Delgado, M.R.; i Xamena, F.L.; García-Raso, A.; Morterra, C. The combined use of acetonitrile and adamantane–carbonitrile as IR spectroscopic probes to discriminate between external and internal surfaces of medium pore zeolites. Microporous Mesoporous Mater. 2000, 34, 55–60. [Google Scholar] [CrossRef]
- Giordanino, F.; Vennestrøm, P.N.R.; Lundegaard, L.F.; Stappen, F.N.; Mossin, S.; Beato, P.; Bordiga, S.; Lamberti, C. Characterization of Cu-exchanged SSZ-13: A comparative FTIR, UV-Vis, and EPR study with Cu-ZSM-5 and Cu-β with similar Si/Al and Cu/Al ratios. Dalton Trans. 2013, 42, 12741–12761. [Google Scholar] [CrossRef]
- Deka, U.; Lezcano-Gonzalez, I.; Weckhuysen, B.M.; Beale, A.M. Local Environment and Nature of Cu Active Sites in Zeolite-Based Catalysts for the Selective Catalytic Reduction of NOx. ACS Catal. 2013, 3, 413–427. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| SAMPLE | SBET (m2g−1) | Vtotal a (cm3g−1) | Vmicro b (cm3g−1) | Smicro b (m2g−1) | Sext b (m2g−1) | Vmeso c (cm3g−1) | Si/Al d | M/Al d |
|---|---|---|---|---|---|---|---|---|
| HB-0 | 548 | 0.271 | 0.183 | 478 | 71 | 0.088 | 25.4 | / |
| HB-1 | 674 | 0.527 | 0.118 | 278 | 397 | 0.409 | 13.9 | / |
| MgHB-1 | 645 | 0.491 | 0.121 | 286 | 358 | 0.370 | 20.8 | 0.54 |
| CoHB-1 | 658 | 0.525 | 0.123 | 293 | 365 | 0.402 | 20.6 | 0.58 |
| NiHB-1 | 620 | 0.518 | 0.119 | 249 | 370 | 0.399 | 20.9 | 0.51 |
| CuHB-1 | 618 | 0.503 | 0.115 | 275 | 343 | 0.388 | 21.2 | 0.95 |
| ZnHB-1 | 621 | 0.489 | 0.122 | 291 | 330 | 0.367 | 20.2 | 0.60 |
| HB-C | 558 | 0.486 | 0.084 | 202 | 369 | 0.402 | 31.3 | / |
| MgHB-C | 471 | 0.407 | 0.055 | 131 | 340 | 0.352 | 31.6 | 0.50 |
| CoHB-C | 509 | 0.437 | 0.077 | 185 | 324 | 0.360 | 31.5 | 0.59 |
| NiHB-C | 519 | 0.443 | 0.084 | 199 | 320 | 0.359 | 31.9 | 0.50 |
| CuHB-C | 478 | 0.415 | 0.073 | 173 | 305 | 0.342 | 32.0 | 1.00 |
| ZnHB-C | 429 | 0.382 | 0.050 | 119 | 309 | 0.332 | 29.6 | 0.58 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Landripet, I.; Puškarić, A.; Robić, M.; Bronić, J. Fine Tuning of Hierarchical Zeolite Beta Acid Sites Strength. Crystals 2024, 14, 53. https://doi.org/10.3390/cryst14010053
Landripet I, Puškarić A, Robić M, Bronić J. Fine Tuning of Hierarchical Zeolite Beta Acid Sites Strength. Crystals. 2024; 14(1):53. https://doi.org/10.3390/cryst14010053
Chicago/Turabian StyleLandripet, Ivana, Andreas Puškarić, Marko Robić, and Josip Bronić. 2024. "Fine Tuning of Hierarchical Zeolite Beta Acid Sites Strength" Crystals 14, no. 1: 53. https://doi.org/10.3390/cryst14010053
APA StyleLandripet, I., Puškarić, A., Robić, M., & Bronić, J. (2024). Fine Tuning of Hierarchical Zeolite Beta Acid Sites Strength. Crystals, 14(1), 53. https://doi.org/10.3390/cryst14010053