

Crystal Structure of Aspartate Semialdehyde Dehydrogenase from Porphyromonas gingivalis



Abstract
:1. Introduction
2. Materials and Methods
2.1. Plasmid Construction and Protein Expression and Purification
2.2. Crystallization and Data Collection of Apo and Complex Forms
2.3. Structure Determination and Refinement of the Apo- and Cofactor-Complexed Structures
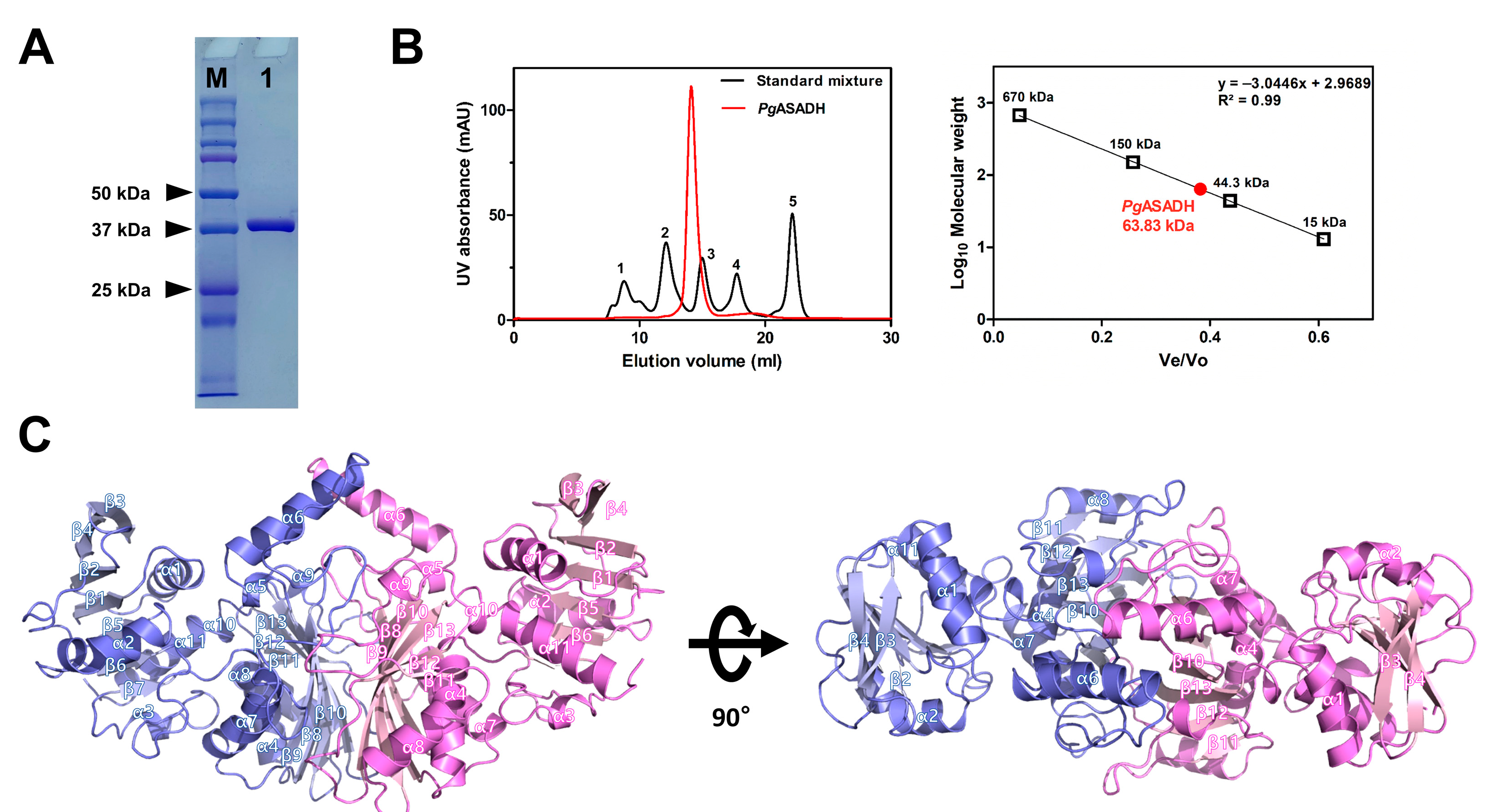
2.4. Analytical SEC
2.5. Comparative Bioinformatics Analysis
3. Results and Discussion
3.1. Apo Structure of PgASADH
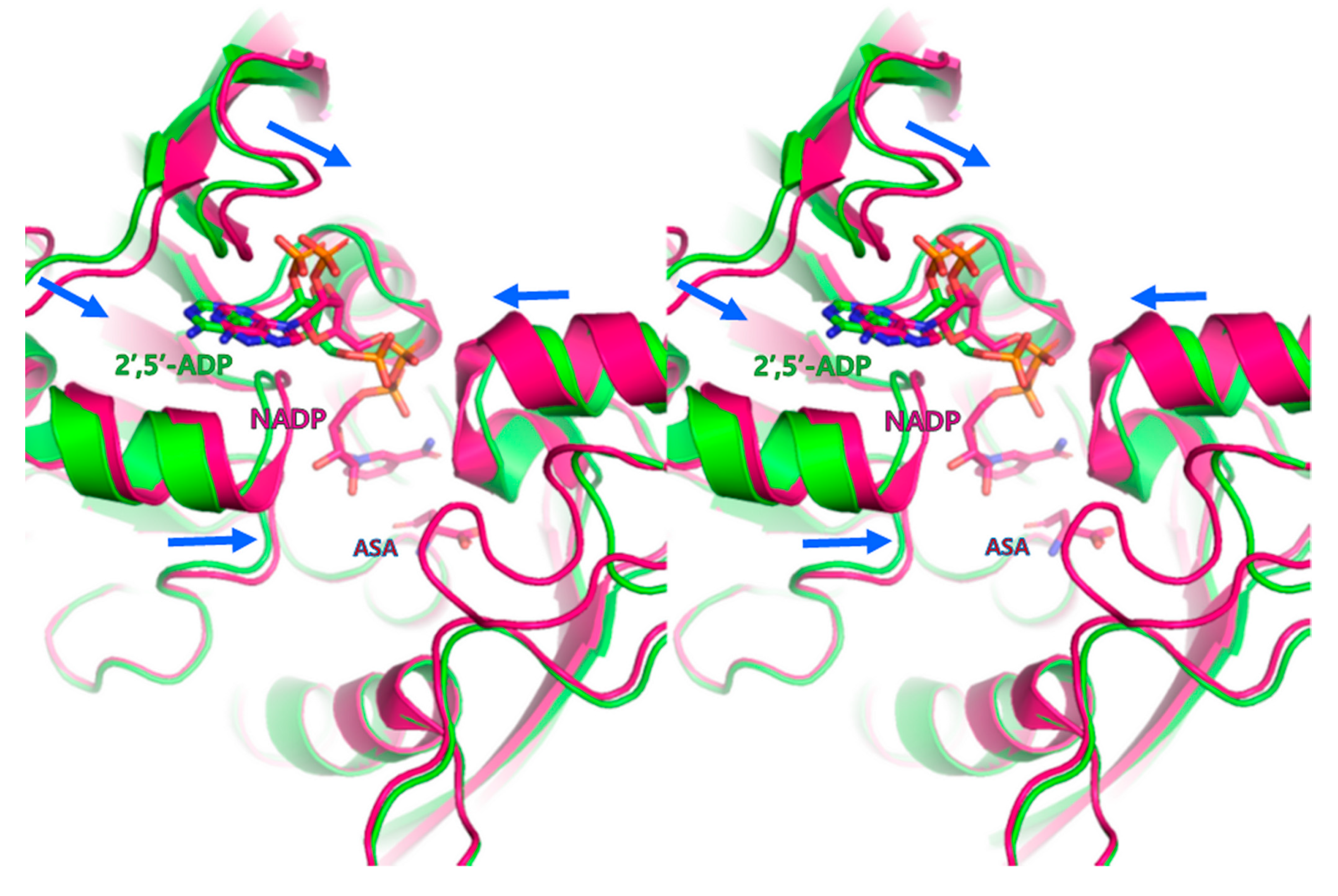
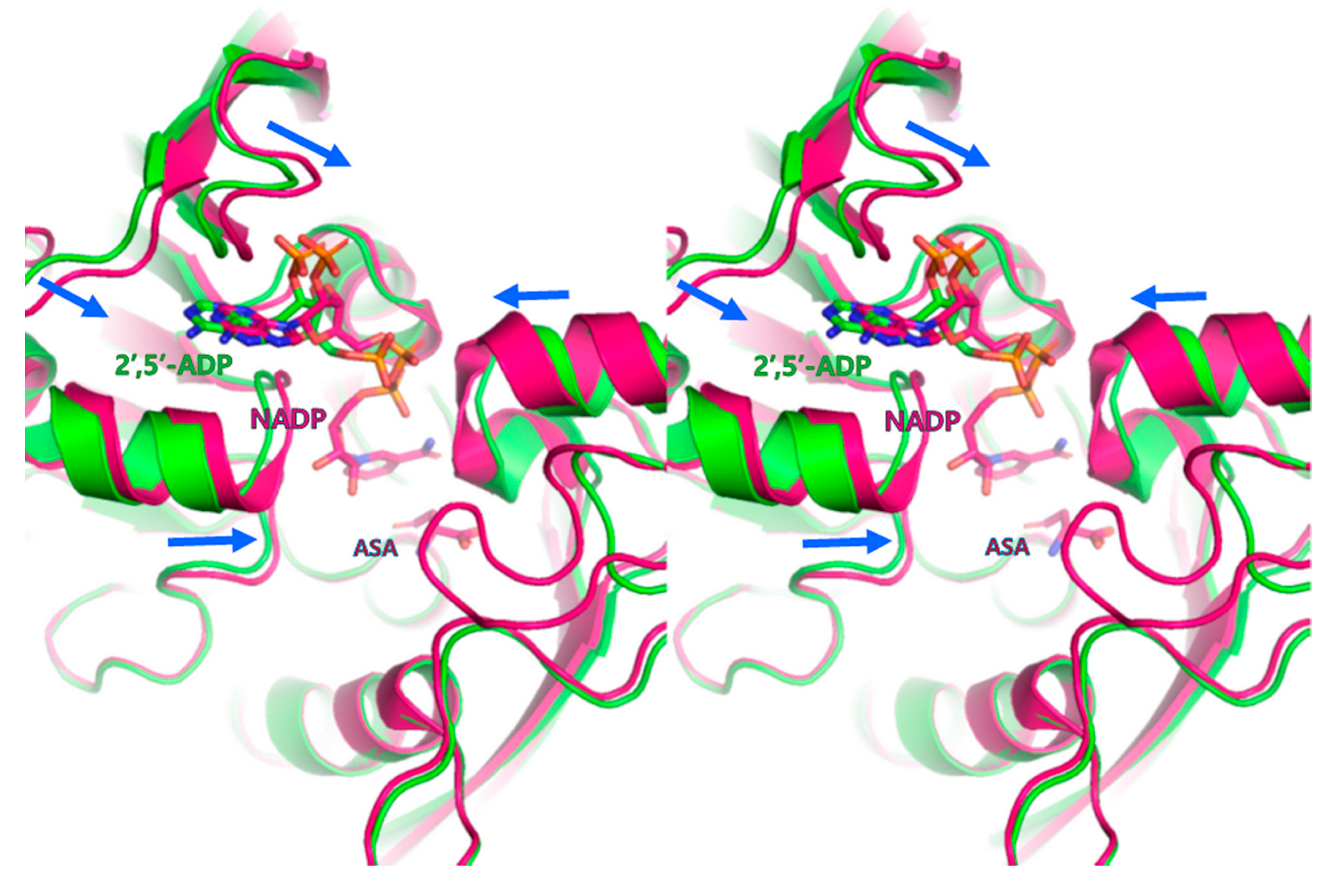
3.2. Cofactor-Bound Structure of PgASADH
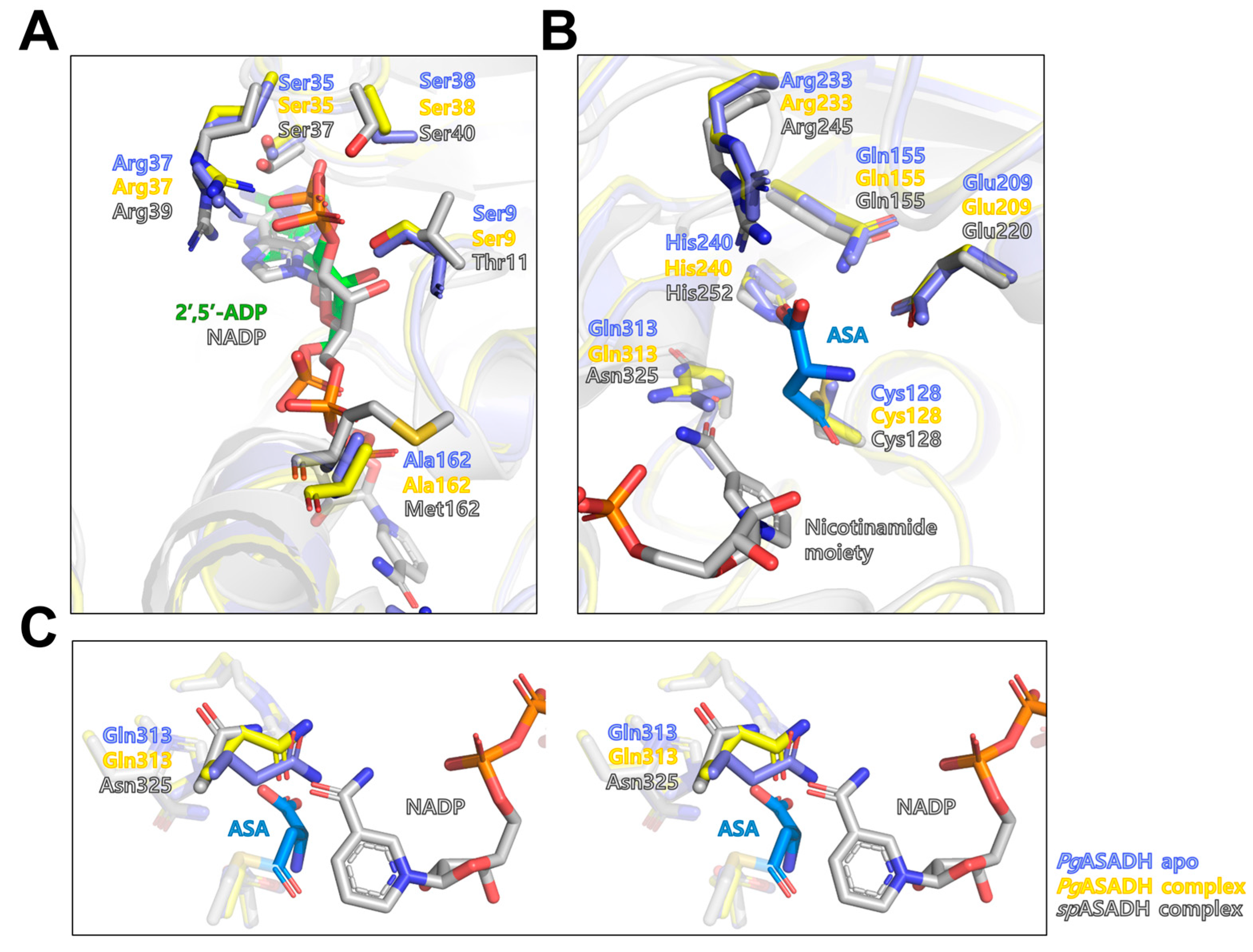
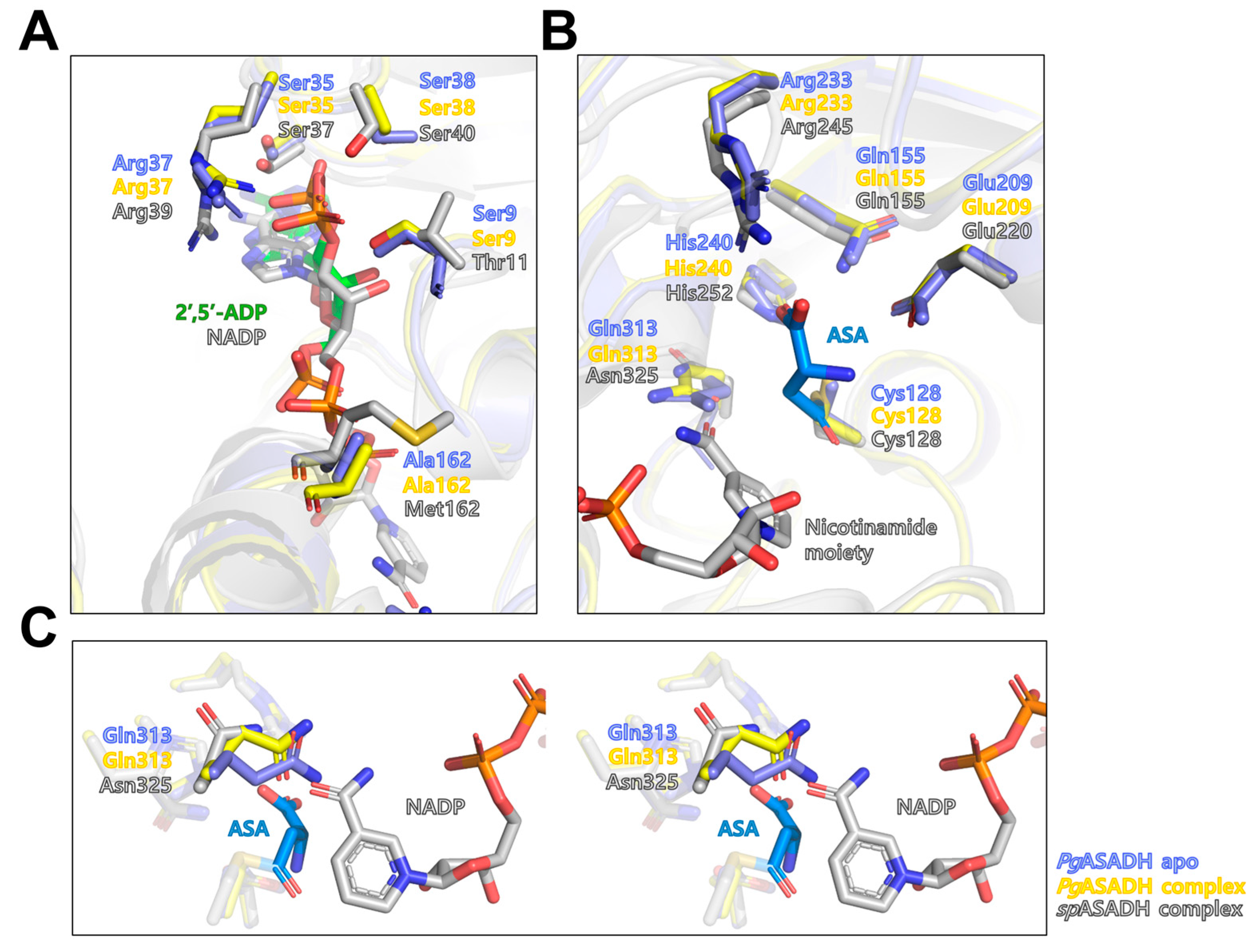
3.3. Active Site of PgASADH
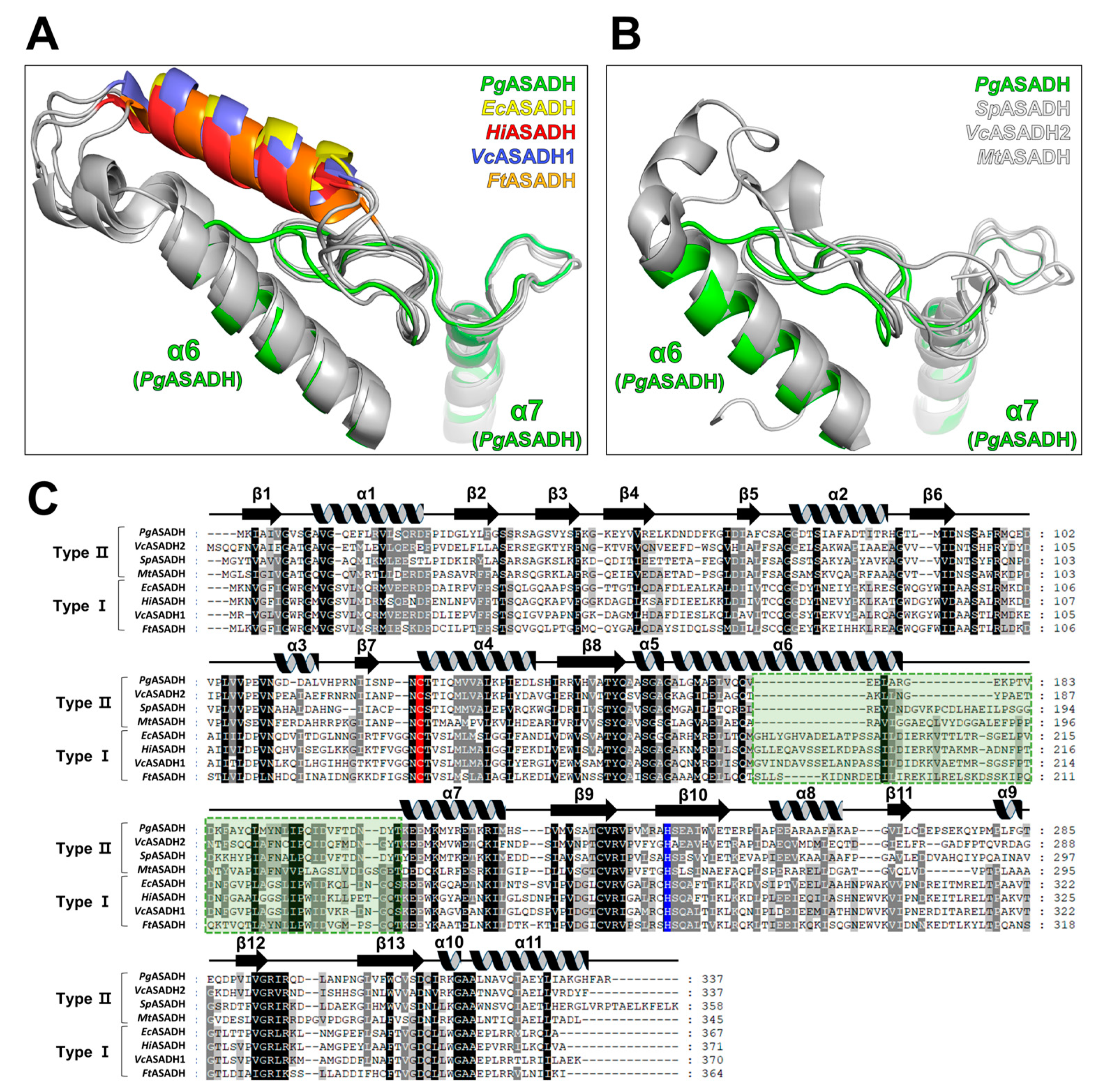
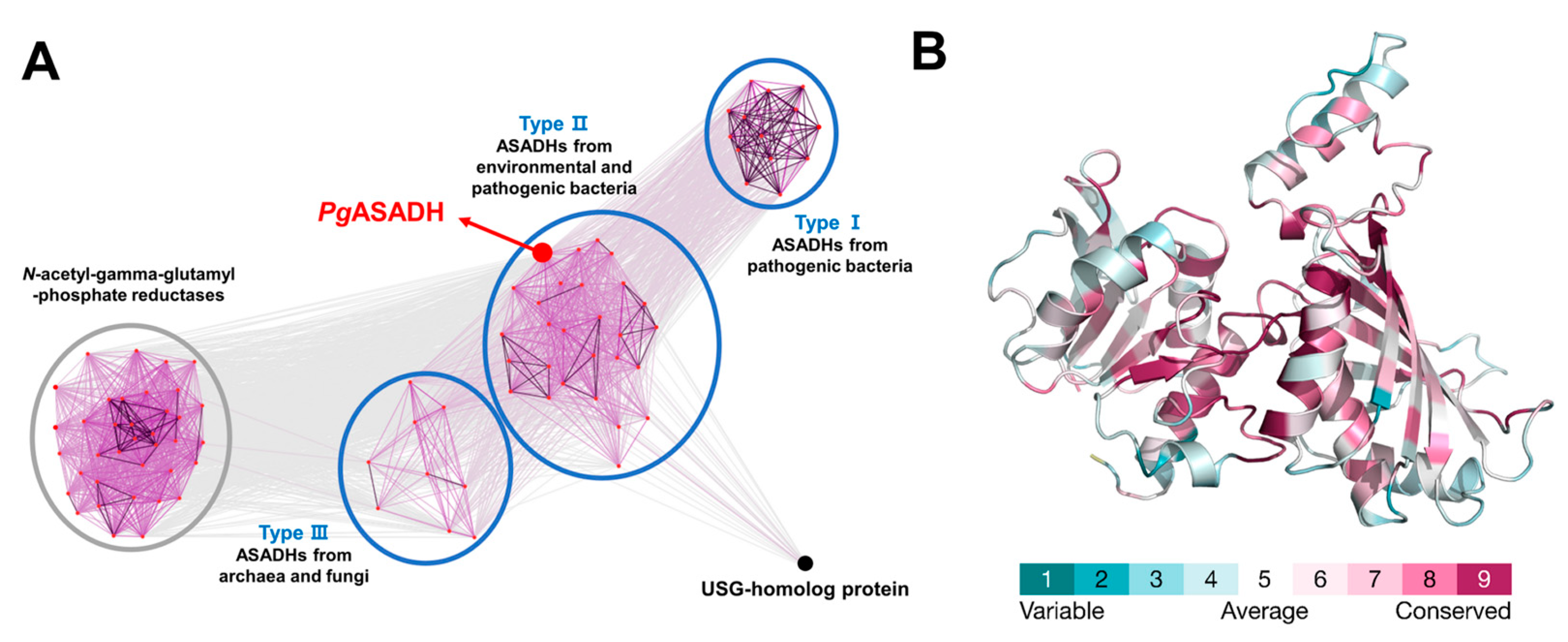
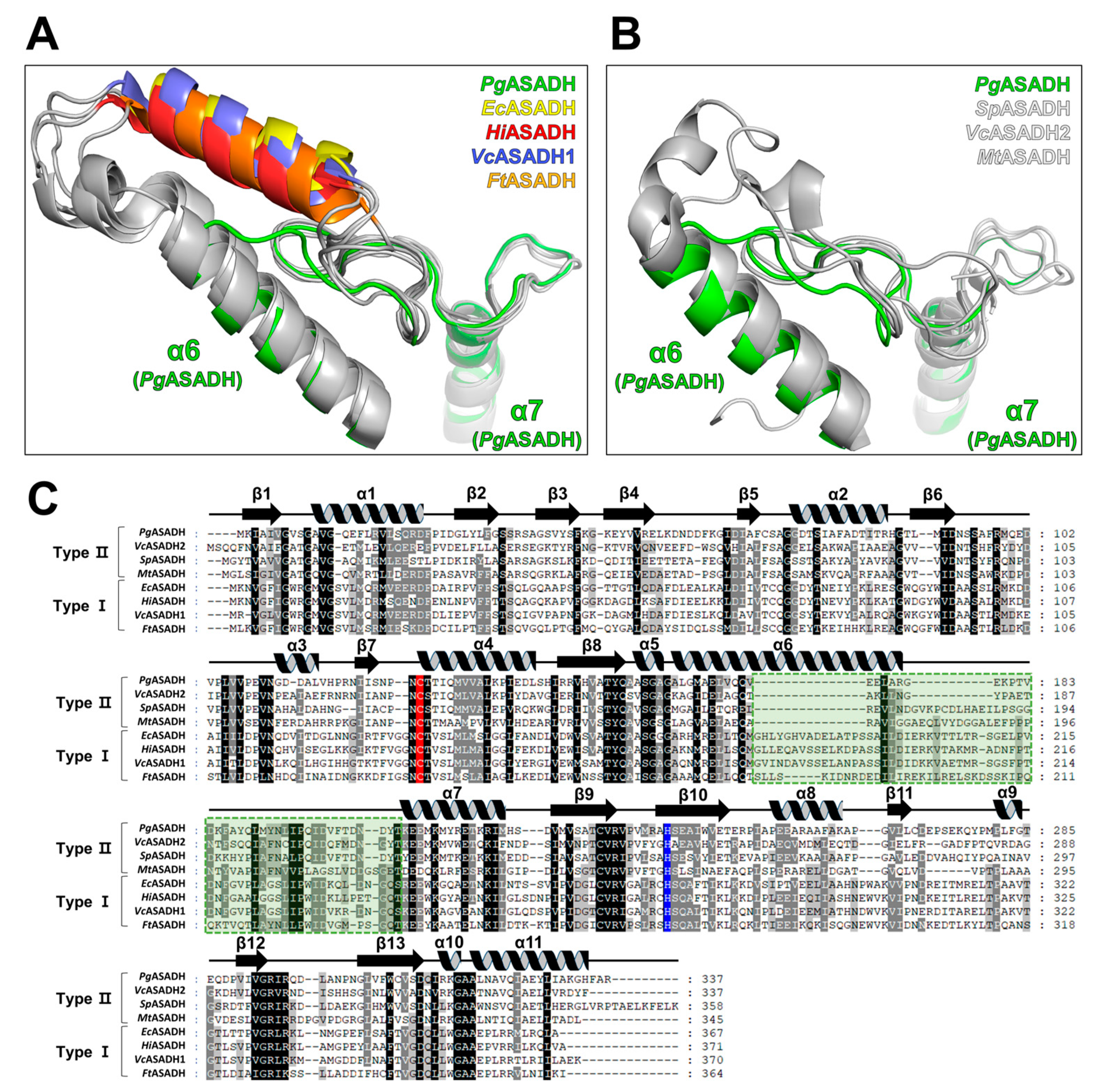
3.4. Comparative Sequential and Structural Analysis of ASADHs
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Elwishahy, A.; Antia, K.; Bhusari, S.; Ilechukwu, N.C.; Horstick, O.; Winkler, V. Porphyromonas gingivalis as a risk factor to Alzheimer’s disease: A systematic review. J. Alzheimer’s Dis. Rep. 2021, 5, 721–732. [Google Scholar] [CrossRef] [PubMed]
- Dominy, S.S.; Lynch, C.; Ermini, F.; Benedyk, M.; Marczyk, A.; Konradi, A.; Nguyen, M.; Haditsch, U.; Raha, D.; Griffin, C.; et al. Porphyromonas gingivalis in Alzheimer’s disease brains: Evidence for disease causation and treatment with small-molecule inhibitors. Sci. Adv. 2019, 5, eaau3333. [Google Scholar] [CrossRef] [PubMed]
- Ryder, M.I. Porphyromonas gingivalis and Alzheimer disease: Recent findings and potential therapies. J. Periodontol. 2020, 91 (Suppl. 1), S45–S49. [Google Scholar] [CrossRef]
- Viola, R.E. The central enzymes of the aspartate family of amino acid biosynthesis. Acc. Chem. Res. 2001, 34, 339–349. [Google Scholar] [CrossRef]
- Paidhungat, M.; Setlow, B.; Driks, A.; Setlow, P. Characterization of spores of Bacillus subtilis which lack dipicolinic acid. J. Bacteriol. 2000, 182, 5505–5512. [Google Scholar] [CrossRef]
- Pavelka, M.S., Jr.; Jacobs, W.R., Jr. Biosynthesis of diaminopimelate, the precursor of lysine and a component of peptidoglycan, is an essential function of Mycobacterium smegmatis. J. Bacteriol. 1996, 178, 6496–6507. [Google Scholar] [CrossRef]
- Cardineau, G.A.; Curtiss, R., 3rd. Nucleotide sequence of the asd gene of Streptococcus mutans. Identification of the promoter region and evidence for attenuator-like sequences preceding the structural gene. J. Biol. Chem. 1987, 262, 3344–3353. [Google Scholar] [CrossRef] [PubMed]
- Galan, J.E.; Nakayama, K.; Curtiss, R., 3rd. Cloning and characterization of the asd gene of Salmonella typhimurium: Use in stable maintenance of recombinant plasmids in Salmonella vaccine strains. Gene 1990, 94, 29–35. [Google Scholar] [CrossRef]
- Harb, O.S.; Abu Kwaik, Y. Identification of the aspartate-beta-semialdehyde dehydrogenase gene of Legionella pneumophila and characterization of a null mutant. Infect. Immun. 1998, 66, 1898–1903. [Google Scholar] [CrossRef]
- Hadfield, A.; Kryger, G.; Ouyang, J.; Petsko, G.A.; Ringe, D.; Viola, R. Structure of aspartate-beta-semialdehyde dehydrogenase from Escherichia coli, a key enzyme in the aspartate family of amino acid biosynthesis. J. Mol. Biol. 1999, 289, 991–1002. [Google Scholar] [CrossRef]
- Hadfield, A.; Shammas, C.; Kryger, G.; Ringe, D.; Petsko, G.A.; Ouyang, J.; Viola, R.E. Active site analysis of the potential antimicrobial target aspartate semialdehyde dehydrogenase. Biochemistry 2001, 40, 14475–14483. [Google Scholar] [CrossRef]
- Nichols, C.E.; Dhaliwal, B.; Lockyer, M.; Hawkins, A.R.; Stammers, D.K. High-resolution structures reveal details of domain closure and “half-of-sites-reactivity” in Escherichia coli aspartate beta-semialdehyde dehydrogenase. J. Mol. Biol. 2004, 341, 797–806. [Google Scholar] [CrossRef]
- Blanco, J.; Moore, R.A.; Kabaleeswaran, V.; Viola, R.E. A structural basis for the mechanism of aspartate-beta-semialdehyde dehydrogenase from Vibrio cholerae. Protein Sci. 2003, 12, 27–33. [Google Scholar] [CrossRef] [PubMed]
- Viola, R.E.; Liu, X.; Ohren, J.F.; Faehnle, C.R. The structure of a redundant enzyme: A second isoform of aspartate beta-semialdehyde dehydrogenase in Vibrio cholerae. Acta Crystallogr. D Biol. Crystallogr. 2008, 64, 321–330. [Google Scholar] [CrossRef] [PubMed]
- Kabsch, W. XDS. Acta Crystallogr. D Biol. Crystallogr. 2010, 66, 125–132. [Google Scholar] [CrossRef]
- Winn, M.D.; Ballard, C.C.; Cowtan, K.D.; Dodson, E.J.; Emsley, P.; Evans, P.R.; Keegan, R.M.; Krissinel, E.B.; Leslie, A.G.; McCoy, A.; et al. Overview of the CCP4 suite and current developments. Acta Crystallogr. D Biol. Crystallogr. 2011, 67, 235–242. [Google Scholar] [CrossRef]
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; ZiŽídek, A.; Potapenko, A.; et al. Highly accurate protein structure prediction with AlphaFold. Nature 2021, 596, 583–589. [Google Scholar] [CrossRef]
- Emsley, P.; Cowtan, K. Coot: Model-building tools for molecular graphics. Acta Crystallogr. D Biol. Crystallogr. 2004, 60, 2126–2132. [Google Scholar] [CrossRef]
- Murshudov, G.N.; Skubaák, P.; Lebedev, A.A.; Pannu, N.S.; Steiner, R.A.; Nicholls, R.A.; Winn, M.D.; Long, F.; Vagin, A.A. REFMAC5 for the refinement of macromolecular crystal structures. Acta Crystallogr. D Biol. Crystallogr. 2011, 67, 355–367. [Google Scholar] [CrossRef]
- Afonine, P.V.; Grosse-Kunstleve, R.W.; Echols, N.; Headd, J.J.; Moriarty, N.W.; Mustyakimov, M.; Terwilliger, T.C.; Urzhumtsev, A.; Zwart, P.H.; Adams, P.D. Towards automated crystallographic structure refinement with phenix.refine.refine. Acta Crystallogr. D Biol. Crystallogr. 2012, 68, 352–367. [Google Scholar] [CrossRef]
- Chen, V.B.; Arendall, W.B., 3rd.; Headd, J.J.; Keedy, D.A.; Immormino, R.M.; Kapral, G.J.; Murray, L.W.; Richardson, J.S.; Richardson, D.C. MolProbity: All-atom structure validation for macromolecular crystallography. Acta Crystallogr. D Biol. Crystallogr. 2010, 66, 12–21. [Google Scholar] [CrossRef]
- Frickey, T.; Lupas, A. CLANSlans: A Java application for visualizing protein families based on pairwise similarity. Bioinformatics 2004, 20, 3702–3704. [Google Scholar] [CrossRef]
- Yariv, B.; Yariv, E.; Kessel, A.; Masrati, G.; Chorin, A.B.; Martz, E.; Mayrose, I.; Pupko, T.; Ben-Tal, N. Using evolutionary data to make sense of macromolecules with a “face-lifted”. ConSurf. Protein Sci. 2023, 32, e4582. [Google Scholar] [CrossRef] [PubMed]
- Matthews, B.W. Solvent content of protein crystals. J. Mol. Biol. 1968, 33, 491–497. [Google Scholar] [CrossRef]
- Krissinel, E.; Henrick, K. Inference of macromolecular assemblies from crystalline state. J. Mol. Biol. 2007, 372, 774–797. [Google Scholar] [CrossRef] [PubMed]
- Dahal, G.P.; Viola, R.E. Structure of a fungal form of aspartate-semialdehyde dehydrogenase from Aspergillus fumigatus. Acta Crystallogr. F Struct. Biol. Commun. 2017, 73, 36–44. [Google Scholar] [CrossRef]
- Dahal, G.P.; Viola, R.E. Structural insights into inhibitor binding to a fungal ortholog of aspartate semialdehyde dehydrogenase. Biochem. Biophys. Res. Commun. 2018, 503, 2848–2854. [Google Scholar] [CrossRef] [PubMed]
- Holm, L. Dali server: Structural unification of protein families. Nucleic Acids Res. 2022, 50, W210–W215. [Google Scholar] [CrossRef]
- Pavlovsky, A.G.; Liu, X.; Faehnle, C.R.; Potente, N.; Viola, R.E. Structural characterization of inhibitors with selectivity against members of a homologous enzyme family. Chem. Biol. Drug Des. 2012, 79, 128–136. [Google Scholar] [CrossRef]
- Pavlovsky, A.G.; Thangavelu, B.; Bhansali, P.; Viola, R.E. A cautionary tale of structure-guided inhibitor development against an essential enzyme in the aspartate-biosynthetic pathway. Acta Crystallogr. D Biol. Crystallogr. 2014, 70, 3244–3252. [Google Scholar] [CrossRef]
- Vyas, R.; Tewari, R.; Weiss, M.S.; Karthikeyan, S. Structures of ternary complexes of aspartate-semialdehyde dehydrogenase (Rv3708c) from Mycobacterium tuberculosis H37Rv. Acta Crystallogr. D Biol. Crystallogr. 2012, 68, 671–679. [Google Scholar] [CrossRef] [PubMed]
- Mank, N.J.; Pote, S.; Majorek, K.A.; Arnette, A.K.; Klapper, V.G.; Hurlburt, B.K.; Chruszcz, M. Structure of aspartate beta-semialdehyde dehydrogenase from Francisella tularensis. Acta Crystallogr. F Struct. Biol. Commun. 2018, 74, 14–22. [Google Scholar] [CrossRef] [PubMed]
- Blanco, J.; Moore, R.A.; Faehnle, C.R.; Coe, D.M.; Viola, R.E. The role of substrate-binding groups in the mechanism of aspartate-beta-semialdehyde dehydrogenase. Acta Crystallogr. D Biol. Crystallogr. 2004, 60, 1388–1395. [Google Scholar] [CrossRef]
- Johnson, M.; Zaretskaya, I.; Raytselis, Y.; Merezhuk, Y.; McGinnis, S.; Madden, T.L.; NCBI. NCBI BLAST: A better web interface. Nucleic Acids Res. 2008, 36, W5–W9. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Data Set | PgASADH | 2′,5′-ADP-Bound PgASADH |
|---|---|---|
| X-ray source | BL-5C (PAL) | BL-5C (PAL) |
| Space group | I212121 | C2221 |
| Unit-cell parameters (Å, °) | a = 74.86, b = 108.86, c = 162.07 α = β = γ = 90 | a = 74.99, b = 108.22, c = 160.4 α = β = γ = 90 |
| Wavelength (Å) | 0.9796 | 0.9796 |
| Resolution (Å) | 29.75–1.73 (1.79–1.73) | 28.46–1.73 (1.79–1.73) |
| Total reflections | 920,322 (91,009) | 929,371 (90,382) |
| Unique reflections | 69,004 (6845) | 68,236 (6742) |
| Average I/σ (I) | 19.59 (2.03) | 29.89 (7.46) |
| Rmergea | 0.073 (1.269) | 0.060 (0.306) |
| Redundancy | 13.3 (13.3) | 13.6 (13.4) |
| Completeness (%) | 99.5 (99.9) | 100 (100) |
| Refinement | ||
| Resolution range (Å) | 29.75–1.73 (1.75–1.73) | 28.46–1.73 (1.79–1.73) |
| No. of reflections of the working set | 68,955 (6841) | 68,224 (6742) |
| No. of reflections of the test set | 3363 (310) | 3412 (337) |
| No. of amino acid residues | 670 | 670 |
| No. of water molecules | 346 | 419 |
| Rcrystb | 0.217 (0.321) | 0.184 (0.204) |
| Rfreec | 0.238 (0.333) | 0.206 (0.244) |
| R.m.s. bond length (Å) | 0.008 | 0.009 |
| R.m.s. bond angle (°) | 0.95 | 1.01 |
| Average B value (Å2) (protein) | 28.25 | 17.84 |
| Average B value (Å2) (solvent) | 28.30 | 25.14 |
| Average B value (Å2) (ligand) | - | 24.09 |
| Protein | PDB Code | DALI Z-Score | UniProtKB Code | Sequence % ID with PgASADH (Aligned Residue Number) | Reference |
|---|---|---|---|---|---|
| ASASDH from Vibrio cholerae | 2QZ9 | 49.8 | P23247 | 46 (330/336) | [14] |
| ASASDH from Thermus thermophilus HB8 | 2YV3 | 47.8 | Q5SKU8 | 46 (327/328) | Not yet published |
| ASASDH from Streptococcus pneumoniae SP23-BS72 | 3PYL | 47.7 | a N/A | 44 (333/362) | [29] |
| ASASDH from Mycobacterium tuberculosis H37Rv | 3VOS | 47.6 | P9WNX5 | 42 (324/343) | [31] |
| ASASDH from Pseudomonas aeruginosa PAO1 | 2HJS | 42.5 | O87014 | 42.5 (328/334) | Not yet published |
| ASASDH from Francisella tularensis | 4WOJ | 40.0 | Q8G8T7 | 26 (329/365) | [32] |
| ASASDH from Acinetobacter baumannii | 7SKB | 39.9 | V5VGT0 | 27 (332/374) | Not yet published |
| ASASDH from Escherichia coli | 1GL3 | 39.4 | P0A9Q9 | 27 (327/368) | [11] |
| ASASDH fromHaemophilus influenzae Rd KW20 | 1PQU | 39.1 | P44801 | 30 (328/372) | [33] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hwang, J.; Do, H.; Shim, Y.-S.; Lee, J.H. Crystal Structure of Aspartate Semialdehyde Dehydrogenase from Porphyromonas gingivalis. Crystals 2023, 13, 1274. https://doi.org/10.3390/cryst13081274
Hwang J, Do H, Shim Y-S, Lee JH. Crystal Structure of Aspartate Semialdehyde Dehydrogenase from Porphyromonas gingivalis. Crystals. 2023; 13(8):1274. https://doi.org/10.3390/cryst13081274
Chicago/Turabian StyleHwang, Jisub, Hackwon Do, Youn-Soo Shim, and Jun Hyuck Lee. 2023. "Crystal Structure of Aspartate Semialdehyde Dehydrogenase from Porphyromonas gingivalis" Crystals 13, no. 8: 1274. https://doi.org/10.3390/cryst13081274
APA StyleHwang, J., Do, H., Shim, Y.-S., & Lee, J. H. (2023). Crystal Structure of Aspartate Semialdehyde Dehydrogenase from Porphyromonas gingivalis. Crystals, 13(8), 1274. https://doi.org/10.3390/cryst13081274