
Energetics of Interfaces and Strain Partition in GaN/AlN Pseudomorphic Superlattices


Abstract
1. Introduction
2. Materials and Methods
2.1. Experimental
2.2. Computational Method
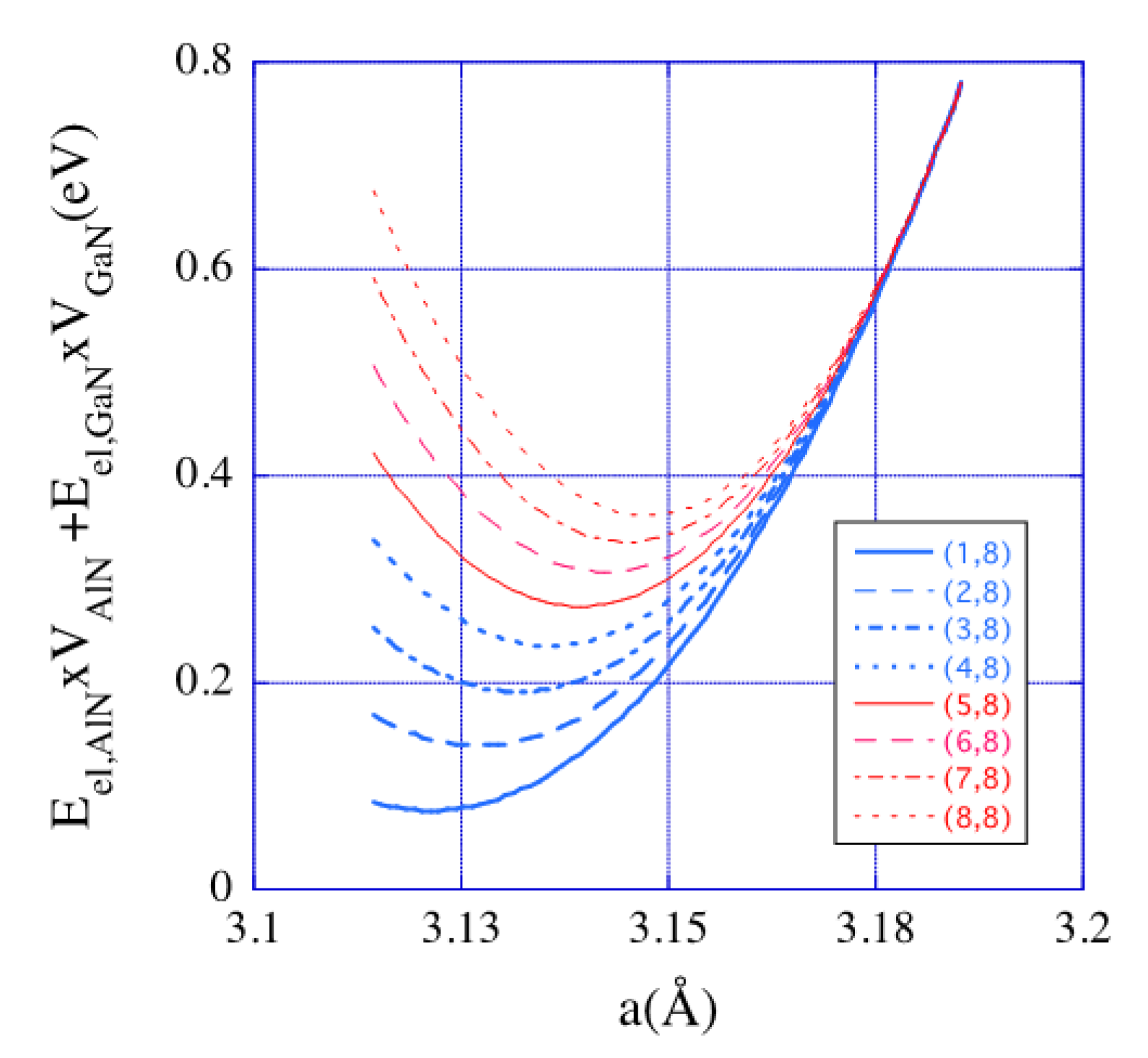
2.2.1. Geometrical Model
2.2.2. DFT Calculations
3. Results
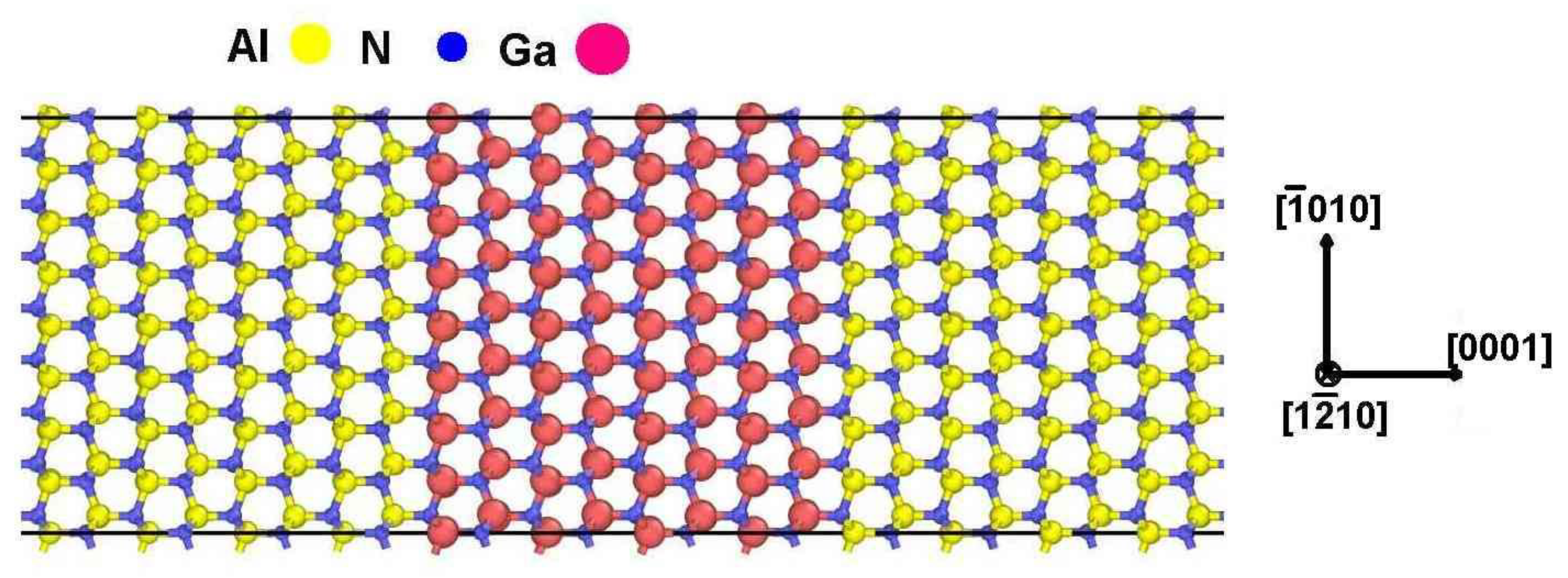
3.1. TEM Observations
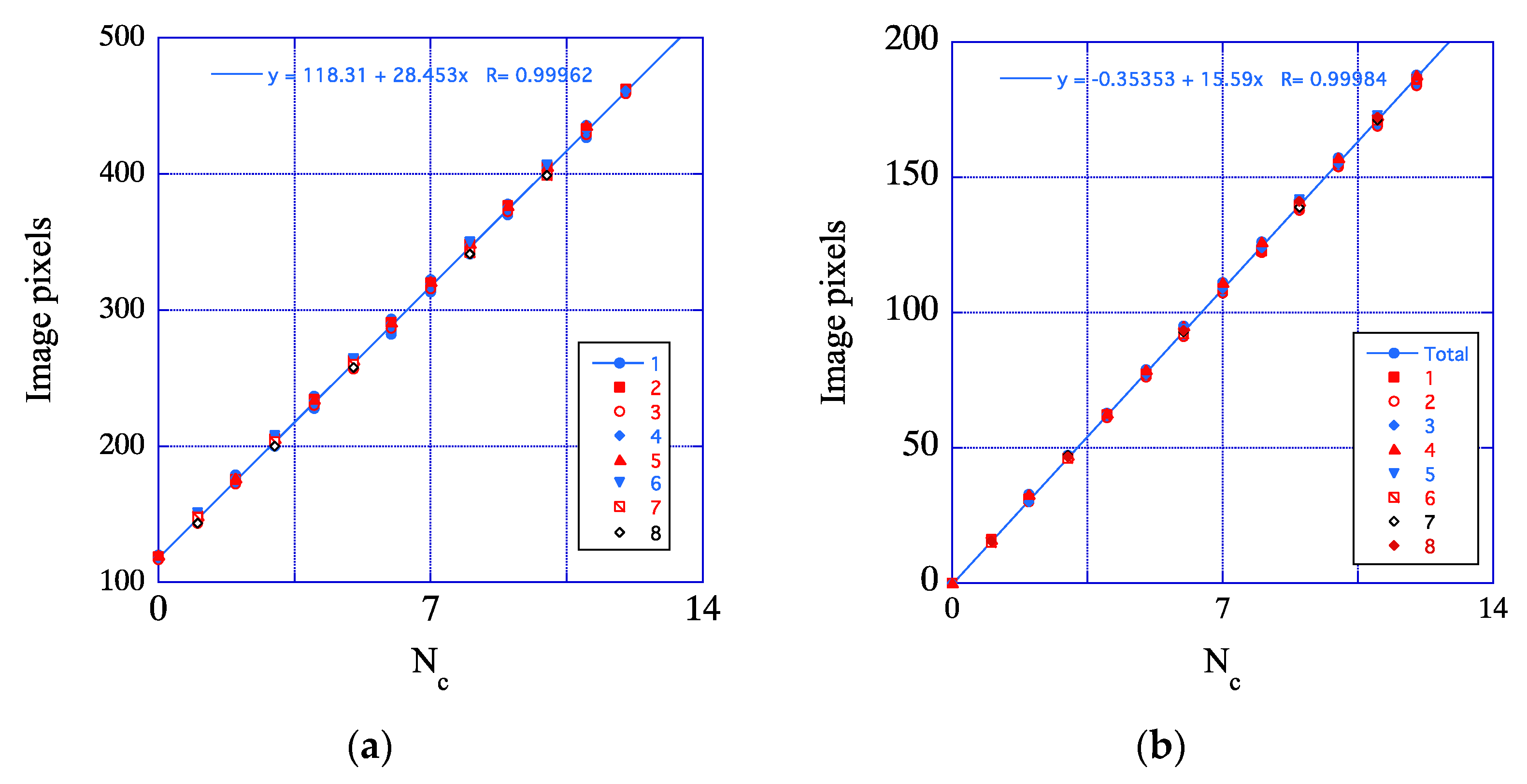
3.2. HRTEM Data Deconvolution
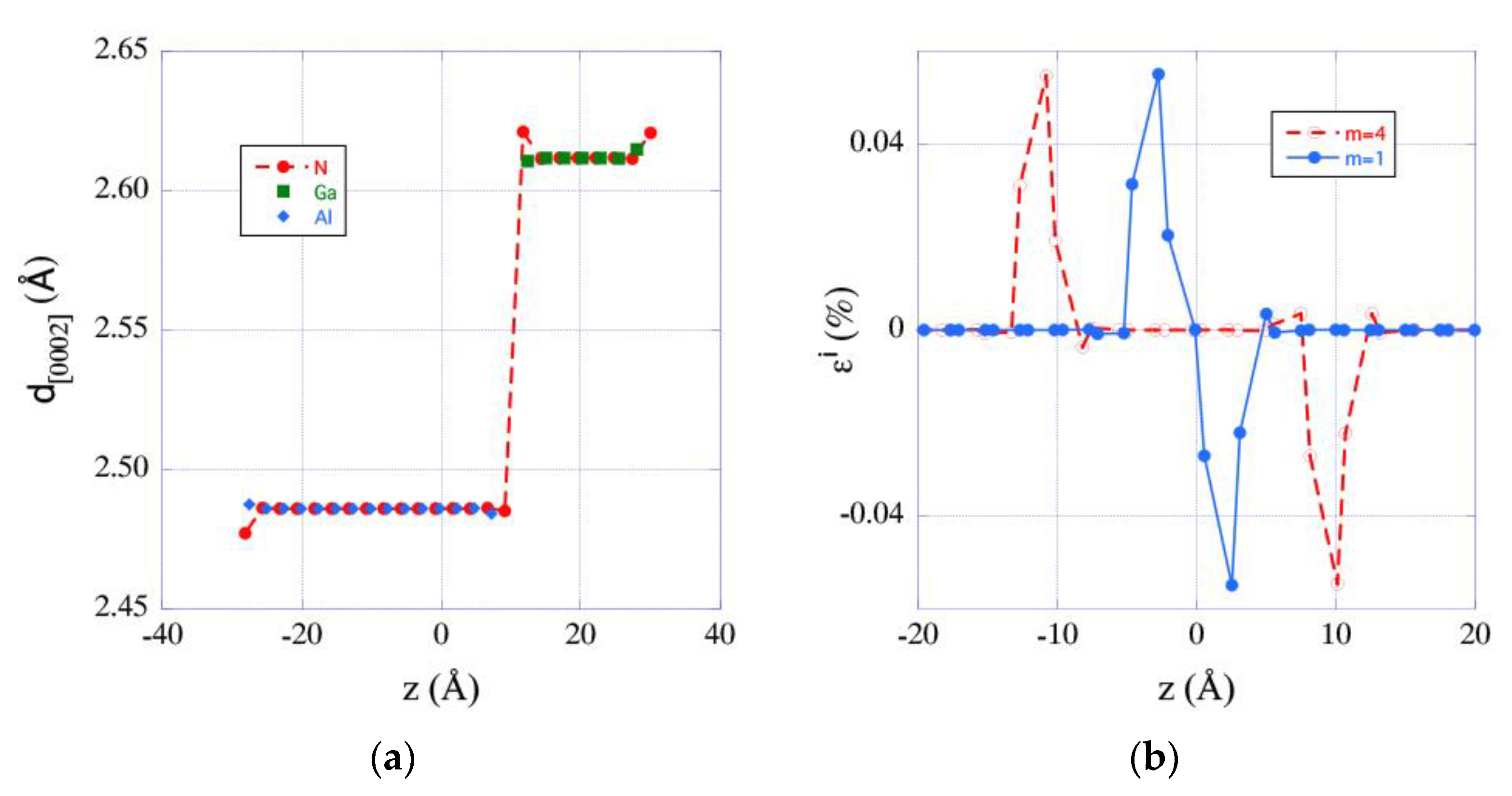
3.3. Spatial Extension and Deformation States of Interfaces
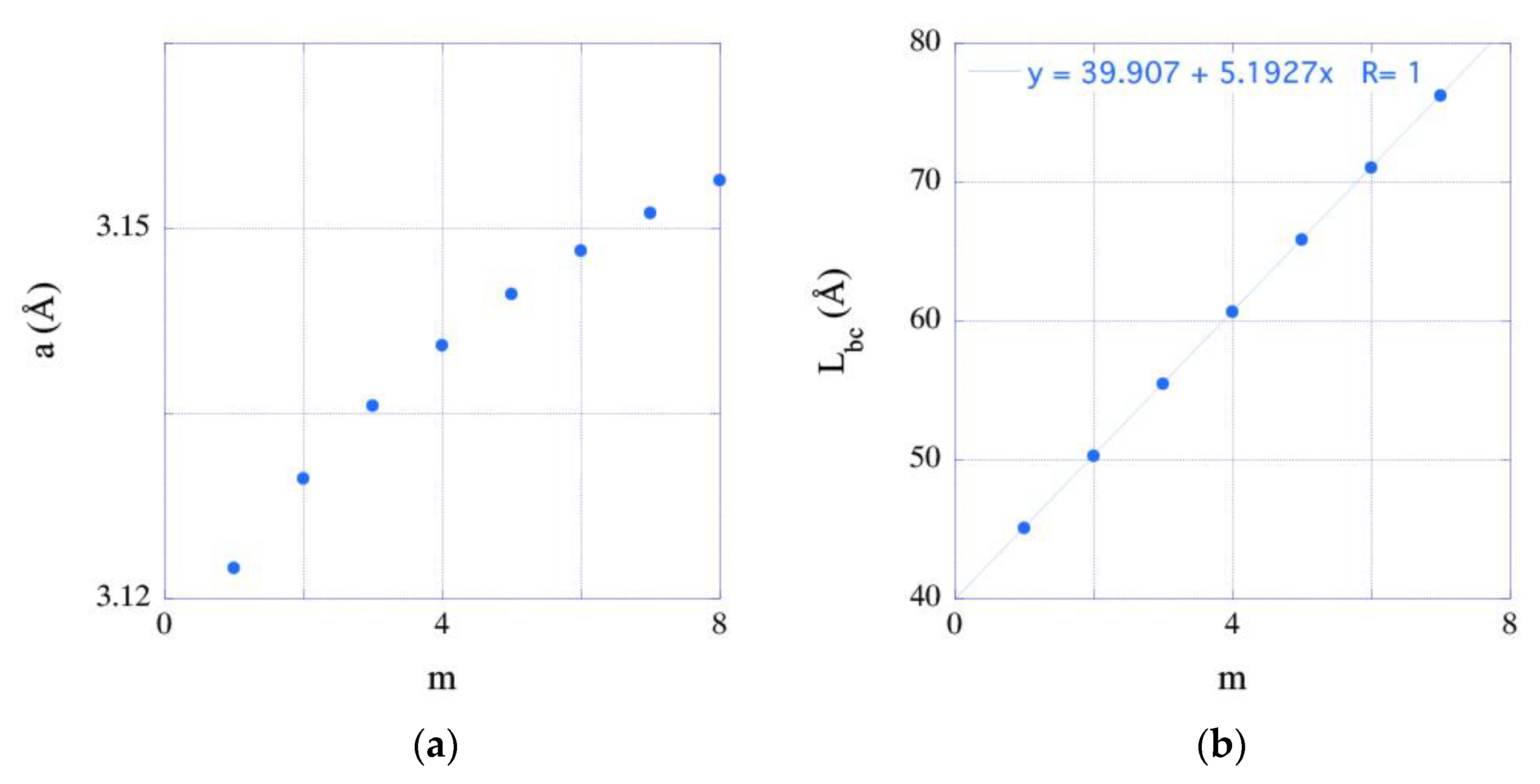
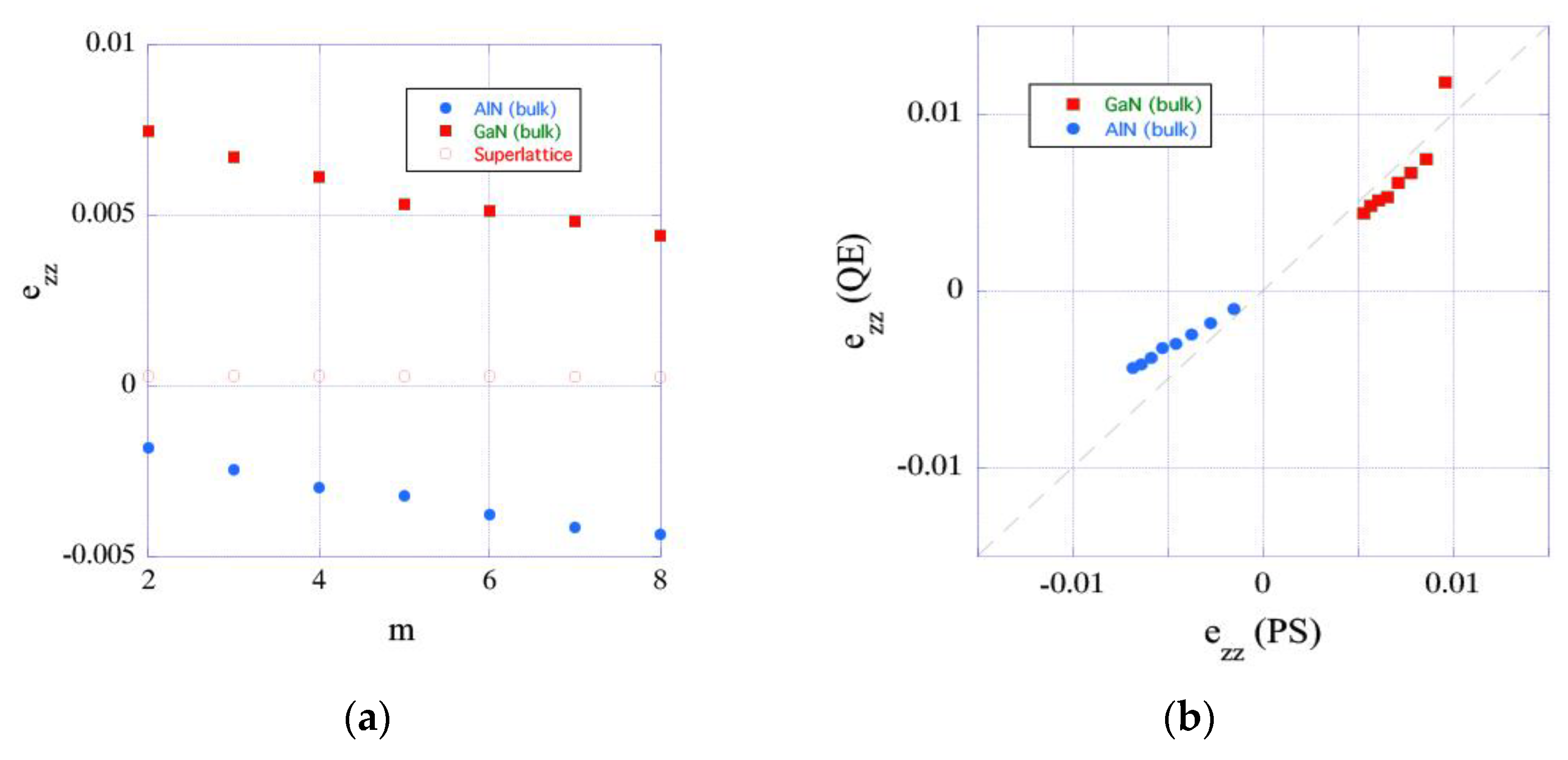
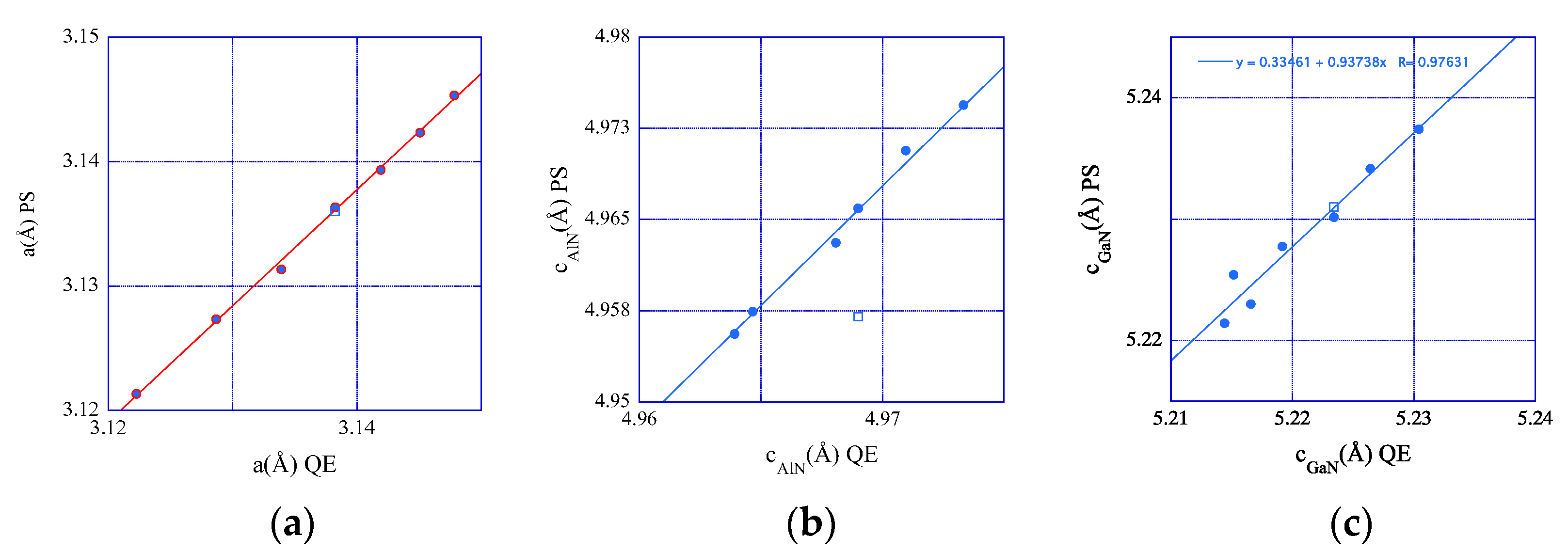
3.4. Deformation States of Relaxed Superlattices and Zero-Stress
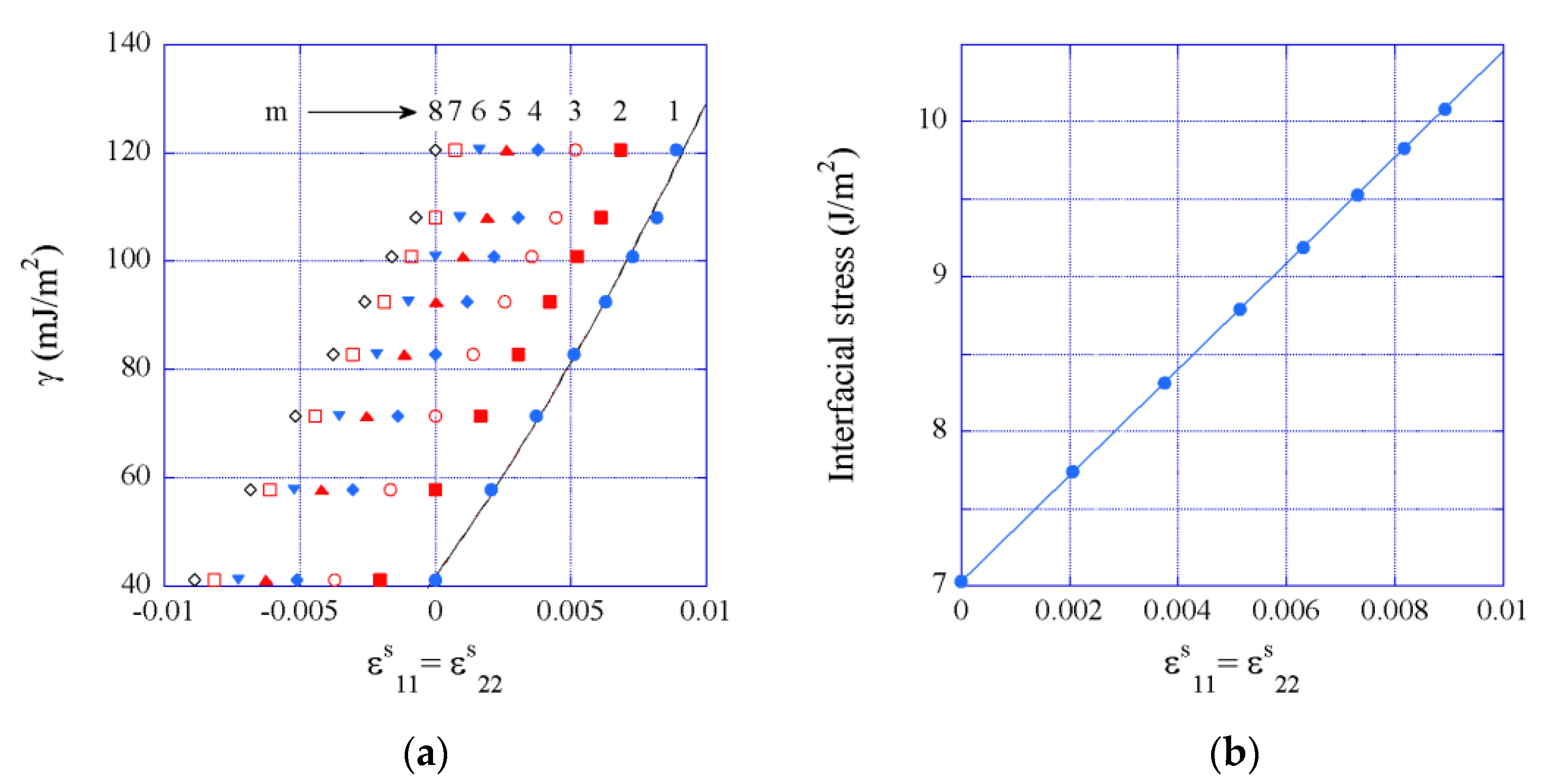
3.5. Interfacial Excess Energy
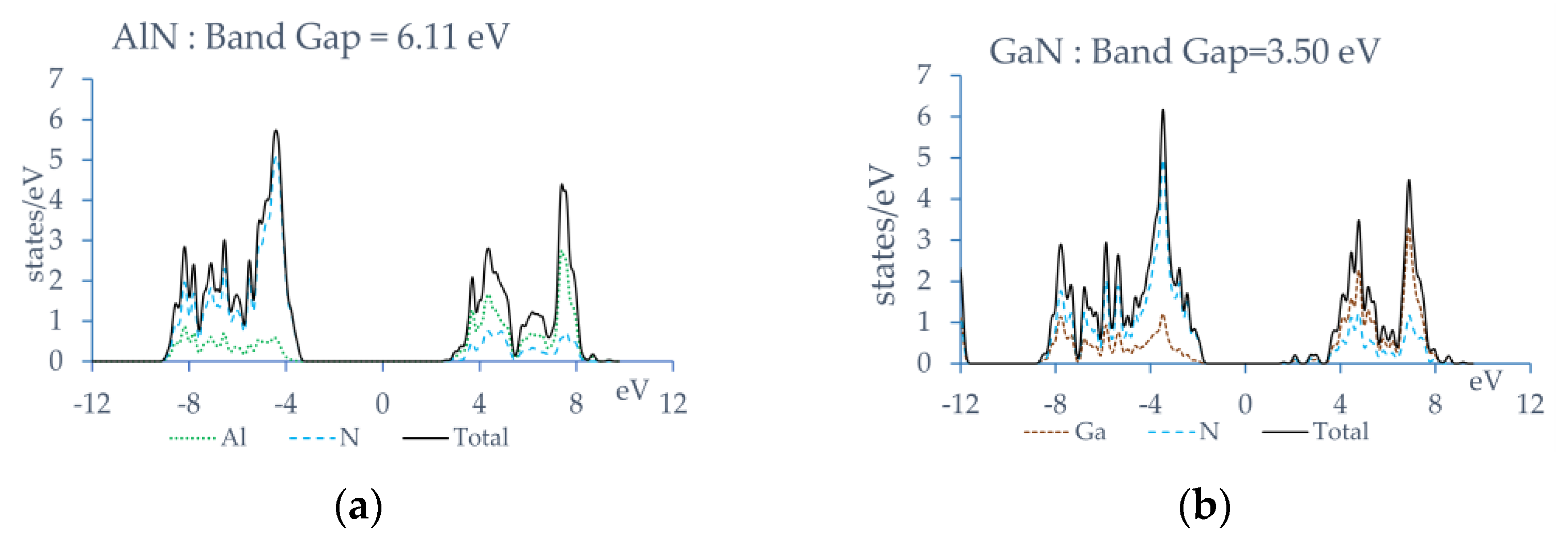
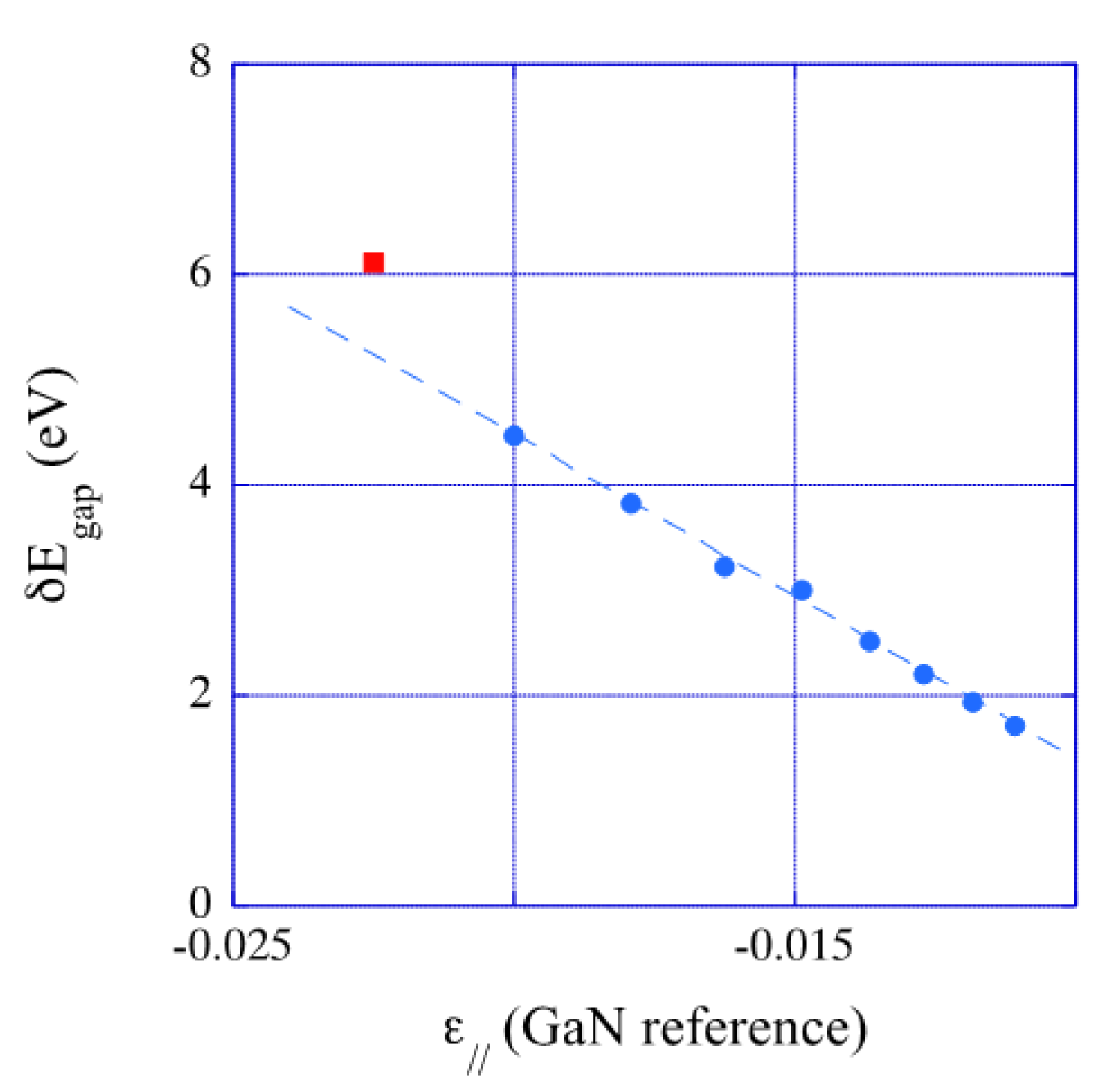
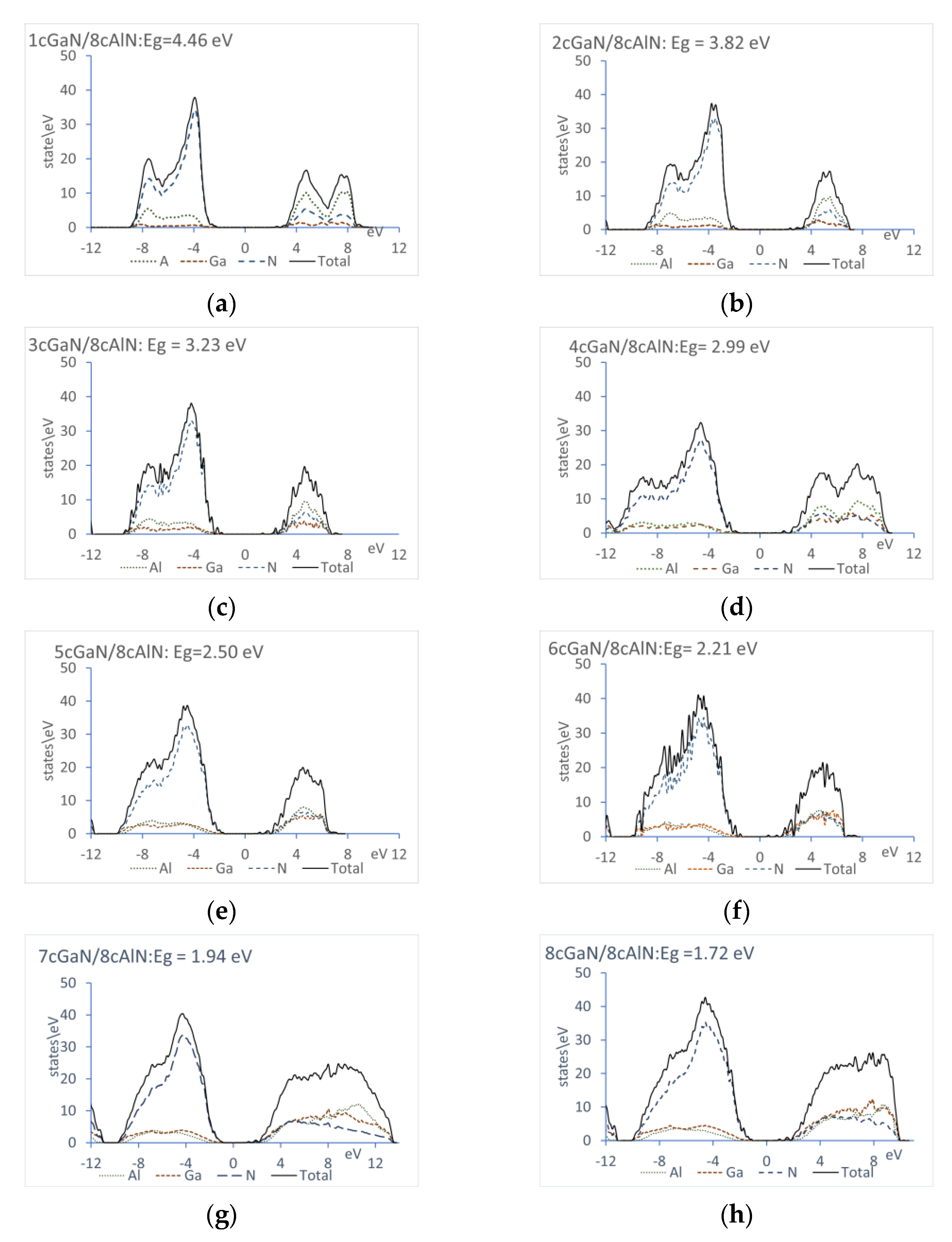
3.6. Energy Gap
4. Discussion and Conclusions
- (i).
- The spatial extension of the interfaces is very short-ranged and remains practically unchanged when the thickness of the GaN QW changes.
- (ii).
- Energy-minimized configurations of the studied systems adopt different deformation states. It has been shown that the elastic prediction of the lattice constants of elastically strained bulk compounds reasonably agrees with the predictions of total energy minimizations.
- (iii).
- A no-trivial result is that the interfacial excess energy is a function of the thicknesses of the superlattice constituents, which relates to the evolving deformation states of the interfaces with changing the respective thicknesses. Works in the literature have not accounted for such effects.
- (iv).
- Similar to structural properties and the interfacial excess energy, the superlattice electronic properties are tightly deformation state dependent. Indeed, it has been established that the valence and conduction bands offsets in strained superlattices and the energy gaps as well are in a linear relationship with the tangential deformation component of the bulk constituents, [56]. We have verified that energy gaps calculated in the present work obey this linear relationship.
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
Appendix A

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | a (Å) | c (Å) | c/a | E (eV) |
|---|---|---|---|---|
| AlN | 3.114 (3.113) | 4.9869 (4.9816) | 1.601 (1.601) | 1831.51 |
| GaN | 3.185 (3.189) | 5.1913 (5.185) | 1.630 (1.626) | 8302.31 |
| Compound | C11 | C12 | C13 | C33 | C44 | C66 |
|---|---|---|---|---|---|---|
| AlN | 382.1 (411 ± 10) | 137.3 (149 ± 10) | 107.0 (99 ± 4) | 358.5 (389 ± 10) | 111.9 (125 ± 5) | 122.4 (131 ± 10) |
| GaN | 355.3 (390) | 126.0 (145) | 88.6 (106) | 394.2 (398) | 93.3 (105) | 114.6 (123) |
| m | abc (Å) | Lbc (Å) | Ebc (eV) |
|---|---|---|---|
| 1 | 3.122 | 45.087 | 22,954.292 |
| 2 | 3.129 | 50.278 | 31,256.541 |
| 3 | 3.134 | 55.469 | 39,558.801 |
| 4 | 3.138 | 60.661 | 47,861.066 |
| 5 | 3.142 | 65.852 | 56,163.338 |
| 6 | 3.145 | 71.043 | 64,465.616 |
| 7 | 3.148 | 76.235 | 72,767.895 |
| 8 | 3.150 | 81.426 | 81,070.175 |

References
- Kehagias, T.; Komninou, P.; Dimitrakopulos, G. Intergranular and Intephase Boundaries in Materials; Springer Science & Business Media: New York, NY, USA, 2014; Volume 49, p. 11. [Google Scholar]
- Hardouin Duparc, O.B.M.; Lartigue-Korinek, S. Interface science in JMS. J. Mater. Sci. 2020, 55, 16861–16863. [Google Scholar] [CrossRef] [PubMed]
- Ayers, J.E. Heteroepitaxy of Semiconductors: Theory, Growth, and Characterization, 1st ed.; Taylor & Francis Group: Boca Raton, FL, USA, 2007; pp. 1–480. [Google Scholar] [CrossRef]
- Rüdiger, Q. Gallium Nitride Electronics, 2008th ed.; Springer Series in Materials Science; Springer: Berlin/Heidelberg, Germany, 2008; p. 96. [Google Scholar]
- Kasap, S.; Capper, P. Handbook of Electronic and Photonic Materials; Springer International Publishing AG: Berlin, Germany, 2017. [Google Scholar]
- Alkauskas, A.; Deák, P.; Neugebauer, J.; Pasquarello, A.; Van de Walle, C.G. Advanced Calculations for Defects in Materials Electronic Structure Methods; Wiley-VCH Verlag & Co. KGaA: Weinheim, Germany, 2011. [Google Scholar]
- Matsuoka, T.; Kangawa, Y. Epitaxial Growth of III-Nitride Compounds; Computational Approach. Springer Series in Materials Science; Springer International Publishing: New York, NY, USA, 2018; p. 269. [Google Scholar]
- Morkoç, H. Handbook of Nitride Semiconductors and Devices: Materials Properties, Physics and Growth; WILEY-VCH Verlag GmbH & Co., Ltd.: Weinheim, Germany, 2008. [Google Scholar]
- Gil, B. III-Nitride Semiconductors and their Modern Devices; Oxford University Press: Oxford, UK, 2013. [Google Scholar]
- Dimitrakopulos, G.P.; Vasileiadis, I.G.; Bazioti, C.; Smalc-Koziorowska, J.; Kret, S.; Dimakis, E.; Florini, N.; Kehagias, T.; Suski, T.; Karakostas, T.; et al. Compositional and strain analysis of In Ga N/GaN short period superlattices. J. Appl. Phys. 2018, 123, 024304-12. [Google Scholar] [CrossRef]
- Bykhovski, A.D.; Gelmont, B.L.; Shur, M.S. Elastic strain relaxation and piezoeffect in GaN-AlN, GaN-AlGaN and GaN-InGaN superlattices. J. Appl. Phys. 1997, 81, 6332–6338. [Google Scholar] [CrossRef]
- Kladko, V.; Kuchuk, A.; Lytvyn, P.; Yefanov, O.; Safriuk, N.; Belyaev, A.; I Mazur, Y.; A DeCuir, E.; E Ware, M.; Salamo, G.J. Substrate effects on the strain relaxation in GaN/AlN short-period superlattices. Nanoscale Res. Lett. 2012, 7, 289. [Google Scholar] [CrossRef] [PubMed]
- Sohi, P.; Martin, D.; Grandjean, N. Critical thickness of GaN on AlN: Impact of growth temperature and dislocation density. Semicond. Sci. Technol. 2017, 32, 075010. [Google Scholar] [CrossRef]
- Davydov, V.Y.; Roginskii, E.M.; Kitaev, Y.E.; Smirnov, A.N.; Eliseyev, I.A.; Yagovkina, M.A.; Nechaev, D.V.; Jmerik, V.N.; Smirnov, M.B. Structural and dynamic properties of short-period GaN/AlN superlattices grown by submonolayer digital epitaxy. J. Phys. Conf. Ser. 2020, 1697, 012155. [Google Scholar] [CrossRef]
- Kioseoglou, J.; Komninou, P.; Chen, J.; Nouet, G.; Kalesaki, E.; Karakostas, T. Structural and electronic properties of elastically strained InN/GaN quantum well multilayer heterostructures. Phys. Status Solidi C 2014, 11, 289. [Google Scholar] [CrossRef]
- Coppeta, R.A.; Ceric, H.; Holec, D.; Gracer, T. Critical thickness for GaN thin film on AlN substrate. In IEEE International Integrated Reliability Workshop Final Report; IEEE: South Lake Tahoe, CA, USA, 2013; pp. 133–136. [Google Scholar] [CrossRef]
- Kryvy, S.B.; Lytvyn, P.M.; Kladko, V.P.; Stanchu, H.V.; Kuchuk, A.V.; Mazur, Y.I.; Salamo, G.I.; Li, S.; Kogutyuk, P.P.; Belyaev, A.E. Effect of well/barrier thickness ratio on strain relaxation in GaN/AlN superlattices grown on GaN/sapphire template. J. Vac. Sci. Technol. B 2017, 1, 062902. [Google Scholar] [CrossRef]
- Kioseoglou, J.; Kalessaki, E.; Dimitrakopulos, G.P.; Komninou, P.; Karakostas, T. Study of InN/GaN interfaces using molecular dynamics. J. Mater. Sci. 2008, 43, 3982. [Google Scholar] [CrossRef]
- Kaminska, A.; Strak, P.; Borysiuk, J.; Sobczak, K.; Domagala, J.Z.; Beeler, M.; Grzanka, E.; Sakowski, K.; Krukowski, S.; Monroy, E. Correlation of optical and structural properties of GaN/AlN multi-quantum wells—Ab initio and experimental study. J. Appl. Phys. 2016, 119, 015703. [Google Scholar] [CrossRef]
- Gorczyca, I.; Skrobas, K.; Suski, T.; Christensen, N.E.; Svane, A. Influence of strain and internal electric fields on band gaps in short period nitride based superlattices. Superlattices Microstruct. 2015, 82, 438. [Google Scholar] [CrossRef]
- Gorczyca, I.; Suski, T.; Christensen, N.E.; Svane, A. Theoretical study of nitride short period superlattices. J. Phys. Condens. Matter 2018, 30, 063001. [Google Scholar] [CrossRef] [PubMed]
- Gorczyca, I.; Suski, T.; Strak, P.; Staszczak, G.; Christensen, N.E. Band gap engineering of In(Ga)N/GaN short period superlattices. Sci. Rep. 2017, 7, 16055. [Google Scholar] [CrossRef] [PubMed]
- Kandaswamy, P.K.; Guillot, F.E.; Bellet-Amalric, E.; Monroy, E.; Nevou, L.; Tchernycheva, M.; Michon, A.; Julien, F.H.; Baumann, E.; Giorgetta, F.R.; et al. GaN/AlN short-period superlattices for intersubband optoelectronics: A systematic study of their epitaxial growth, design, and performance. J. Appl. Phys. 2008, 104, 093501. [Google Scholar] [CrossRef]
- Kuchuk, A.V.; Kryvyi, S.; Lytvyn, P.M.; Li, S.; Kladko, V.P.; Ware, M.E.; Mazur, Y.I.; Safryuk, N.V.; Stanchu, H.V.; Belyaev, A.E.; et al. The Peculiarities of Strain Relaxation in GaN/AlN Superlattices Grown on Vicinal GaN (0001) Substrate: Comparative XRD and AFM Study. Nanoscale Res. Lett. 2016, 11, 252. [Google Scholar] [CrossRef]
- Stanchu, H.V.; Kuchuk, A.V.; Lytvyn, P.M.; Mazur, Y.I.; Maidaniuk, Y.; Benamara, M.; Shibin, L.; Kryvyi, S.; Kladko, V.P.; Belyaev, A.E.; et al. Strain relaxation in GaN/AlN superlattices on GaN(0001) substrate: Combined superlattice-to-substrate lattice misfit and thickness dependent effects. Mater. Des. 2018, 157, 141–150. [Google Scholar] [CrossRef]
- Aleksandrov, I.A.; Malin, T.V.; Zhuravlev, K.S.; Trubina, S.V.; Erenburg, S.B.; Pecz, B.; Lebiadok, Y.V. Diffusion in GaN/AlN superlattices: DFT and EXAFS study Diffusion in GaN/AlN superlattices: DFT and EXAFS study. App. Surf. Sci. 2020, 515, 146001. [Google Scholar] [CrossRef]
- Dimitrakopulos, G.P.; Komninou, P.; Kehagias, T.; Sahonta, S.-L.; Kioseoglou, J.; Vouroutzis, N.; Hausler, I.; Neumann, W.; Iliopoulos, E.; Georgakilas, A.; et al. Strain relaxation in AlN/GaN heterostructures grown by molecular beam epitaxy. Phys. Status Solidi A 2008, 205, 2569. [Google Scholar] [CrossRef]
- Dimitrakopulos, G.P.; Kalesaki, E.; Komninou, P.; Kehagias, T.; Kioseoglou, J.; Karakostas, T. Strain ac-commodation and interfacial structure of AlN interlayers in GaN. Cryst. Res. Technol. 2009, 44, 1170–1180. [Google Scholar] [CrossRef]
- Dimitrakopulos, G.P.; Sanchez, A.M.; Komninou, P.; Ruterana, P.; Nouet, G.; Kehagias, T.; Karakostas, T. Disconnections and inversion domain formation in GaN/AlN heteroepitaxy on (111) silicon. Phys. Status Solidi C 2005, 1, 4. [Google Scholar] [CrossRef]
- Kioseoglou, J.; Kalesaki, E.; Lymperakis, L.; Dimitrakopulos, G.P.; Komninou, P.; Karakostas, T. Polar AlN/GaN interfaces: Structures and energetics. Phys. Status Solidi A 2009, 206, 1892–1897. [Google Scholar] [CrossRef]
- Friel, I.; Driscoll, K.; Kulenica, E.; Dutta, M.; Paiella, R.; Moustakas, T.D. Investigation of the design parameters of AlN/GaN multiple quantum wells grown by molecular beam epitaxy for intersubband absorption. J. Cryst. Growth 2005, 278, 387–392. [Google Scholar] [CrossRef]
- Available online: https://www.gatan.com/products/tem-analysis (accessed on 21 January 2021).
- Stukowski, A. Visualization and analysis of atomistic simulation data with OVITO– the Open Visualization Tool. Model. Simul. Mater. Sci. Eng. 2010, 18, 015012. [Google Scholar] [CrossRef]
- Giannozzi, P.; Baroni, S.; Bonini, N.; Calandra, M.; Car, R.; Cavazzoni, C.; Ceresoli, D.; Chiarotti, G.L.; Cococcioni, M.; Dabo, I.; et al. QUANTUM ESPRESSO: A modular and open-source software project for quantum simulations of materials. J. Phys. Condens. Matter 2009, 21, 395502. [Google Scholar] [CrossRef]
- Giannozzi, P.; Andreussi, O.; Brumme, T.; Bunau, O.; Buongiorno Nardelli, M.; Calandra, M.; Car, R.; Cavazzoni, C.; Ceresoli, D.; Cococcioni, M.; et al. Advanced capabilities for materials modelling with Quantum ESPRESSO. J. Phys. Condens. Matter 2017, 29, 465901. [Google Scholar] [CrossRef]
- Giannozzi, P.; Baseggio, O.; Bonfà, P.; Brunato, D.; Car, R.; Carnimeo, I.; Cavazzoni, C.; de Gironcoli, S.; Delugas, P.; Ferrari Ruffino, F.; et al. Quantum ESPRESSO toward the exascale. J. Chem. Phys. 2020, 152, 154105. [Google Scholar] [CrossRef] [PubMed]
- Available online: https://dalcorso.github.io/thermo_pw/ (accessed on 7 July 2022).
- Wu, Z.; Cohen, R.E. More accurate generalized gradient approximation for solids. Phys. Rev. B 2006, 73, 235116. [Google Scholar] [CrossRef]
- Dal Corso, A. Pseudopotentials periodic table: From H to Pu. Comput. Mater. Sci. 2014, 95, 337. [Google Scholar] [CrossRef]
- Fletcher, R. Practical Methods of Optimization, 2nd ed.; John Wiley & Sons: New York, NY, USA, 1987; ISBN 978-0-471-91547-8. [Google Scholar] [CrossRef]
- Dudarev, S.L.; Botton, G.A.; Savrasov, S.Y.; Humphreys, C.J.; Sutton, A.P. Electron-energy-loss spectra and the structural stability of nickel oxide: An LSDA+U study. Phys. Rev. B 1998, 57, 1505. [Google Scholar] [CrossRef]
- Timrov, I.; Marzari, N.; Cococcioni, M. Hubbard parameters from density-functional perturbation theory. Phys. Rev. B 2018, 98, 085127. [Google Scholar] [CrossRef]
- Timrov, I.; Marzari, N.; Cococcioni, M. HP–A code for the calculation of Hubbard parameters using densi-ty-functional perturbation theory. Comput. Phys. Commun. 2022, 279, 108455. [Google Scholar] [CrossRef]
- Angerer, H.; Brunner, D.; Freudenberg, F.; Ambacher, O.; Stutzmann, M. Determination of the Al mole fraction and the band gap bowing of epitaxial AlxGa1−xN films. Appl. Phys. Lett. 1997, 71, 1504–1506. [Google Scholar] [CrossRef]
- Leszczynski, M.; Teisseyre, H.; Suski, T.; Grzegory, I.; Bockowski, M.; Jun, J.; Porowski, S. Lattice parameters of gallium nitride. Appl. Phys. Lett. 1996, 69, 73–75. [Google Scholar] [CrossRef]
- Papageorgiou, D.G.; Demetropoulos, I.N.; Lagaris, I.E. Merlin-3.1.1. a new version of the merlin optimization environment. Comput. Phys. Commun. 2004, 159, 70–71. [Google Scholar] [CrossRef]
- Dingreville, R.; Hallil, A.; Berbenni, S. From coherent to incoherent mismatched interfaces: A generalized continuum formulation of surface stresses. J. Mech. Phys. Solids 2014, 72, 40–60. [Google Scholar] [CrossRef]
- Dingreville, R.; Qu, J. Interfacial excess energy, excess stress and excess strain in elastic solids: Planar interfaces. J. Mech. Phys. Solids 2008, 56, 1944–1954. [Google Scholar] [CrossRef]
- Dingreville, R.; Qu, J. A semi-analytical method to estimate interface elastic properties. Comp. Mater. Sci. 2009, 46, 83–91. [Google Scholar] [CrossRef]
- McNeil, L.E.; Grimsditch, M.; Fresh, R.H. Vibrational Spectroscopy of Aluminum Nitride. J. Am. Ceram. Soc. 1993, 76, 1132–1136. [Google Scholar] [CrossRef]
- Polian, A.; Grimsditch, M.; Grzegory, I. Elastic constants of gallium nitride. J. Appl. Phys. 1996, 79, 3343. [Google Scholar] [CrossRef]
- Ekins-Daukes, N.J.; Kawaguchi, K.; Zhang, J. Strain-Balanced Criteria for Multiple Quantum Well Structures and Its Signature in X-ray Rocking Curves. Cryst. Growth Des. 2002, 2, 287. Available online: https://pubs.acs.org/doi/pdf/10.1021/cg025502y (accessed on 20 July 2023).
- Kaptay, G.; Bader, E.; Bolyan, L. Interfacial Forces and Energy Relevant to Production of Metal Matrix Composites. Mater. Sci. Forum 2000, 329–330, 151–156. [Google Scholar] [CrossRef]
- Holec, D.; Mayrhofer, P.H. Surface energies of AlN allotropes from first principles. Scr. Mater. 2012, 67, 760–762. [Google Scholar] [CrossRef]
- Dreyer, C.E.; Janotti, A.; Van de Walle, C.G. Absolute surface energies of polar and nonpolar planes of GaN. Phys. Rev. B 2014, 89, 081305. [Google Scholar] [CrossRef]
- Northrup, J.E.; Neugebauer, J. Indium-induced changes in GaN(0001) surface morphology. Phys. Rev. B 1999, 60, R8473. [Google Scholar] [CrossRef]
- Razia Chugh, M.; Ranganathan, M. Surface energy and surface stress of polar GaN(0001). Appl. Surf. Sci. 2021, 566, 150627. [Google Scholar] [CrossRef]
- Van de Walle, C.G.; McCluskey, M.; Master, C.; Romano, L.; Johnson, N. Large and composition-dependent band gap bowing in In x Ga 1− x N alloys. Mater. Sci. Eng. 1999, B59, 274. [Google Scholar] [CrossRef]
- Peressi, M.; Binggeli, N.; Baldereschi, A. Band engineering at interfaces:theory and numerical experiments. J. Phys. D Appl. Phys. 1998, 31, 1273–1299. [Google Scholar] [CrossRef]












| Compound | a (Å) | ε// | c (Å) | c/a | Eel (MPa) | |
|---|---|---|---|---|---|---|
| AlN | 3.136 | 0.008 | 4.957 | −0.004 | 1.58 | 33.5 |
| GaN | 3.136 | −0.017 | 5.23 | 0.009 | 1.67 | 132.9 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Karakostas, T.; Komninou, P.; Pontikis, V. Energetics of Interfaces and Strain Partition in GaN/AlN Pseudomorphic Superlattices. Crystals 2023, 13, 1272. https://doi.org/10.3390/cryst13081272
Karakostas T, Komninou P, Pontikis V. Energetics of Interfaces and Strain Partition in GaN/AlN Pseudomorphic Superlattices. Crystals. 2023; 13(8):1272. https://doi.org/10.3390/cryst13081272
Chicago/Turabian StyleKarakostas, Theodoros, Philomela Komninou, and Vassilis Pontikis. 2023. "Energetics of Interfaces and Strain Partition in GaN/AlN Pseudomorphic Superlattices" Crystals 13, no. 8: 1272. https://doi.org/10.3390/cryst13081272
APA StyleKarakostas, T., Komninou, P., & Pontikis, V. (2023). Energetics of Interfaces and Strain Partition in GaN/AlN Pseudomorphic Superlattices. Crystals, 13(8), 1272. https://doi.org/10.3390/cryst13081272