
Fixed-Target Pink-Beam Serial Synchrotron Crystallography at Pohang Light Source II



Abstract
:1. Introduction
2. Materials and Methods
2.1. Protein Crystal Preparation
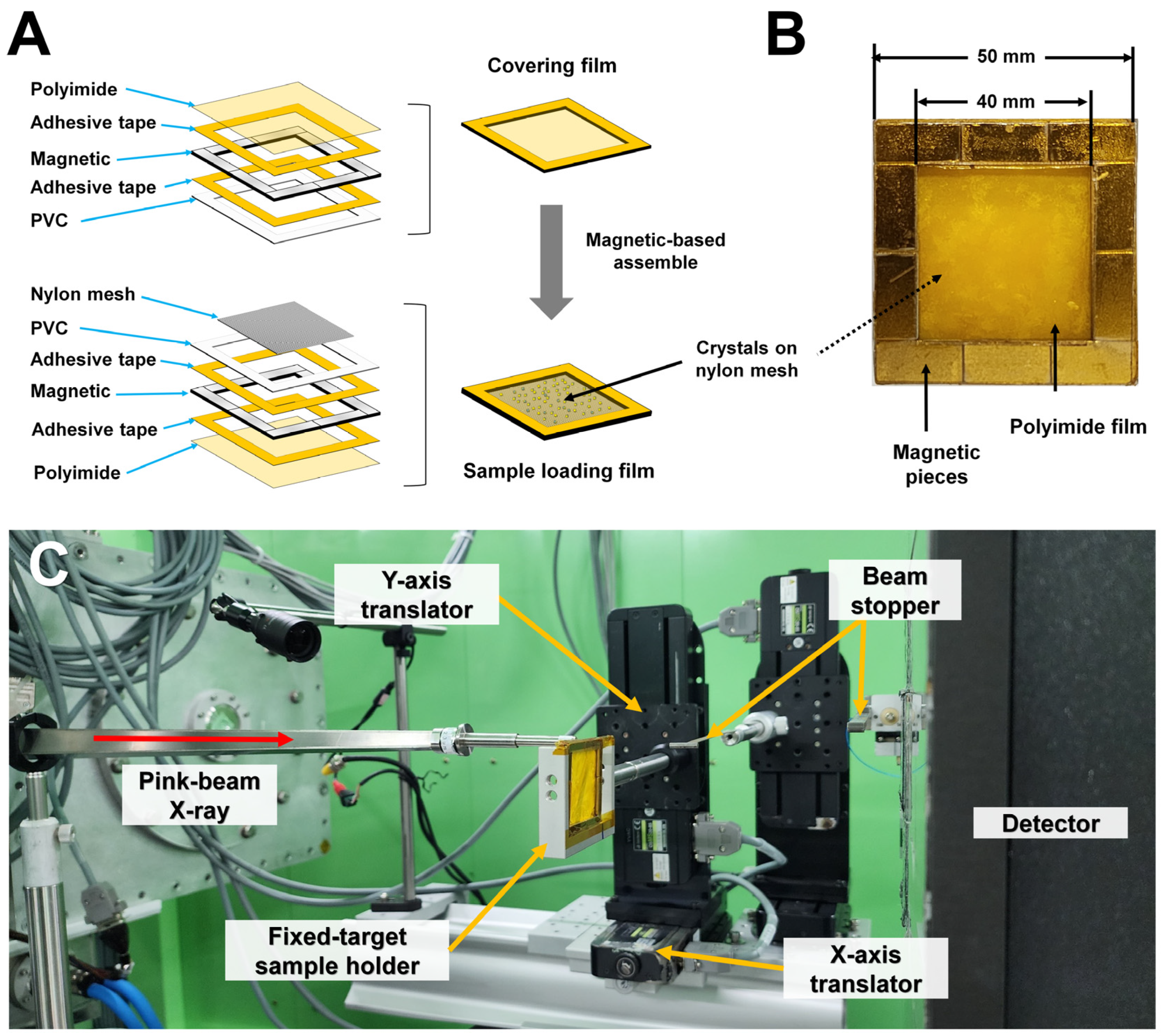
2.2. Sample Holder Preparation
2.3. Data Collection
2.4. Data Processing
2.5. Structure Determination
3. Results
3.1. Experimental Setup for FT Pink-Beam SSX
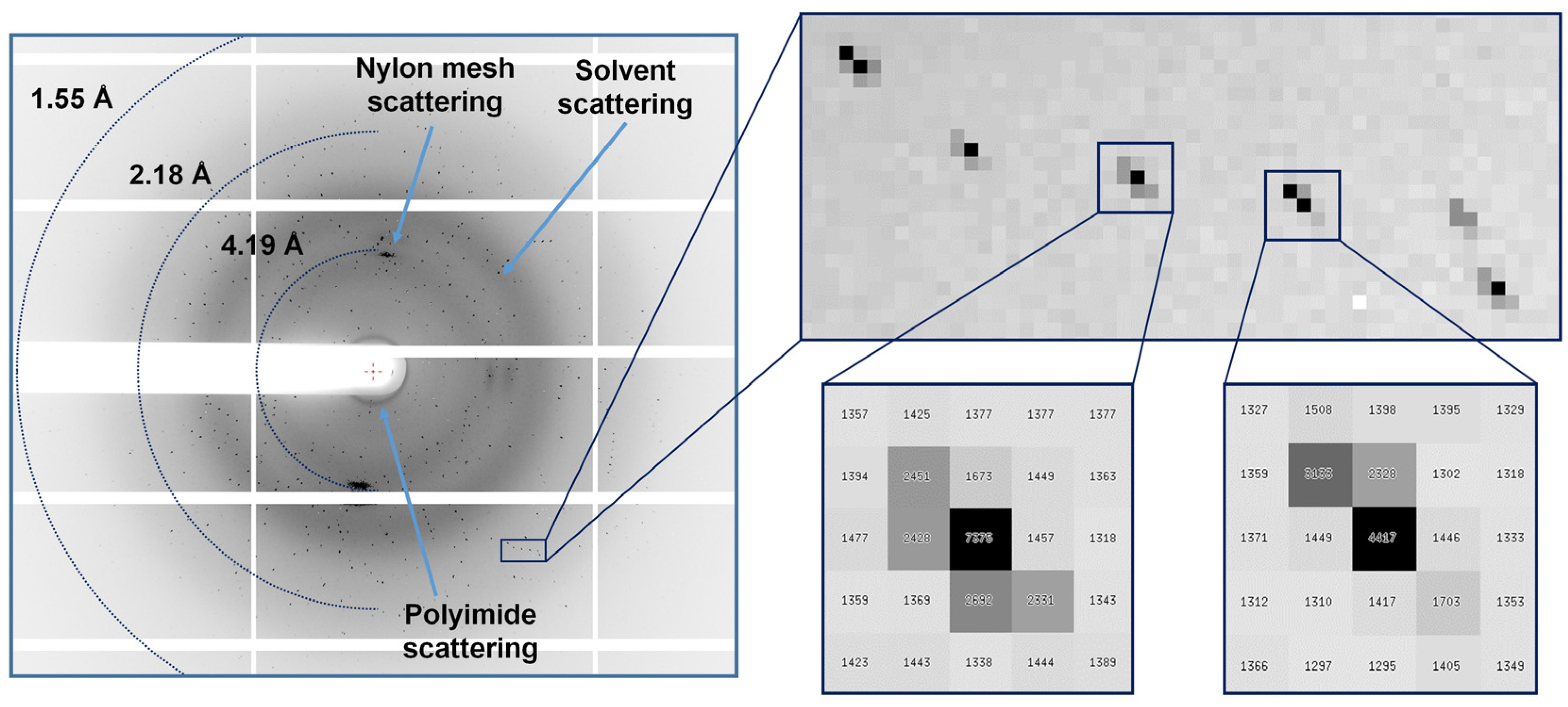
3.2. FT Pink-Beam SSX Data Collection
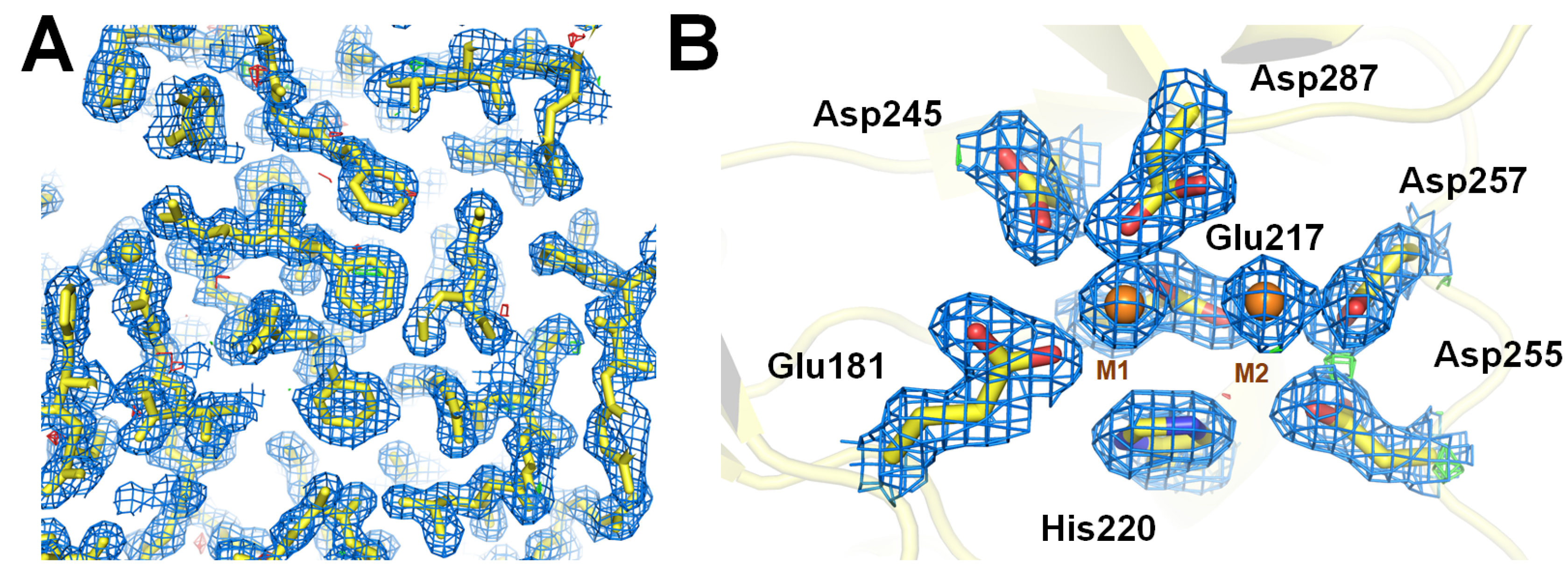
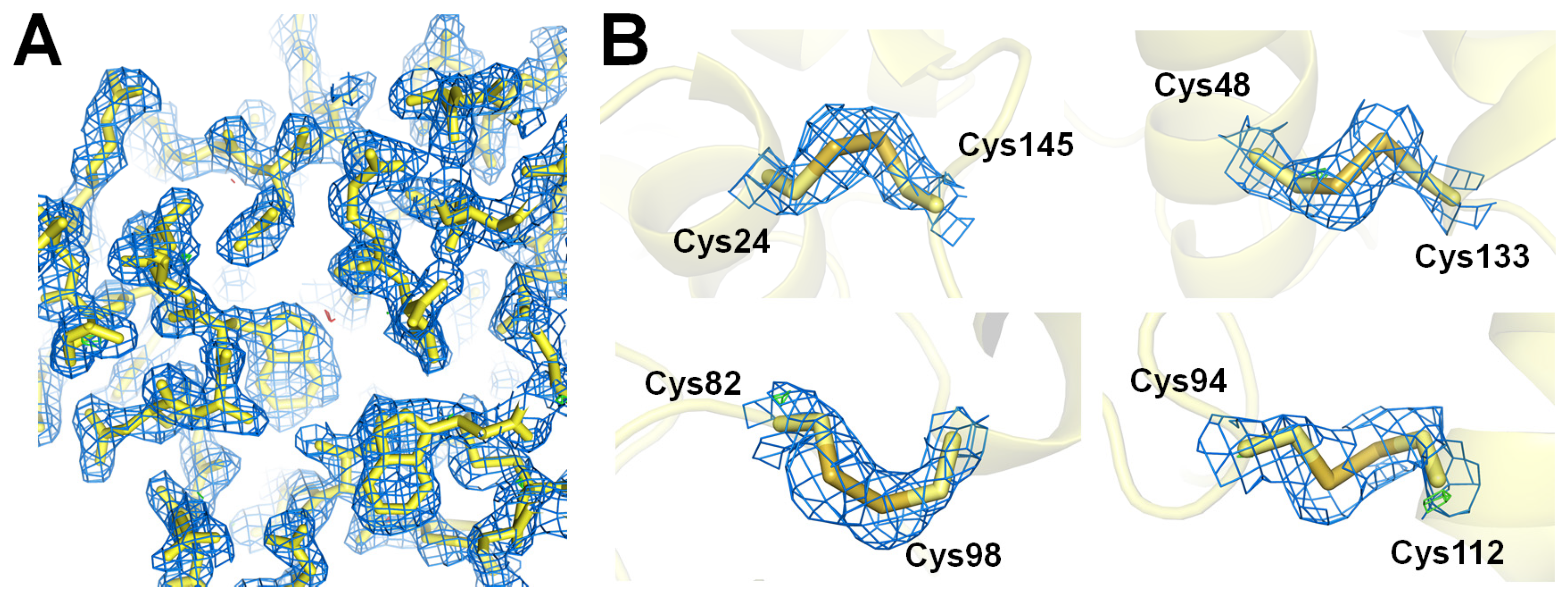
3.3. Structure Determination
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Chapman, H.N.; Fromme, P.; Barty, A.; White, T.A.; Kirian, R.A.; Aquila, A.; Hunter, M.S.; Schulz, J.; DePonte, D.P.; Weierstall, U.; et al. Femtosecond X-ray protein nanocrystallography. Nature 2011, 470, 73–77. [Google Scholar] [CrossRef] [PubMed]
- Chapman, H.N.; Caleman, C.; Timneanu, N. Diffraction before destruction. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2014, 369, 20130313. [Google Scholar] [CrossRef]
- Boutet, S.; Lomb, L.; Williams, G.J.; Barends, T.R.M.; Aquila, A.; Doak, R.B.; Weierstall, U.; DePonte, D.P.; Steinbrener, J.; Shoeman, R.L.; et al. High-Resolution Protein Structure Determination by Serial Femtosecond Crystallography. Science 2012, 337, 362–364. [Google Scholar] [CrossRef] [PubMed]
- Durdagi, S.; Dag, C.; Dogan, B.; Yigin, M.; Avsar, T.; Buyukdag, C.; Erol, I.; Ertem, F.B.; Calis, S.; Yildirim, G.; et al. Near-physiological-temperature serial crystallography reveals conformations of SARS-CoV-2 main protease active site for improved drug repurposing. Structure 2021, 29, 1382–1396. [Google Scholar] [CrossRef] [PubMed]
- Schriber, E.A.; Paley, D.W.; Bolotovsky, R.; Rosenberg, D.J.; Sierra, R.G.; Aquila, A.; Mendez, D.; Poitevin, F.; Blaschke, J.P.; Bhowmick, A.; et al. Chemical crystallography by serial femtosecond X-ray diffraction. Nature 2022, 601, 360–365. [Google Scholar] [CrossRef]
- Takaba, K.; Maki-Yonekura, S.; Inoue, I.; Tono, K.; Hamaguchi, T.; Kawakami, K.; Naitow, H.; Ishikawa, T.; Yabashi, M.; Yonekura, K. Structural resolution of a small organic molecule by serial X-ray free-electron laser and electron crystallography. Nat. Chem. 2023, 15, 491–497. [Google Scholar] [CrossRef]
- Grünbein, M.L.; Nass Kovacs, G. Sample delivery for serial crystallography at free-electron lasers and synchrotrons. Acta Crystallogr. D Biol. Crystallogr. 2019, 75, 178–191. [Google Scholar] [CrossRef]
- Martiel, I.; Muller-Werkmeister, H.M.; Cohen, A.E. Strategies for sample delivery for femtosecond crystallography. Acta Crystallogr. D Struct. Biol. 2019, 75, 160–177. [Google Scholar] [CrossRef]
- Shelley, K.L.; Garman, E.F. Quantifying and comparing radiation damage in the Protein Data Bank. Nat. Commun. 2022, 13, 1314. [Google Scholar] [CrossRef]
- Weinert, T.; Olieric, N.; Cheng, R.; Brunle, S.; James, D.; Ozerov, D.; Gashi, D.; Vera, L.; Marsh, M.; Jaeger, K.; et al. Serial millisecond crystallography for routine room-temperature structure determination at synchrotrons. Nat. Commun. 2017, 8, 542. [Google Scholar] [CrossRef]
- Pearson, A.R.; Mehrabi, P. Serial synchrotron crystallography for time-resolved structural biology. Curr. Opin. Struct. Biol. 2020, 65, 168–174. [Google Scholar] [CrossRef] [PubMed]
- White, T.A.; Barty, A.; Stellato, F.; Holton, J.M.; Kirian, R.A.; Zatsepin, N.A.; Chapman, H.N. Crystallographic data processing for free-electron laser sources. Acta Crystallogr. D Biol. Crystallogr. 2013, 69, 1231–1240. [Google Scholar] [CrossRef]
- Zhao, F.Z.; Zhang, B.; Yan, E.K.; Sun, B.; Wang, Z.J.; He, J.H.; Yin, D.C. A guide to sample delivery systems for serial crystallography. FEBS J. 2019, 286, 4402–4417. [Google Scholar] [CrossRef] [PubMed]
- Sierra, R.G.; Weierstall, U.; Oberthuer, D.; Sugahara, M.; Nango, E.; Iwata, S.; Meents, A. Sample Delivery Techniques for Serial Crystallography. In X-ray Free Electron Lasers; Springer: Cham, Switzerland, 2018; pp. 109–184. [Google Scholar] [CrossRef]
- DePonte, D.P.; Weierstall, U.; Schmidt, K.; Warner, J.; Starodub, D.; Spence, J.C.H.; Doak, R.B. Gas dynamic virtual nozzle for generation of microscopic droplet streams. J. Phys. D 2008, 41, 195505. [Google Scholar] [CrossRef]
- Nam, K.H. Sample delivery media for serial crystallography. Int. J. Mol. Sci. 2019, 20, 1094. [Google Scholar] [CrossRef]
- Weierstall, U.; James, D.; Wang, C.; White, T.A.; Wang, D.; Liu, W.; Spence, J.C.; Bruce Doak, R.; Nelson, G.; Fromme, P.; et al. Lipidic cubic phase injector facilitates membrane protein serial femtosecond crystallography. Nat. Commun. 2014, 5, 3309. [Google Scholar] [CrossRef] [PubMed]
- Sugahara, M.; Mizohata, E.; Nango, E.; Suzuki, M.; Tanaka, T.; Masudala, T.; Tanaka, R.; Shimamura, T.; Tanaka, Y.; Suno, C.; et al. Grease matrix as a versatile carrier of proteins for serial crystallography. Nat. Methods 2015, 12, 61–63. [Google Scholar] [CrossRef] [PubMed]
- Hunter, M.S.; Segelke, B.; Messerschmidt, M.; Williams, G.J.; Zatsepin, N.A.; Barty, A.; Benner, W.H.; Carlson, D.B.; Coleman, M.; Graf, A.; et al. Fixed-target protein serial microcrystallography with an X-ray free electron laser. Sci. Rep. 2014, 4, 6026. [Google Scholar] [CrossRef]
- Lee, D.; Baek, S.; Park, J.; Lee, K.; Kim, J.; Lee, S.J.; Chung, W.K.; Lee, J.L.; Cho, Y.; Nam, K.H. Nylon mesh-based sample holder for fixed-target serial femtosecond crystallography. Sci. Rep. 2019, 9, 6971. [Google Scholar] [CrossRef]
- Park, S.Y.; Choi, H.; Eo, C.; Cho, Y.; Nam, K.H. Fixed-target serial synchrotron crystallography using nylon mesh and enclosed film-based sample holder. Crystals 2020, 10, 803. [Google Scholar] [CrossRef]
- Tolstikova, A.; Levantino, M.; Yefanov, O.; Hennicke, V.; Fischer, P.; Meyer, J.; Mozzanica, A.; Redford, S.; Crosas, E.; Opara, N.L.; et al. 1 kHz fixed-target serial crystallography using a multilayer monochromator and an integrating pixel detector. IUCrJ 2019, 6, 927–937. [Google Scholar] [CrossRef] [PubMed]
- Gu, K.K.; Liu, Z.; Narayanasamy, S.R.; Shelby, M.L.; Chan, N.; Coleman, M.A.; Frank, M.; Kuhl, T.L. All polymer microfluidic chips—A fixed target sample delivery workhorse for serial crystallography. Biomicrofluidics 2023, 17, 051302. [Google Scholar] [CrossRef]
- Bjelčić, M.; Sigfridsson Clauss, K.G.V.; Aurelius, O.; Milas, M.; Nan, J.; Ursby, T. Anaerobic fixed-target serial crystallography using sandwiched silicon nitride membranes. Acta Crystallogr. D Biol. Crystallogr. 2023, 79. [Google Scholar] [CrossRef] [PubMed]
- Oghbaey, S.; Sarracini, A.; Ginn, H.M.; Pare-Labrosse, O.; Kuo, A.; Marx, A.; Epp, S.W.; Sherrell, D.A.; Eger, B.T.; Zhong, Y.; et al. Fixed target combined with spectral mapping: Approaching 100% hit rates for serial crystallography. Acta Crystallogr. D Struct. Biol. 2016, 72, 944–955. [Google Scholar] [CrossRef]
- Beyerlein, K.R.; Dierksmeyer, D.; Mariani, V.; Kuhn, M.; Sarrou, I.; Ottaviano, A.; Awel, S.; Knoska, J.; Fuglerud, S.; Jonsson, O.; et al. Mix-and-diffuse serial synchrotron crystallography. IUCrJ 2017, 4, 769–777. [Google Scholar] [CrossRef] [PubMed]
- Butryn, A.; Simon, P.S.; Aller, P.; Hinchliffe, P.; Massad, R.N.; Leen, G.; Tooke, C.L.; Bogacz, I.; Kim, I.S.; Bhowmick, A.; et al. An on-demand, drop-on-drop method for studying enzyme catalysis by serial crystallography. Nat. Commun. 2021, 12, 4461. [Google Scholar] [CrossRef]
- Lee, K.; Kim, J.; Baek, S.; Park, J.; Park, S.; Lee, J.-L.; Chung, W.K.; Cho, Y.; Nam, K.H. Combination of an inject-and-transfer system for serial femtosecond crystallography. J. Appl. Crystallogr. 2022, 55, 813–822. [Google Scholar] [CrossRef]
- Zielinski, K.A.; Prester, A.; Andaleeb, H.; Bui, S.; Yefanov, O.; Catapano, L.; Henkel, A.; Wiedorn, M.O.; Lorbeer, O.; Crosas, E.; et al. Rapid and efficient room-temperature serial synchrotron crystallography using the CFEL TapeDrive. IUCrJ 2022, 9, 778–791. [Google Scholar] [CrossRef]
- Stellato, F.; Oberthür, D.; Liang, M.; Bean, R.; Gati, C.; Yefanov, O.; Barty, A.; Burkhardt, A.; Fischer, P.; Galli, L.; et al. Room-temperature macromolecular serial crystallography using synchrotron radiation. IUCrJ 2014, 1, 204–212. [Google Scholar] [CrossRef]
- Ghosh, S.; Zorić, D.; Dahl, P.; Bjelčić, M.; Johannesson, J.; Sandelin, E.; Borjesson, P.; Björling, A.; Banacore, A.; Edlund, P.; et al. A simple goniometer-compatible flow cell for serial synchrotron X-ray crystallography. J. Appl. Crystallogr. 2023, 56, 449–460. [Google Scholar] [CrossRef]
- Meents, A.; Wiedorn, M.O.; Srajer, V.; Henning, R.; Sarrou, I.; Bergtholdt, J.; Barthelmess, M.; Reinke, P.Y.A.; Dierksmeyer, D.; Tolstikova, A.; et al. Pink-beam serial crystallography. Nat. Commun. 2017, 8, 1281. [Google Scholar] [CrossRef] [PubMed]
- Martin-Garcia, J.M.; Zhu, L.; Mendez, D.; Lee, M.-Y.; Chun, E.; Li, C.; Hu, H.; Subramanian, G.; Kissick, D.; Ogata, C.; et al. High-viscosity injector-based pink-beam serial crystallography of microcrystals at a synchrotron radiation source. IUCrJ 2019, 6, 412–425. [Google Scholar] [CrossRef]
- Dejoie, C.; McCusker, L.B.; Baerlocher, C.; Abela, R.; Patterson, B.D.; Kunz, M.; Tamura, N. Using a non-monochromatic microbeam for serial snapshot crystallography. J. Appl. Crystallogr. 2013, 46, 791–794. [Google Scholar] [CrossRef]
- Nakane, T. Pink beam crystallography demonstrated in SFX. IUCrJ 2021, 8, 853–854. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.; Nam, K.H. Pink-Beam Serial Synchrotron Crystallography at Pohang Light Source II. Crystals 2022, 12, 1637. [Google Scholar] [CrossRef]
- Barty, A.; Kirian, R.A.; Maia, F.R.; Hantke, M.; Yoon, C.H.; White, T.A.; Chapman, H. Cheetah: Software for high-throughput reduction and analysis of serial femtosecond X-ray diffraction data. J. Appl. Crystallogr. 2014, 47, 1118–1131. [Google Scholar] [CrossRef] [PubMed]
- White, T.A.; Mariani, V.; Brehm, W.; Yefanov, O.; Barty, A.; Beyerlein, K.R.; Chervinskii, F.; Galli, L.; Gati, C.; Nakane, T.; et al. Recent developments in CrystFEL. J. Appl. Crystallogr. 2016, 49, 680–689. [Google Scholar] [CrossRef]
- Gevorkov, Y.; Yefanov, O.; Barty, A.; White, T.A.; Mariani, V.; Brehm, W.; Tolstikova, A.; Grigat, R.R.; Chapman, H.N. XGANDALF—Extended gradient descent algorithm for lattice finding. Acta Crystallogr. A Found. Adv. 2019, 75, 694–704. [Google Scholar] [CrossRef]
- Battye, T.G.; Kontogiannis, L.; Johnson, O.; Powell, H.R.; Leslie, A.G. iMOSFLM: A new graphical interface for diffraction-image processing with MOSFLM. Acta Crystallogr. D Biol. Crystallogr. 2011, 67, 271–281. [Google Scholar] [CrossRef]
- Yefanov, O.; Mariani, V.; Gati, C.; White, T.A.; Chapman, H.N.; Barty, A. Accurate determination of segmented X-ray detector geometry. Opt. Express 2015, 23, 28459–28470. [Google Scholar] [CrossRef]
- Vagin, A.; Teplyakov, A. Molecular replacement with MOLREP. Acta Crystallogr. D Biol. Crystallogr. 2010, 66, 22–25. [Google Scholar] [CrossRef] [PubMed]
- Nam, K.H. Beef tallow injection matrix for serial crystallography. Sci. Rep. 2022, 12, 694. [Google Scholar] [CrossRef] [PubMed]
- Emsley, P.; Cowtan, K. Coot: Model-building tools for molecular graphics. Acta Crystallogr. D Biol. Crystallogr. 2004, 60, 2126–2132. [Google Scholar] [CrossRef] [PubMed]
- Liebschner, D.; Afonine, P.V.; Baker, M.L.; Bunkoczi, G.; Chen, V.B.; Croll, T.I.; Hintze, B.; Hung, L.W.; Jain, S.; McCoy, A.J.; et al. Macromolecular structure determination using X-rays, neutrons and electrons: Recent developments in Phenix. Acta Crystallogr. D Struct. Biol. 2019, 75, 861–877. [Google Scholar] [CrossRef]
- Williams, C.J.; Headd, J.J.; Moriarty, N.W.; Prisant, M.G.; Videau, L.L.; Deis, L.N.; Verma, V.; Keedy, D.A.; Hintze, B.J.; Chen, V.B.; et al. MolProbity: More and better reference data for improved all-atom structure validation. Protein Sci. 2018, 27, 293–315. [Google Scholar] [CrossRef]
- Martin-Garcia, J.M.; Conrad, C.E.; Nelson, G.; Stander, N.; Zatsepin, N.A.; Zook, J.; Zhu, L.; Geiger, J.; Chun, E.; Kissick, D.; et al. Serial millisecond crystallography of membrane and soluble protein microcrystals using synchrotron radiation. IUCrJ 2017, 4, 439–454. [Google Scholar] [CrossRef]
- Andrews, S.J.; Hails, J.E.; Harding, M.M.; Cruickshank, D.W.J. The mosaic spread of very small crystals deduced from Laue diffraction patterns. Acta Crystallogr. A Found. Adv. 1987, 43, 70–73. [Google Scholar] [CrossRef]
- Bhosale, S.H.; Rao, M.B.; Deshpande, V.V. Molecular and industrial aspects of glucose isomerase. Microbiol. Rev. 1996, 60, 280–300. [Google Scholar] [CrossRef]
- Gopakumar, G.; Unger, I.; Slavíček, P.; Hergenhahn, U.; Öhrwall, G.; Malerz, S.; Céolin, D.; Trinter, F.; Winter, B.; Wilkinson, I.; et al. Radiation damage by extensive local water ionization from two-step electron-transfer-mediated decay of solvated ions. Nat. Chem. 2023, 15, 1408–1414. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Data Collection | Glucose Isomerase | Lysozyme |
|---|---|---|
| X-ray source | 1C beamline, PLS-II | 1C beamline, PLS-II |
| X-ray energy (eV) | 14820 | 14820 |
| X-ray exposure (ms) | 100 | 100 |
| Total images | 20210 | 18959 |
| Hit images | 13592 | 9535 |
| Indexed crystals | 17626 | 3485 |
| Space group | I222 | P43212 |
| Cell dimension (Å) a, b, c | ||
| 94.14, 99.94, 103.16 | 78.83, 78.83, 38.20 | |
| Resolution (Å) | 20.00–1.70 (1.76–1.70) | 20.00–2.20 (2.27–2.20) |
| Unique reflections | 53508 (5307) | 6509 (628) |
| Completeness (%) | 100.0 (100.0) | 100.0 (100.0) |
| Redundancy | 191.5 (256.9) | 488.7 (398.5) |
| SNR | 4.67 (4.39) | 5.43 (3.74) |
| CC | 0.8239 (0.3692) | 0.8737 (0.5245) |
| CC* | 0.9505 (0.7343) | 0.9657 (0.8295) |
| Rsplit (%) | 25.98 (27.16) | 21.21 (33.46) |
| Wilson B factor (Å2) | 11.97 | 23.92 |
| Refinement | Glucose Isomerase | Lysozyme |
|---|---|---|
| Resolution (Å) | 7.0–1.7 | 7.0–2.2 |
| Rwork a | 0.2510 | 0.2512 |
| Rfree b | 0.2779 | 0.2993 |
| R.m.s. deviations | ||
| Bonds (Å) | 0.004 | 0.008 |
| Angles (°) | 0.861 | 0.969 |
| B factors (Å2) | ||
| Protein | 13.42 | 12.11 |
| Water | 7.58 | 16.24 |
| Ramachandran plot (%) | ||
| Favored | 93.46 | 95.28 |
| Allowed | 5.76 | 4.72 |
| Disallowed | 0.79 | 0.00 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kim, Y.; Nam, K.H. Fixed-Target Pink-Beam Serial Synchrotron Crystallography at Pohang Light Source II. Crystals 2023, 13, 1544. https://doi.org/10.3390/cryst13111544
Kim Y, Nam KH. Fixed-Target Pink-Beam Serial Synchrotron Crystallography at Pohang Light Source II. Crystals. 2023; 13(11):1544. https://doi.org/10.3390/cryst13111544
Chicago/Turabian StyleKim, Yongsam, and Ki Hyun Nam. 2023. "Fixed-Target Pink-Beam Serial Synchrotron Crystallography at Pohang Light Source II" Crystals 13, no. 11: 1544. https://doi.org/10.3390/cryst13111544
APA StyleKim, Y., & Nam, K. H. (2023). Fixed-Target Pink-Beam Serial Synchrotron Crystallography at Pohang Light Source II. Crystals, 13(11), 1544. https://doi.org/10.3390/cryst13111544