
Unexpected Formation of a Silicon-Centered Spirocyclic Oligosiloxane Bearing Eight Pendant Ferrocene Units



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
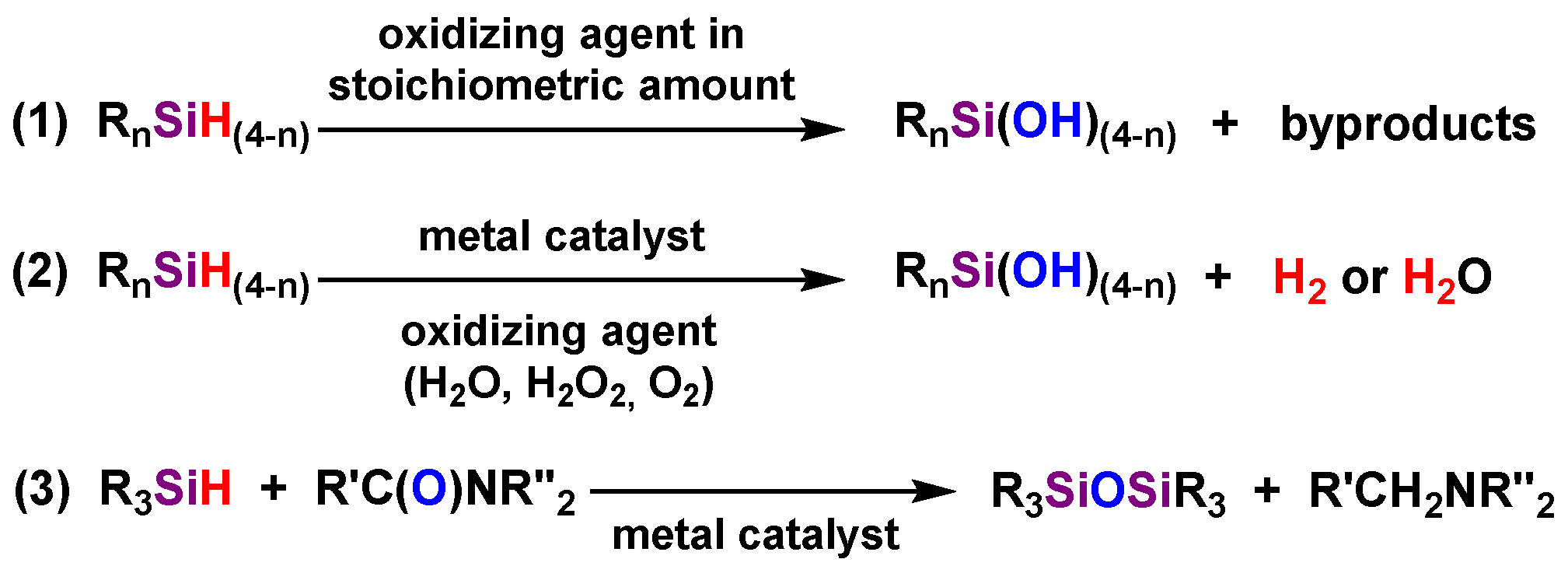
:1. Introduction
2. Materials and Methods
3. Results and Discussion
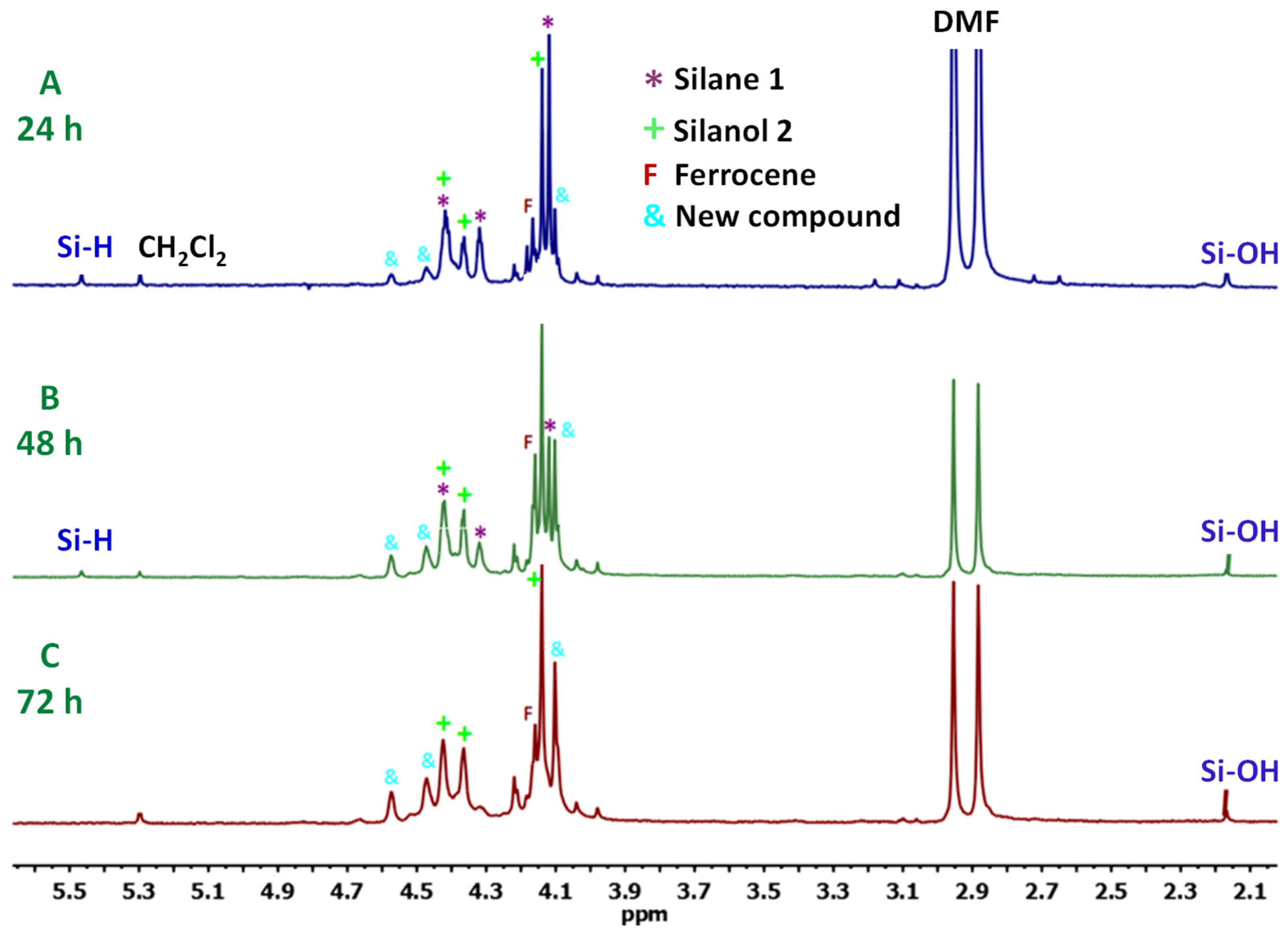
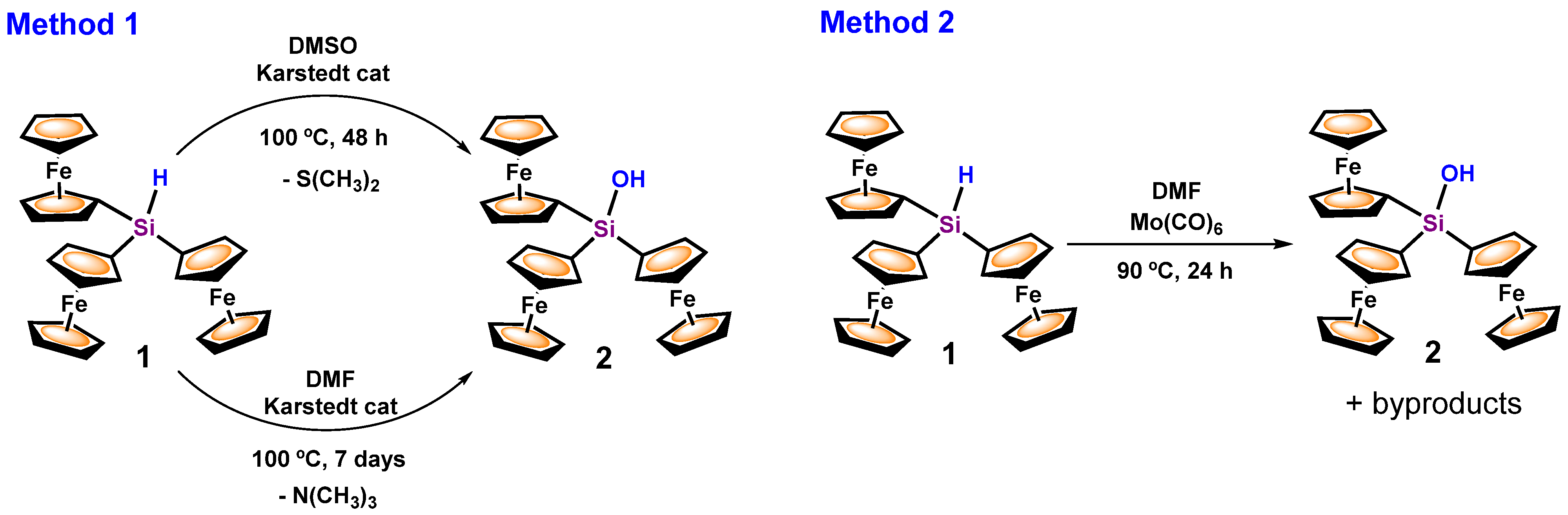
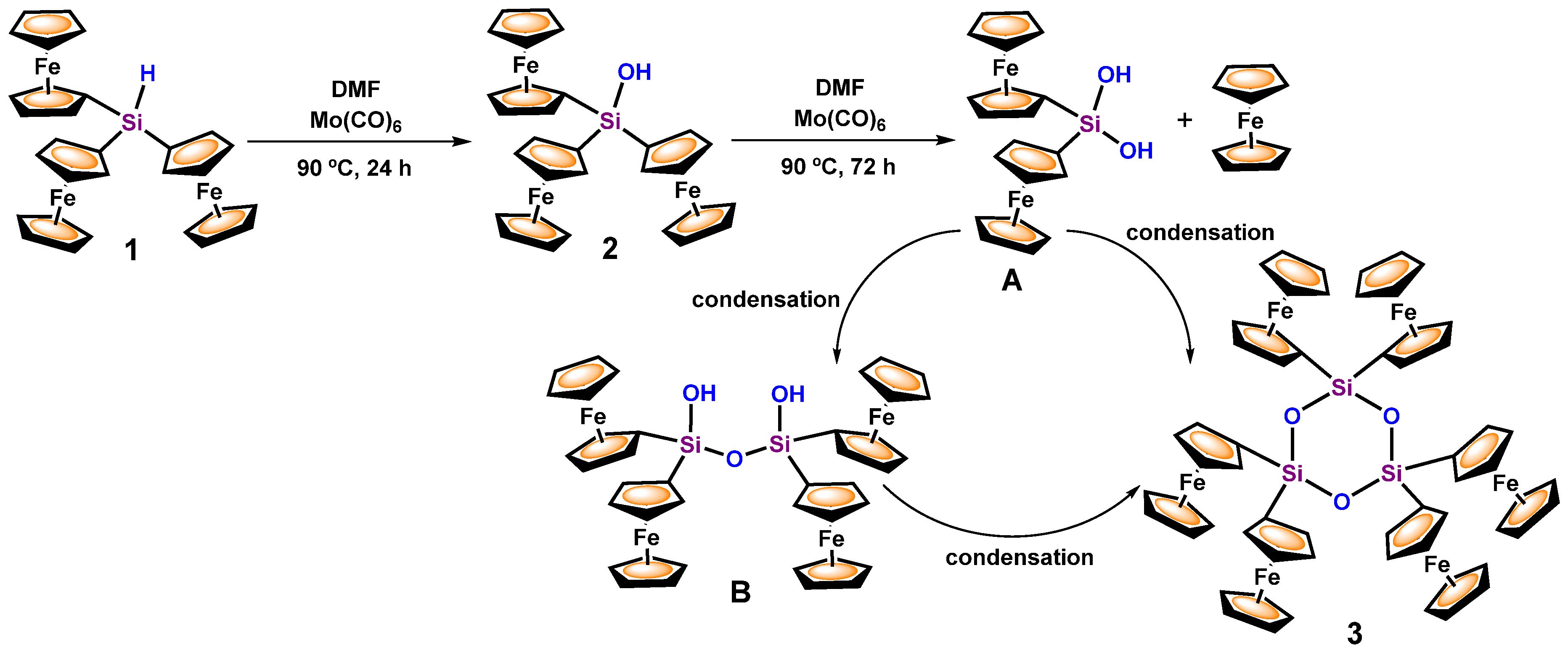
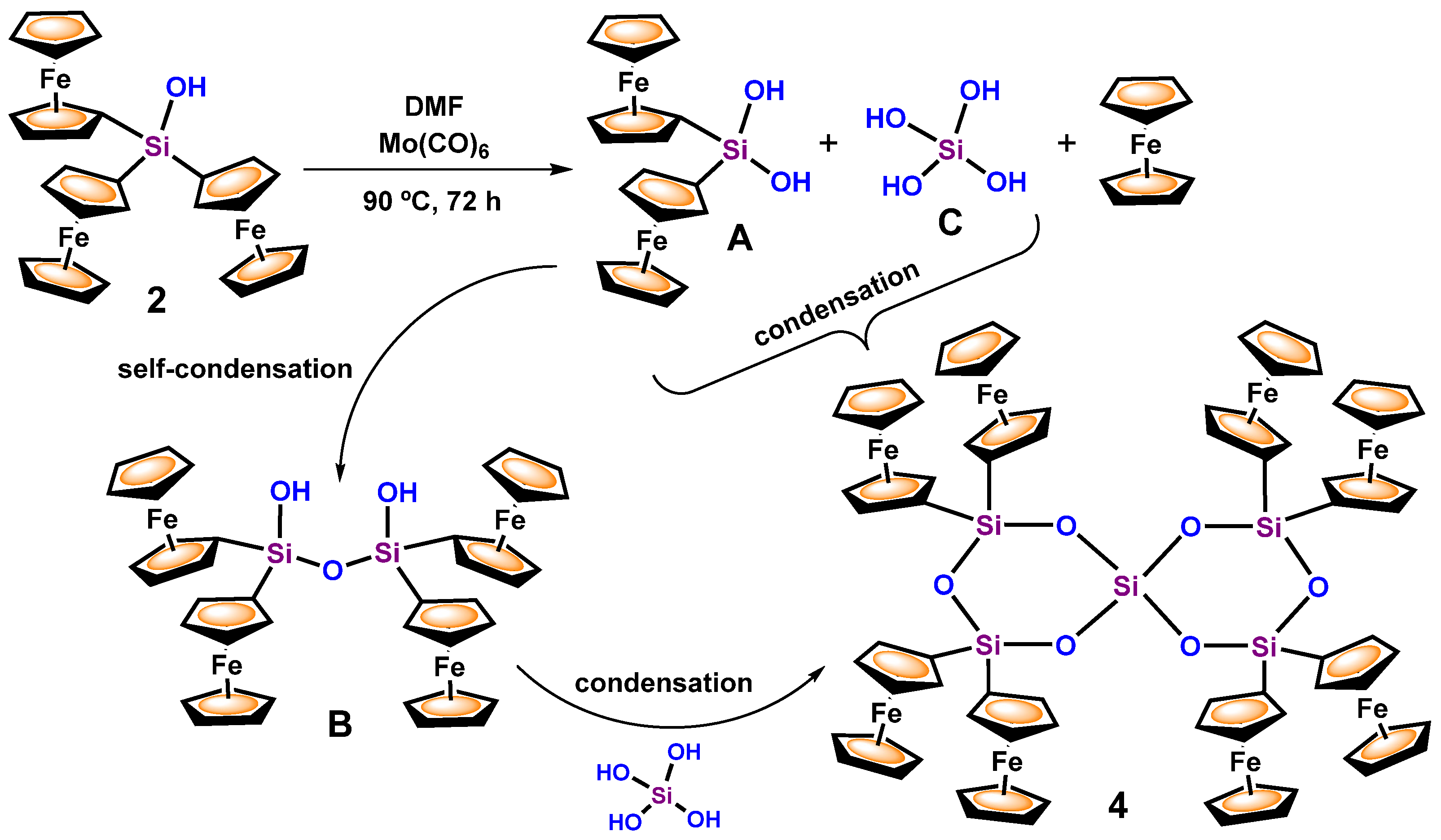
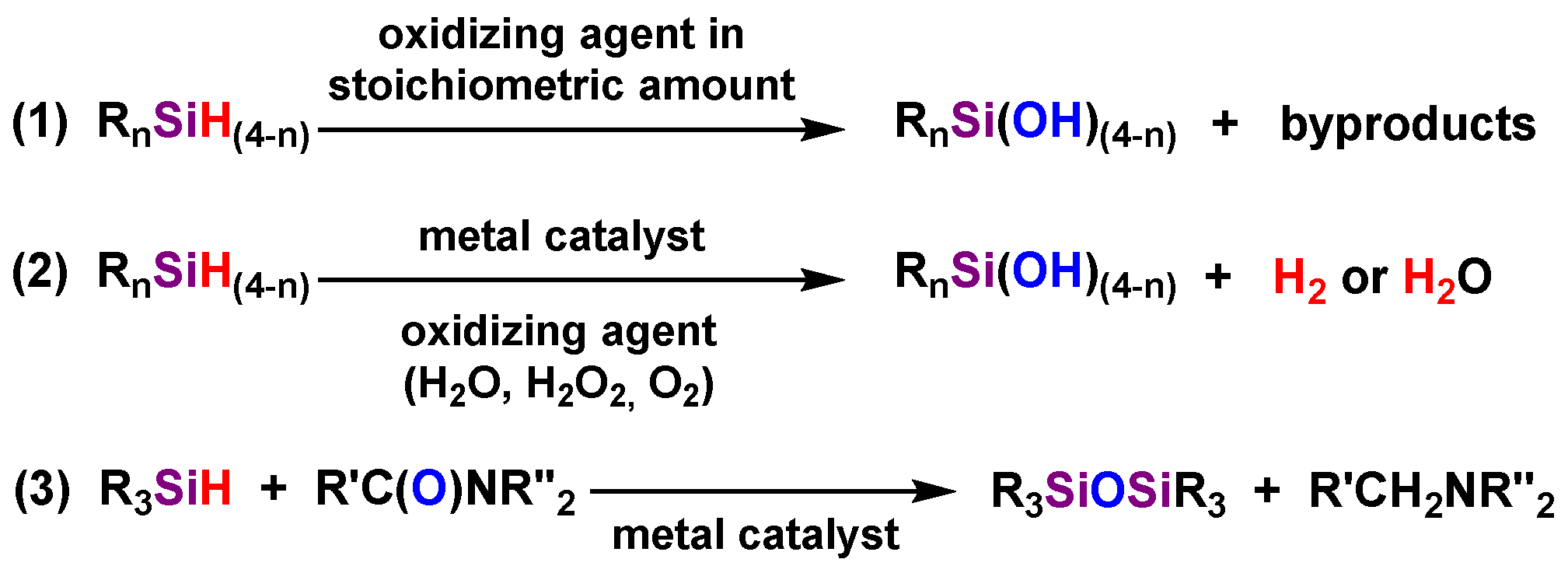
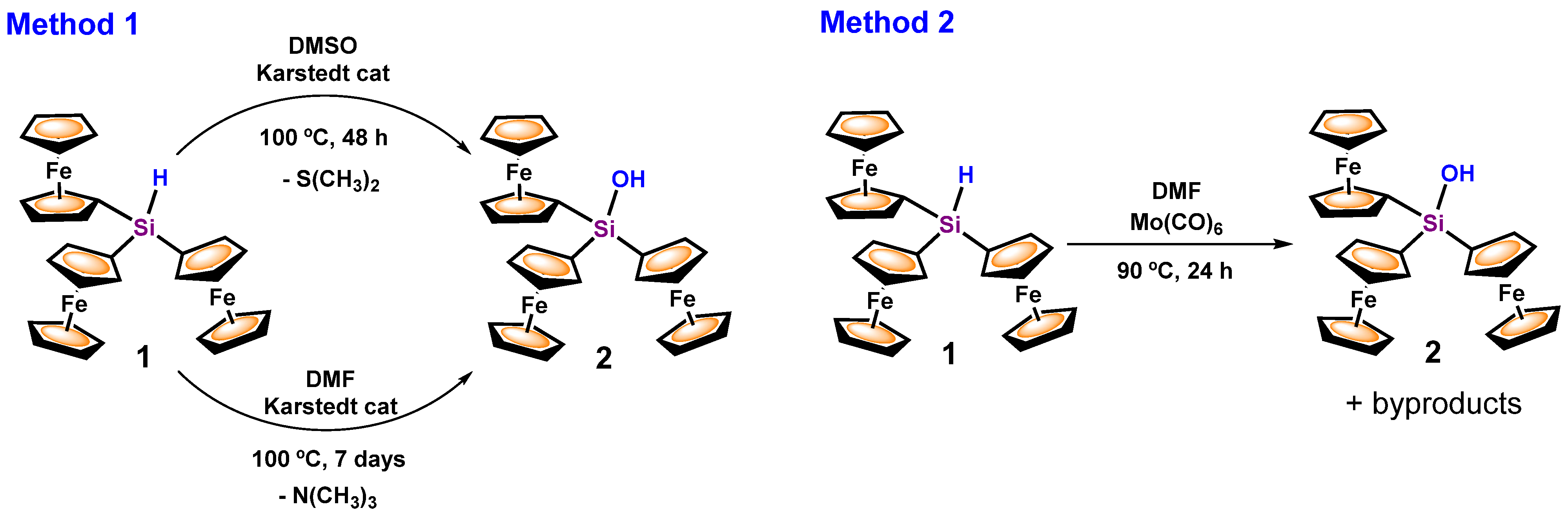
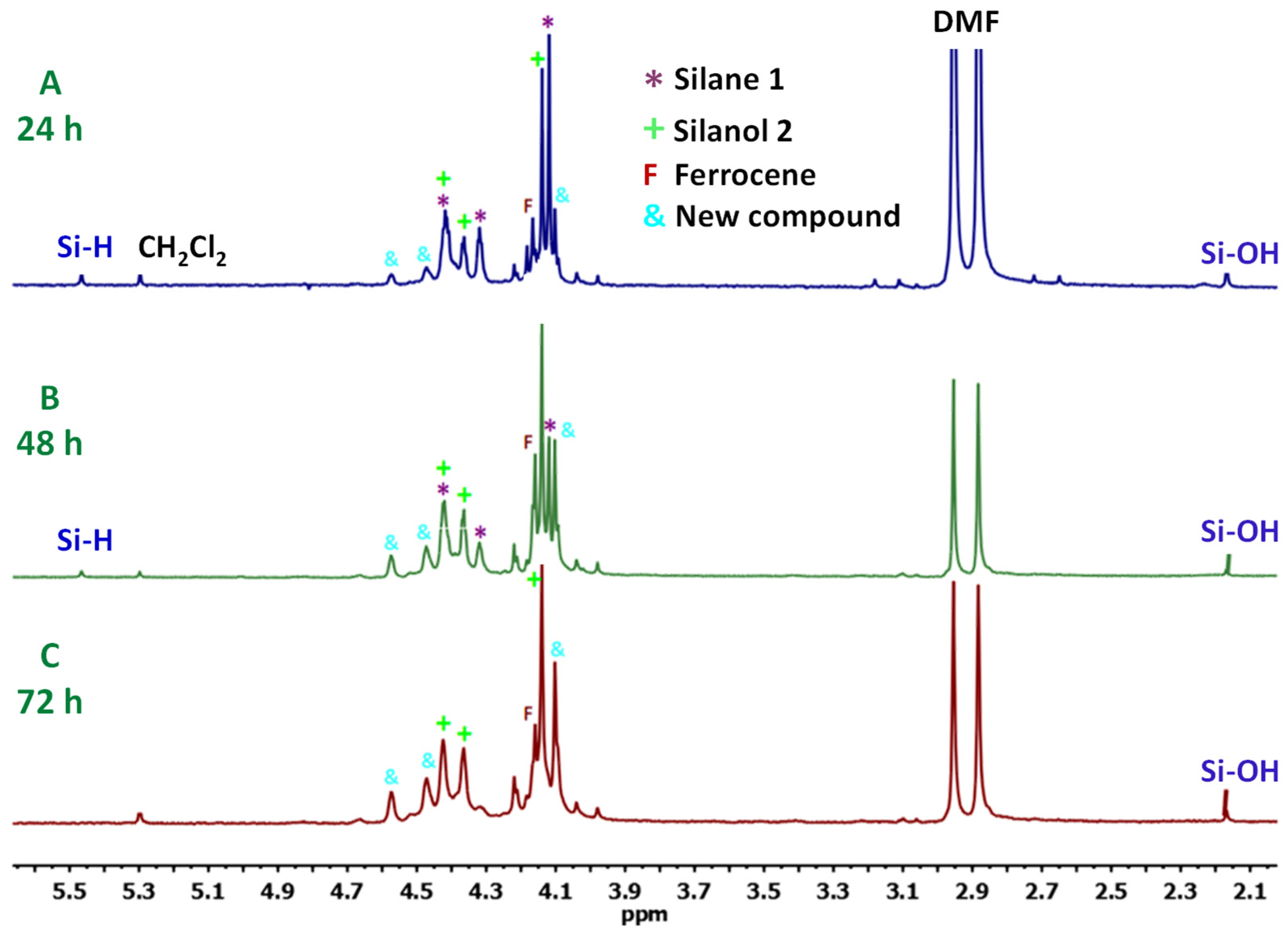
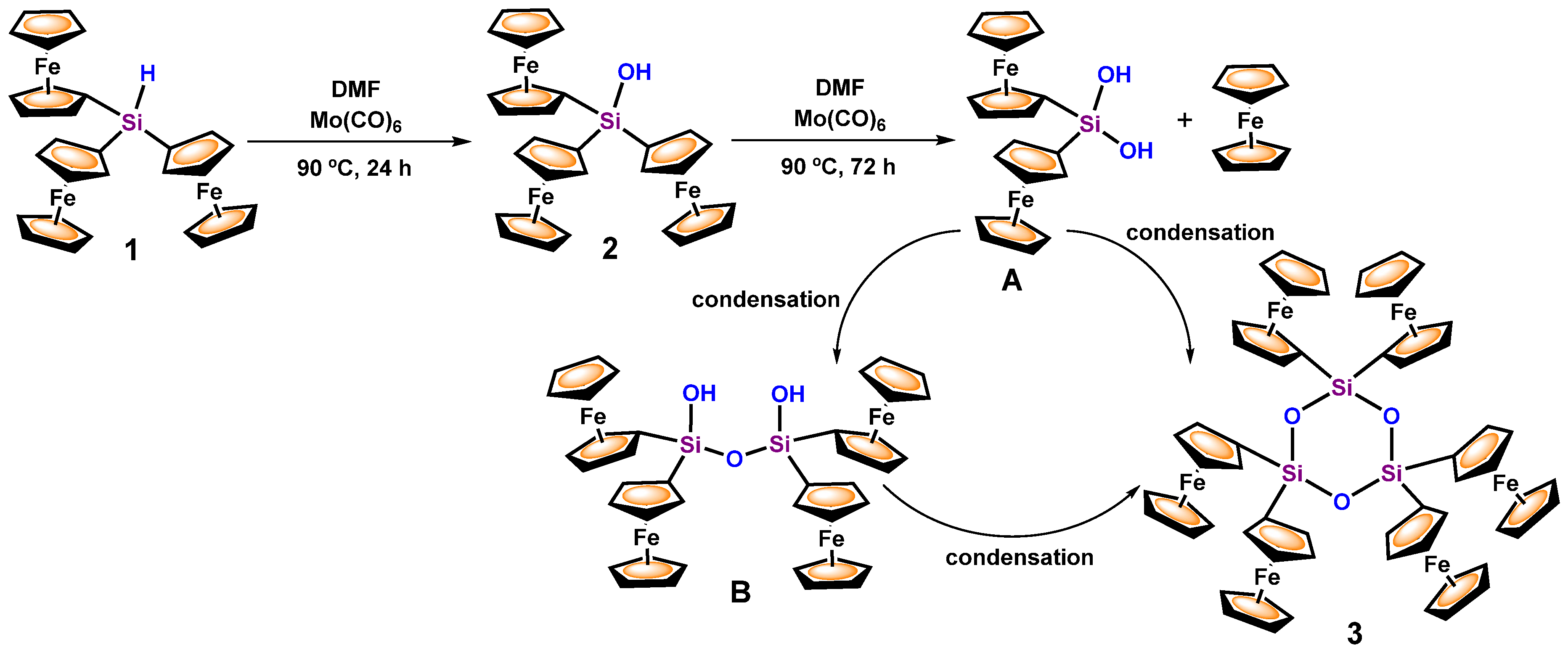
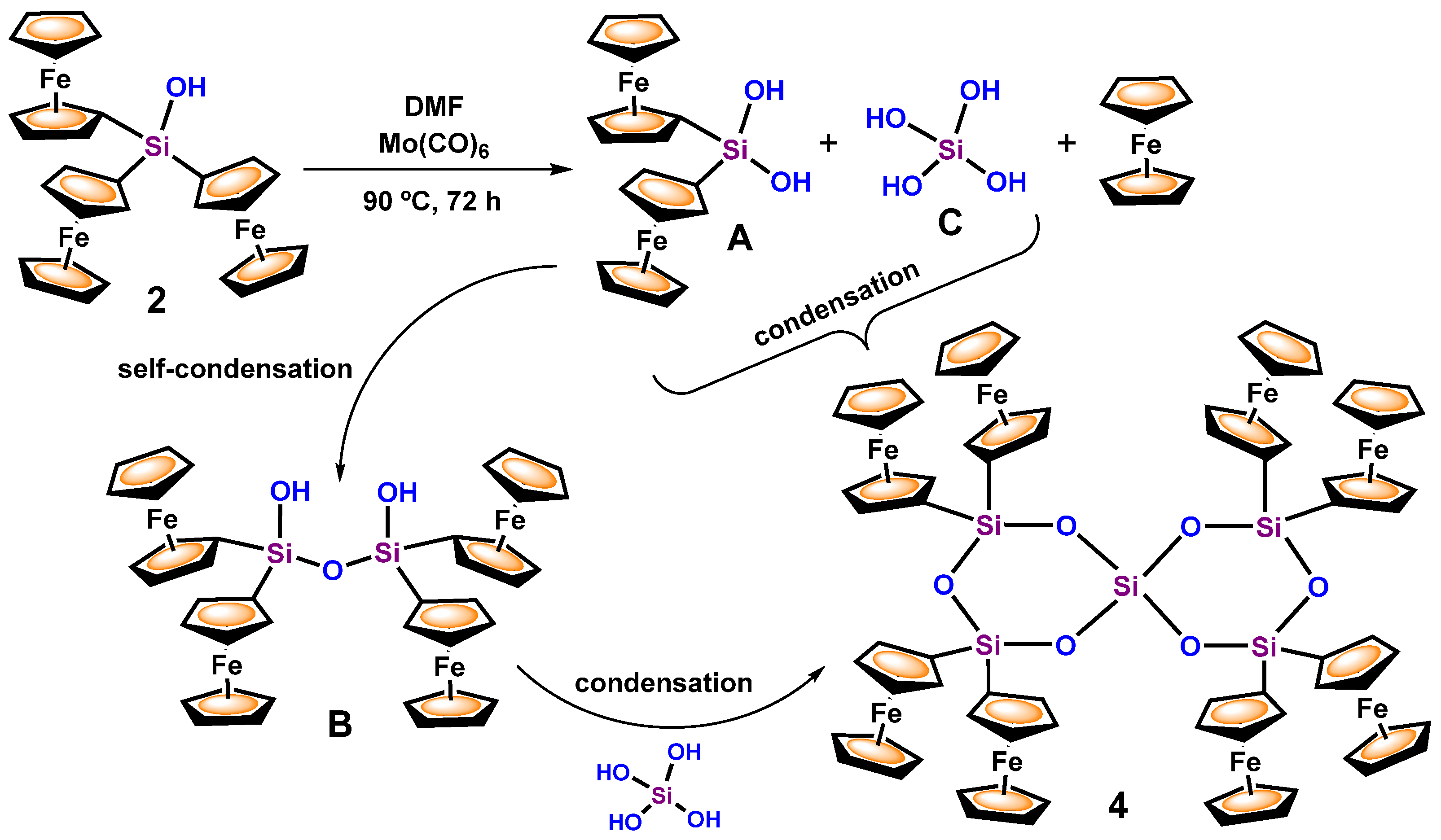
3.1. Transformation of Triferrocenylsilane 1 into Triferrocenylsilanol 2
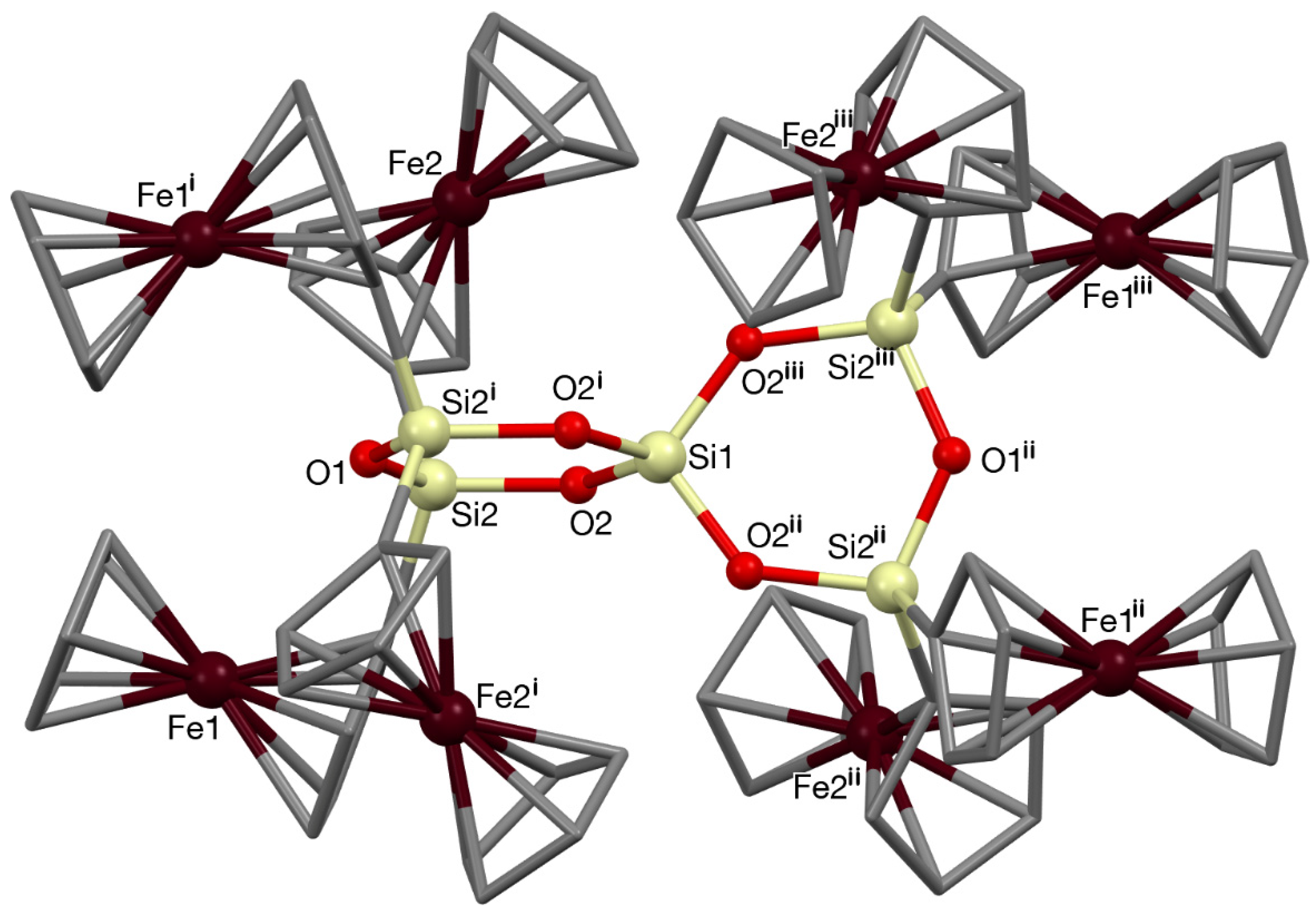
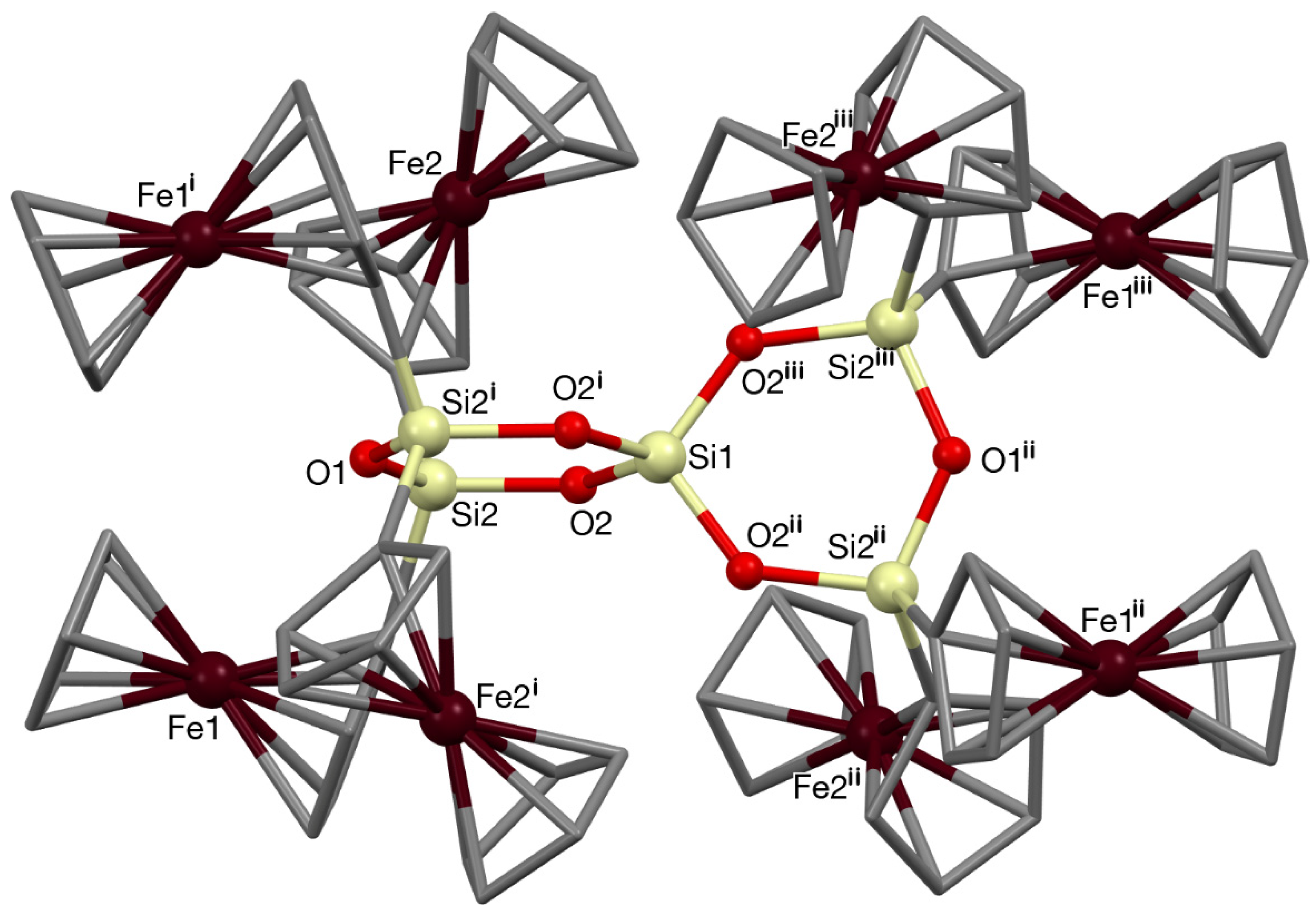
3.2. Single Crystal X-ray Diffraction
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Dvornic, P.R. Thermal Properties of Polysiloxanes. In Silicon-Containing Polymers; Jones, R.G., Ando, W., Chojnowski, J., Eds.; Springer: Dordrecht, The Netherlands, 2000; pp. 185–212. [Google Scholar] [CrossRef]
- Brook, M.A. Silicon in Organic, Organometallic and Polymer Chemistry; Wiley: New York, NY, USA, 2000; ISBN 978-0-471-19658-7. [Google Scholar]
- Noll, W. Chemistry and Technology of Silicones; Academic Press Inc.: New York, NY, USA, 1968. [Google Scholar]
- Kung, M.C.; Riofski, M.V.; Missaghi, M.N.; Kung, H.H. Organosilicon Platforms: Bridging Homogeneous, Heterogeneous, and Bioinspired Catalysis. Chem. Commun. 2014, 50, 3262–3276. [Google Scholar] [CrossRef] [PubMed]
- Dankert, F.; von Hänisch, C. Siloxane Coordination Revisited: Si−O Bond Character, Reactivity and Magnificent Molecular Shapes. Eur. J. Inorg. Chem. 2021, 2021, 2907–2927. [Google Scholar] [CrossRef]
- Churchill, M.R.; Lake, C.H.; Chao, S.-H.L.; Beachley, O.T. Silicone Grease as a Precursor to a Pseudo Crown Ether Ligand: Crystal Structure of [K+]3[K(Me2SiO)7+][InH(CH2CMe3)3−]4. J. Chem. Soc. Chem. Commun. 1993, 20, 1577–1578. [Google Scholar] [CrossRef]
- Eaborn, C.; Hitchcock, P.B.; Izod, K.; Smith, J.D. Two Diorganopotassates: Crystal Structure of [K(C6H6)][K{C(SiMe3)2(SiMe2Ph)}2]. Angew. Chem. Int. Ed. Engl. 1996, 34, 2679–2680. [Google Scholar] [CrossRef]
- Haiduc, I. Silicone Grease: A Serendipitous Reagent for the Synthesis of Exotic Molecular and Supramolecular Compounds. Organometallics 2004, 23, 3–8. [Google Scholar] [CrossRef]
- Padělková, Z.; Švec, P.; Kampová, H.; Sýkora, J.; Semler, M.; Štěpnička, P.; Bakardjieva, S.; Willem, R.; Růžička, A. Unusual Reactivity of a C,N-Chelated Stannylene with Siloxanes and Silanols. Organometallics 2013, 32, 2398–2405. [Google Scholar] [CrossRef]
- Pop, L.C.; Saito, M. Serendipitous Reactions Involving a Silicone Grease. Coord. Chem. Rev. 2016, 314, 64–70. [Google Scholar] [CrossRef]
- Dankert, F.; Erlemeier, L.; Ritter, C.; von Hänisch, C. On the Molecular Architectures of Siloxane Coordination Compounds: (re-)Investigating the Coordination of the Cyclodimethylsiloxanes Dn (n = 5–8) towards Alkali Metal Ions. Inorg. Chem. Front. 2020, 7, 2138–2153. [Google Scholar] [CrossRef]
- Dankert, F.; Richter, R.-M.; Weigend, F.; Xie, X.; Balmer, M.; von Hänisch, C. Construction of Inorganic Crown Ethers by s-Block-Metal-Templated Si–O Bond Activation. Angew. Chem. Int. Ed. 2021, 60, 10393–10401. [Google Scholar] [CrossRef]
- Böhme, U.; Haushälter, J. Surprising Formation of a Silicon Containing Macrocycle. Inorg. Chem. Commun. 2009, 12, 35–37. [Google Scholar] [CrossRef]
- Sheppard, S.A.; Bennett, T.L.R.; Long, N.J. Development and Characterisation of Highly Conjugated Functionalised Ferrocenylene Macrocycles. Eur. J. Inorg. Chem. 2022, 2022, e202200055. [Google Scholar] [CrossRef]
- Scottwella, S.Ø.; Crowley, J.D. Ferrocene-Containing non-Interlocked Molecular Machines. Chem. Commun. 2016, 52, 2451–2464. [Google Scholar] [CrossRef]
- Inkpen, M.; Scheerer, S.; Linseis, M.; White, A.J.P.; Winter, R.F.; Albrecht, T.; Long, N.J. Oligomeric Ferrocene Rings. Nature Chem. 2016, 8, 825–830. [Google Scholar] [CrossRef]
- Xu, L.; Wang, Y.-X.; Chen, L.-J.; Yang, H.-B. Construction of Multiferrocenyl Metallacycles and Metallacages via Coordination-Driven Self-Assembly. Chem. Soc. Rev. 2015, 44, 2148–2167. [Google Scholar] [CrossRef]
- Hildebrandt, A.; Lang, H. (Multi)ferrocenyl Five-Membered Heterocycles: Excellent Connecting Units for Electron Transfer Studies. Organometallics 2013, 32, 5640–5653. [Google Scholar] [CrossRef]
- Gu, H.; Ciganda, R.; Gatard, S.; Lu, F.; Zhao, P.; Ruiz, J.; Astruc, D. On Metallocene-Containing Macromolecules and their Applications. J. Organomet. Chem. 2016, 813, 95–102. [Google Scholar] [CrossRef]
- Rossi, S.; Bisello, A.; Cardena, R.; Santi, S. Testing the Conjugative Properties of Benzodithiophene and Benzotrithiophene in Charge Transfer Multi(ferrocenyl) Systems. Organometallics 2018, 37, 4242–4249. [Google Scholar] [CrossRef]
- Bruña, S.; González-Vadillo, A.M.; Nieto, D.; Pastor, C.; Cuadrado, I. Vinyl-Functionalized Silanes and Disiloxanes with Electronically Communicated Ferrocenes. Organometallics 2010, 29, 2796–2807. [Google Scholar] [CrossRef]
- Bruña, S.; Nieto, D.; González-Vadillo, A.M.; Perles, J.; Cuadrado, I. Cubic Octasilsegsquioxanes, Cyclotetrasiloxanes, and Disiloxanes Maximally Functionalized with Silicon-Bridged Interacting Triferrocenyl Units. Organometallics 2012, 31, 3248–3258. [Google Scholar] [CrossRef]
- Bruña, S.; González-Vadillo, A.M.; Nieto, D.; Pastor, C.J.; Cuadrado, I. Redox-Active Macrocyclic and Linear Oligo-Carbosiloxanes Prepared via Hydrosilylation from 1,3-Divinyl-1,3-Dimethyl-1,3-Diferrocenyldisiloxane. Macromolecules 2012, 45, 781–793. [Google Scholar] [CrossRef]
- Bruña, S.; Perles, J.; Nieto, D.; González-Vadillo, A.M.; Cuadrado, I. Triferrocenylsilane and Unexpected Formation of Diferrocenyl(3,3-dimethylbutyl)silane via Cleavage and Trapping of a Tetrahydrofuran Fragment. Redox Chemistry and Synthesis of (Fc)3Si–Co(CO)4 through Oxidative Addition of the Si–H Bond. J. Organomet. Chem. 2014, 751, 769–780. [Google Scholar] [CrossRef]
- Bruña, S.; González-Vadillo, A.M.; Ferrández, M.; Perles, J.; Montero-Campillo, M.M.; Mó, O.; Cuadrado, I. Formation of Unexpected Silicon- and Disiloxane-bridged Multiferrocenyl Derivatives bearing Si–O–CH=CH2 and Si–(CH2)2C(CH3)3 Substituents via Cleavage of Tetrahydrofuran and Trapping of its Ring Fragments. Dalton Trans. 2017, 46, 11584–11597. [Google Scholar] [CrossRef]
- Bruña, S.; Garrido-Castro, A.F.; Perles, J.; Montero-Campillo, M.M.; Mó, O.; Kaifer, A.E.; Cuadrado, I. Multi-Ferrocene-Containing Silanols as Redox-Active Anion Receptors. Organometallics 2016, 35, 3507–3519. [Google Scholar] [CrossRef]
- Miles, D.; Ward, J.; Foucher, D.A. Polyferrocenyldisiloxane from the Platinum-Catalyzed Reactions of Tertiary Bis(dimethylsilyl)ferrocene in a Polar Aprotic Solvent. Macromolecules 2009, 42, 9199–9203. [Google Scholar] [CrossRef]
- Arias-Ugarte, R.; Sharma, H.K.; Morris, A.L.C.; Pannell, K.H. Metal-Catalyzed Reduction of HCONR′2, R′ = Me (DMF), Et (DEF), by Silanes to Produce R′2NMe and Disiloxanes: A Mechanism Unraveled. J. Am. Chem. Soc. 2012, 134, 848–851. [Google Scholar] [CrossRef] [PubMed]
- Sharma, H.K.; Pannell, K.H. The Photochemical Irradiation of R3SiH in the Presence of [(η5-C5H5)Fe(CO)2CH3] in DMF Leads to Disiloxanes not Disilanes. Angew. Chem. Int. Ed. 2009, 48, 7052–7054. [Google Scholar] [CrossRef] [PubMed]
- Sheldrick, G.M. SADABS, Version 2.03; Bruker AXS Inc.: Madison, WI, USA, 2001. [Google Scholar]
- Sheldrick, G.M. SAINT + NT, Version 6.04, SAX Area-Detector Integration Program; Bruker Analytical X-ray Instruments: Madison, WI, USA, 1997. [Google Scholar]
- Bruker. AXS SHELXTL, Version 6.10, Structure Determination Package; Bruker Analytical X-ray Instruments: Madison, WI, USA, 2000. [Google Scholar]
- Sheldrick, G.M. Phase Annealing in SHELX-90: Direct Methods for Larger Structures. Acta Crystallogr. A 1990, 46, 467–473. [Google Scholar] [CrossRef]
- Sheldrick, G.M. SHELXL97, Program for Crystal Structure Refinement; University of Göttingen: Göttingen, Germany, 1997. [Google Scholar]
- Sollott, G.P.; Peterson, W.R. Germylation of Ferrocene under Friedel-Crafts Conditions. Question of the Existence of Germonium Ions. J. Am. Chem. Soc. 1967, 89, 6783–6784. [Google Scholar] [CrossRef]
- MacLachlan, M.J.; Zheng, J.; Thieme, K.; Lough, A.J.; Manners, I.; Mordas, C.; LeSuer, R.; Geiger, W.E.; Liable-Sands, L.-M.; Rheingold, A.L. Synthesis, Characterization, and Ring-opening Polymerization of a Novel [1]Silaferrocenophane with two Ferrocenyl Substituents at Silicon. Polyhedron 2000, 19, 275–289. [Google Scholar] [CrossRef]
- Kamitani, M.; Fukumoto, K.; Tada, R.; Itazaki, M.; Nakazawa, H. Catalytic Synthesis of Cyclic and Linear Germoxanes Mediated by an Iron Complex. Organometallics 2012, 31, 2957–2960. [Google Scholar] [CrossRef]
- Corey, J.Y. Reactions of Hydrosilanes with Transition Metal Complexes and Characterization of the Products. Chem. Rev. 2011, 111, 863–1071. [Google Scholar] [CrossRef]
- MacLachlan, M.J.; Zheng, J.; Lough, A.J.; Manners, I.; Mordas, C.; LeSuer, R.; Geiger, W.E.; Liable-Sands, L.M.; Rheingold, A.L. Ferrocenylsiloxane Chemistry: Synthesis and Characterization of Hexaferrocenylcyclotrisiloxane and Tetraferrocenyldisiloxanediol. Organometallics 1999, 18, 1337–1345. [Google Scholar] [CrossRef]
- Jitchum, V.; Chivin, S.; Wongkasemjit, S.; Ishida, H. Synthesis of Spirosilicates Directly from Silica and Ethylene Glycol Derivatives. Tetrahedron 2001, 57, 3997–4003. [Google Scholar] [CrossRef]
- Laine, R.M.; Blohowiak, K.Y.; Robinson, T.R.; Hoppe, M.L.; Nardi, P.; Kampf, J.; Uhm, J. Synthesis of Pentacoordinate Silicon Complexes from SiO2. Nature 1991, 353, 642–644. [Google Scholar] [CrossRef]
- Roth, W.L.; Harker, D. The Crystal Structure of Octamethylspiro [5.5] pentasiloxane: Rotation about the Ionic Silicon–Oxygen Bond. Acta Cryst. 1948, 1, 34–42. [Google Scholar] [CrossRef]
- Sarkar, D.; Nesterov, V.; Szilvasi, T.; Altmann, P.J.; Inoue, S. The Quest for Stable Silaaldehydes: Synthesis and Reactivity of a Masked Silacarbonyl. Chem. Eur. J. 2019, 25, 1198–1202. [Google Scholar] [CrossRef]
- Shklover, V.E.; Struchkov, Y.T.; Zachernyuk, A.B.; Andrianov, K.A. The Crystal Structure of Cyclic Siloxanes and Silazanes. VIII. Trans, trans-d,l-2,4,8,10-tetramethyl-2,4,8,10-tetraphenylspiro [5.5] pentasiloxane. J. Struct. Chem. 1978, 19, 98–104. [Google Scholar] [CrossRef]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bruña, S.; Cuadrado, I.; Perles, J. Unexpected Formation of a Silicon-Centered Spirocyclic Oligosiloxane Bearing Eight Pendant Ferrocene Units. Crystals 2022, 12, 1122. https://doi.org/10.3390/cryst12081122
Bruña S, Cuadrado I, Perles J. Unexpected Formation of a Silicon-Centered Spirocyclic Oligosiloxane Bearing Eight Pendant Ferrocene Units. Crystals. 2022; 12(8):1122. https://doi.org/10.3390/cryst12081122
Chicago/Turabian StyleBruña, Sonia, Isabel Cuadrado, and Josefina Perles. 2022. "Unexpected Formation of a Silicon-Centered Spirocyclic Oligosiloxane Bearing Eight Pendant Ferrocene Units" Crystals 12, no. 8: 1122. https://doi.org/10.3390/cryst12081122
APA StyleBruña, S., Cuadrado, I., & Perles, J. (2022). Unexpected Formation of a Silicon-Centered Spirocyclic Oligosiloxane Bearing Eight Pendant Ferrocene Units. Crystals, 12(8), 1122. https://doi.org/10.3390/cryst12081122