
Intermolecular Interactions Drive the Unusual Co-Crystallization of Different Calix[4]arene Conformations




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
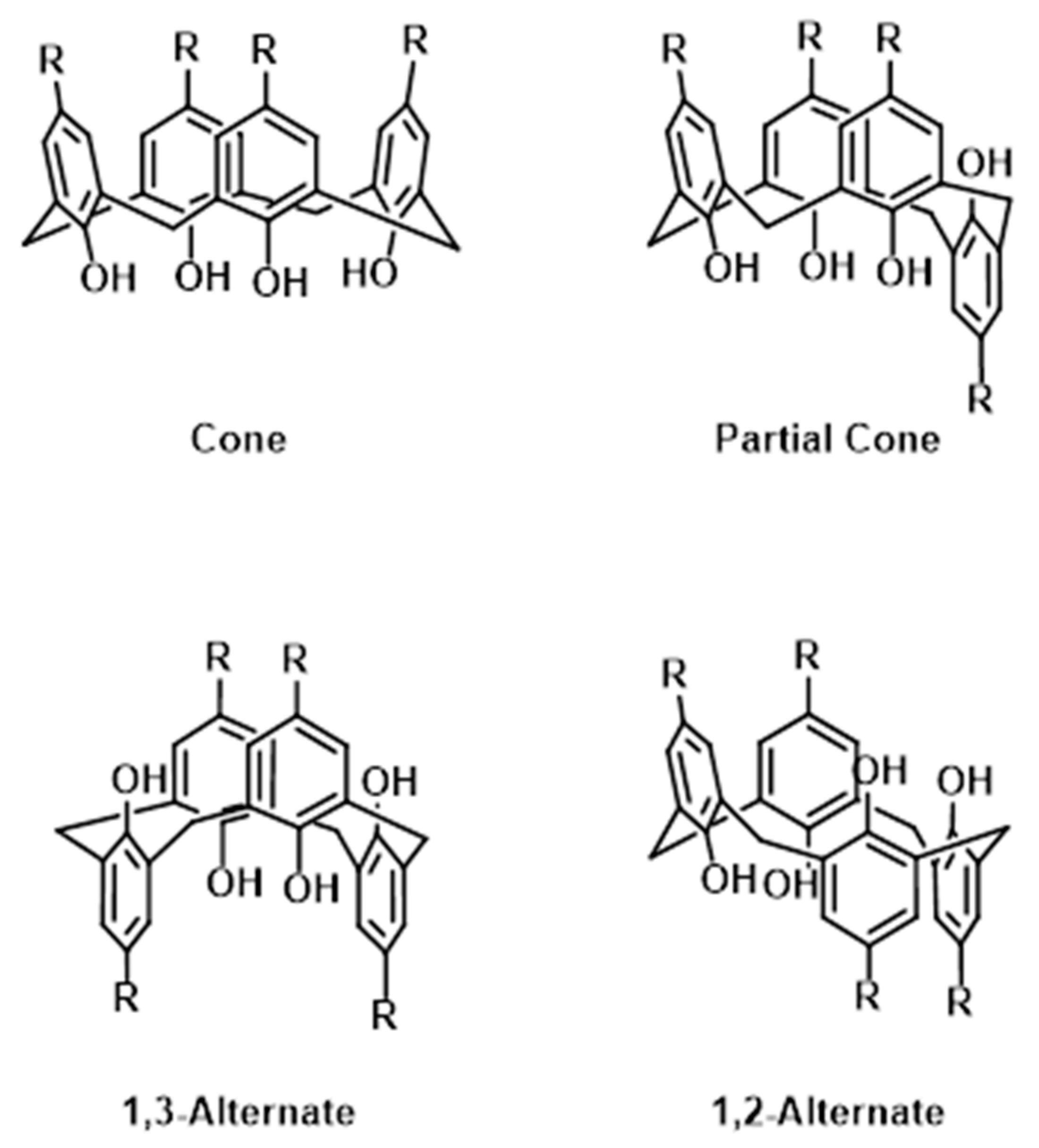
1. Introduction
2. Materials and Methods
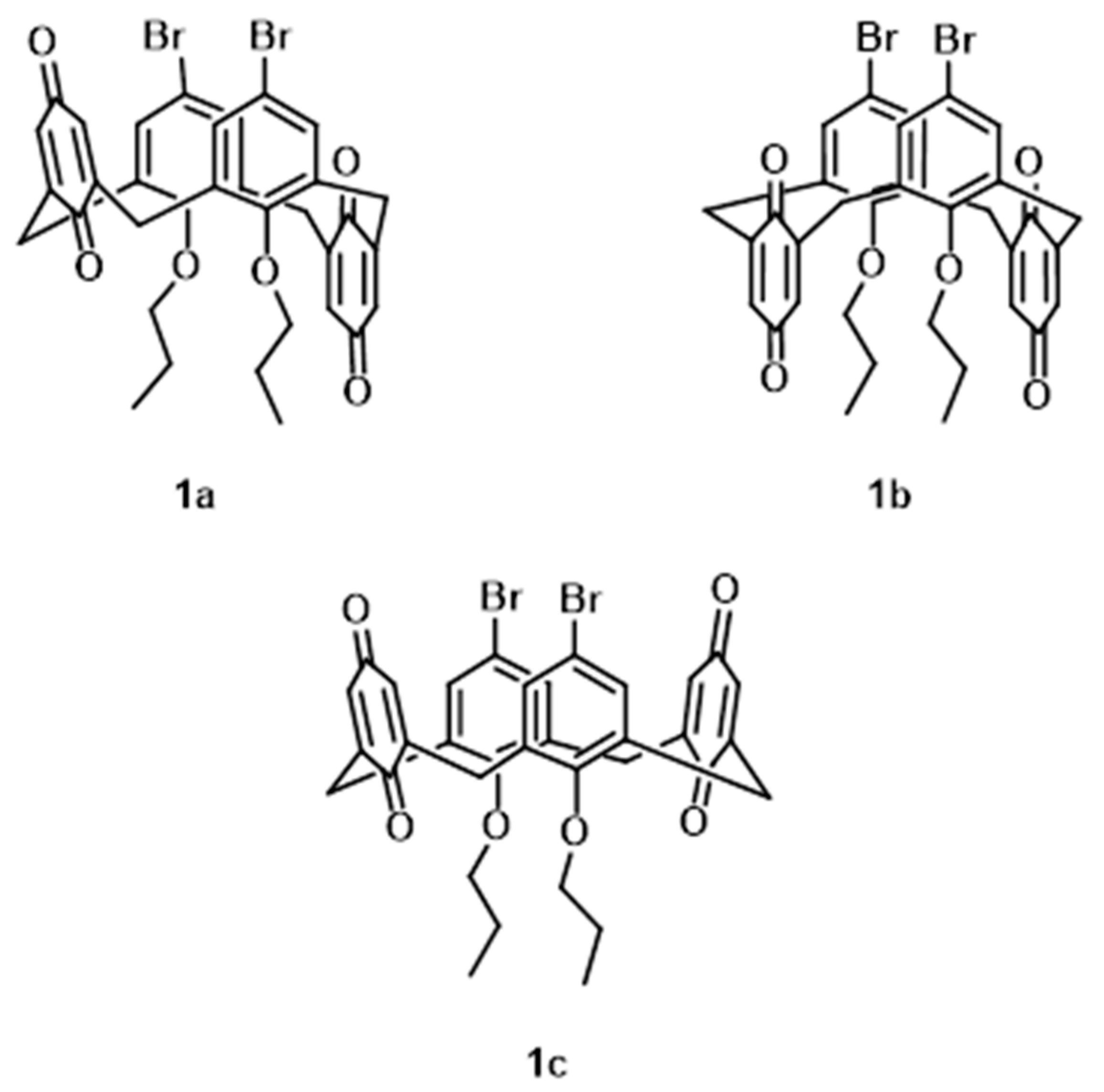
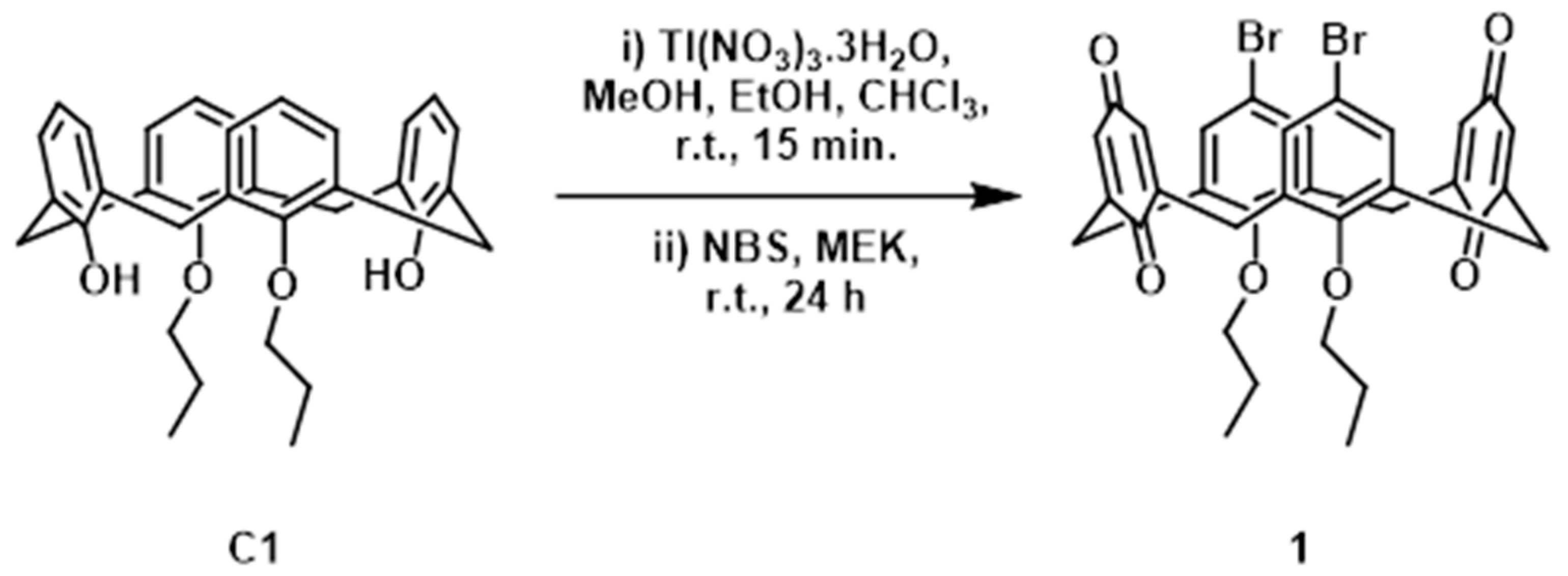
2.1. Synthesis and Characterisation of 1
2.2. Crystallography
2.3. Hirshfeld Surface Analysis
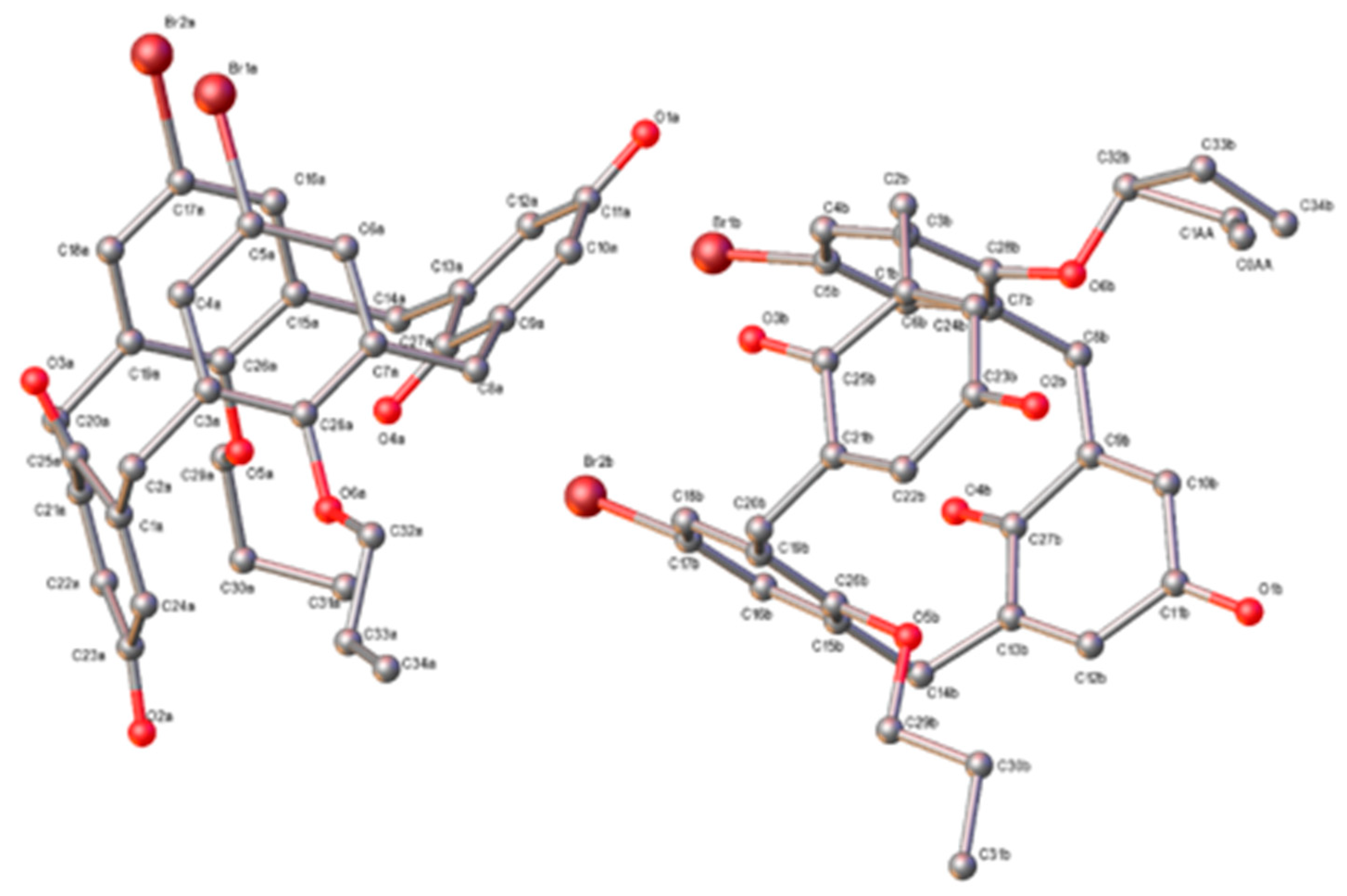
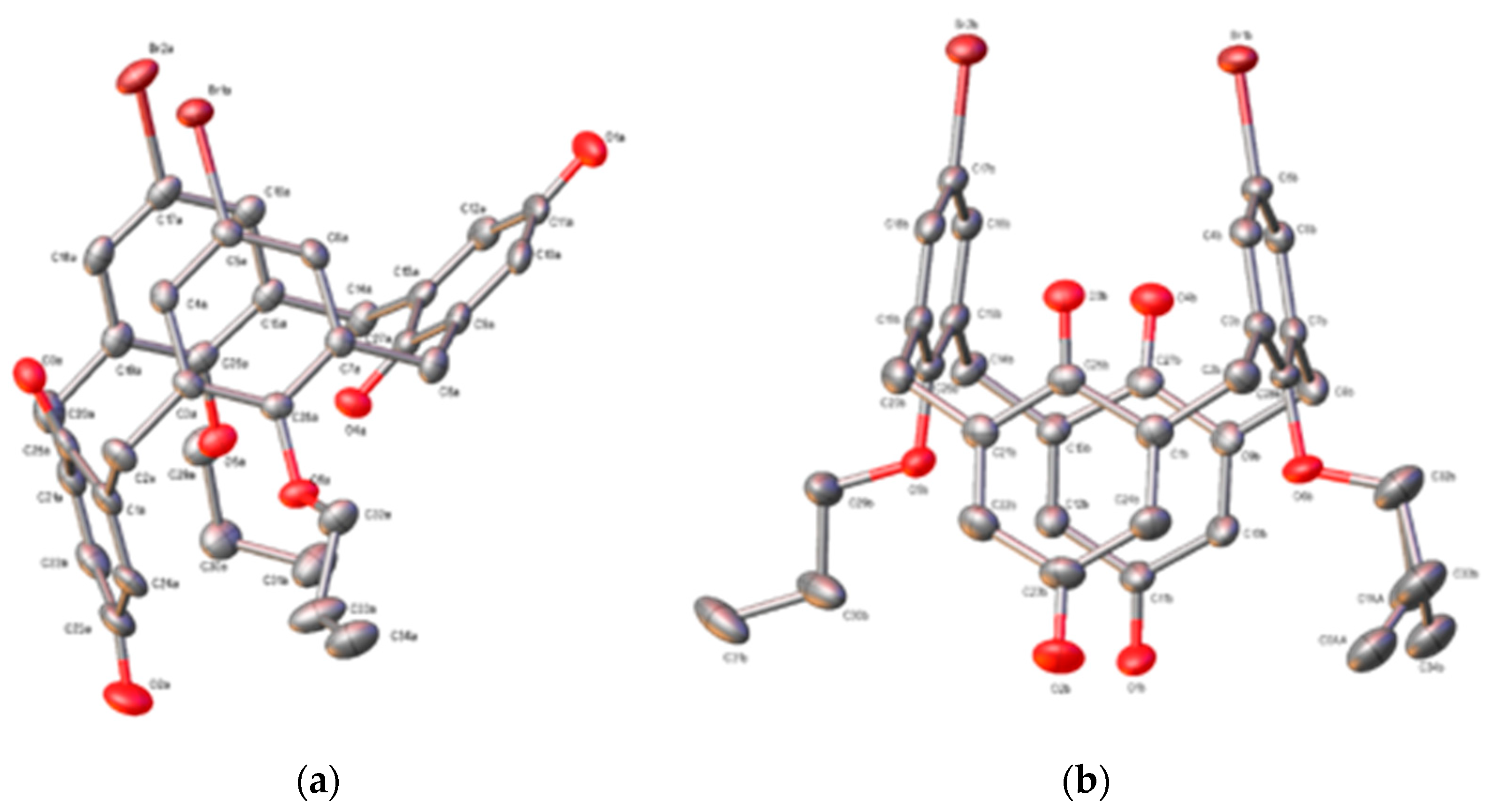
3. Results
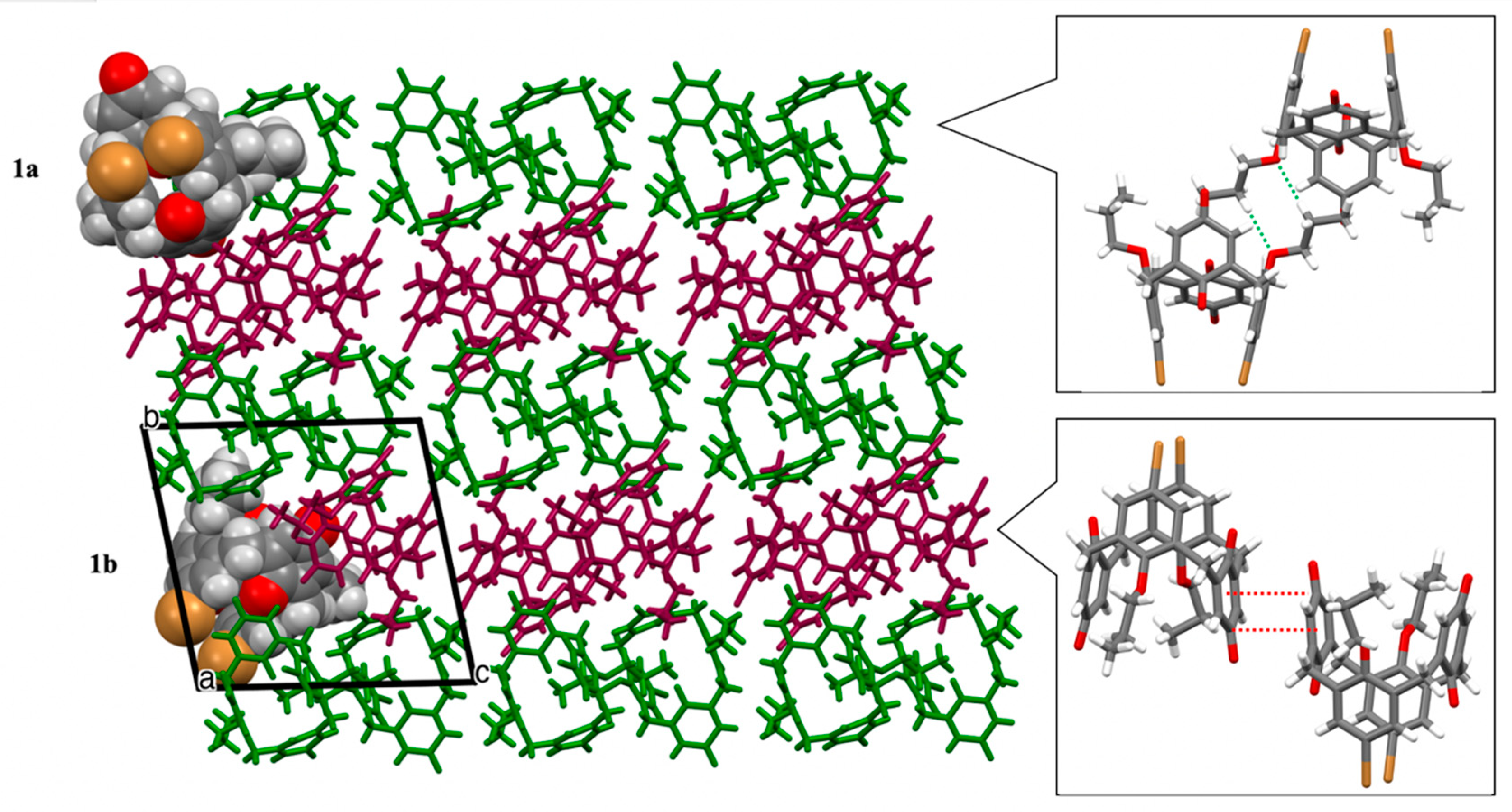
3.1. Structural Description
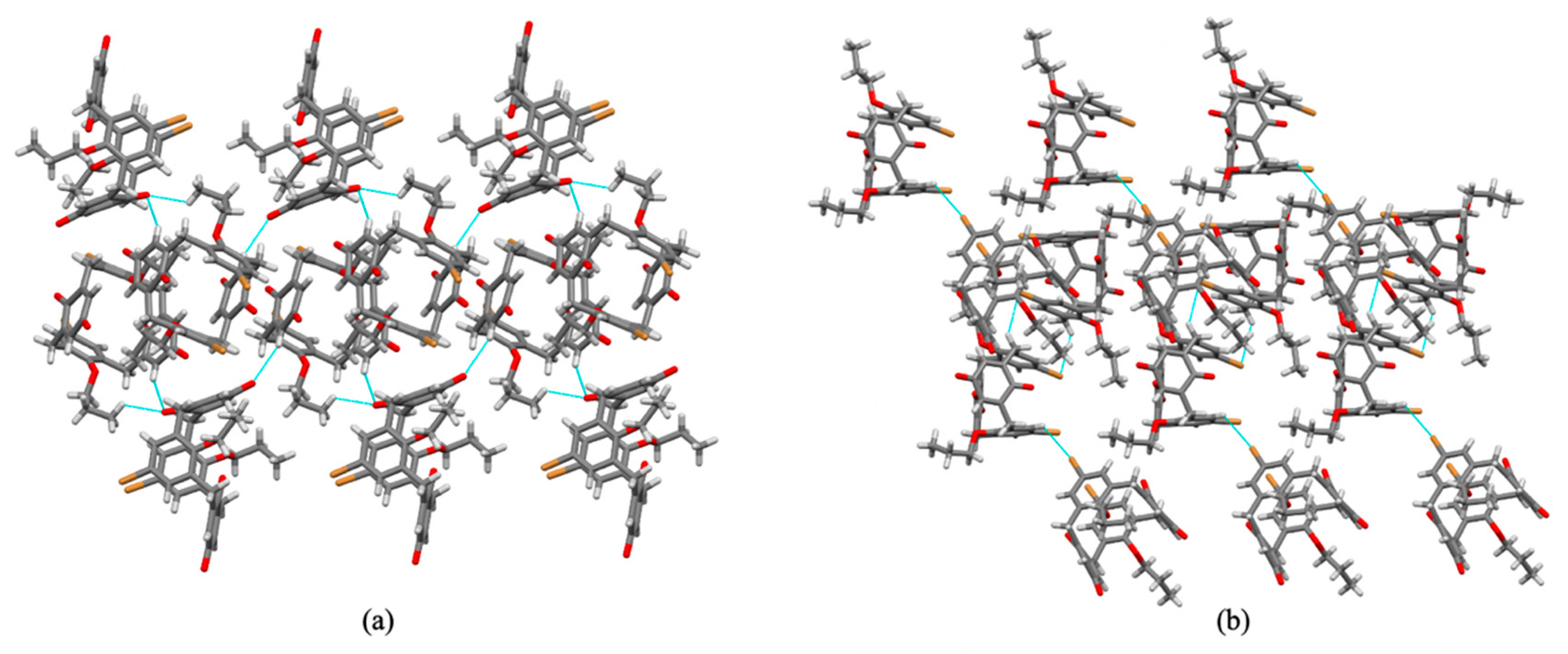
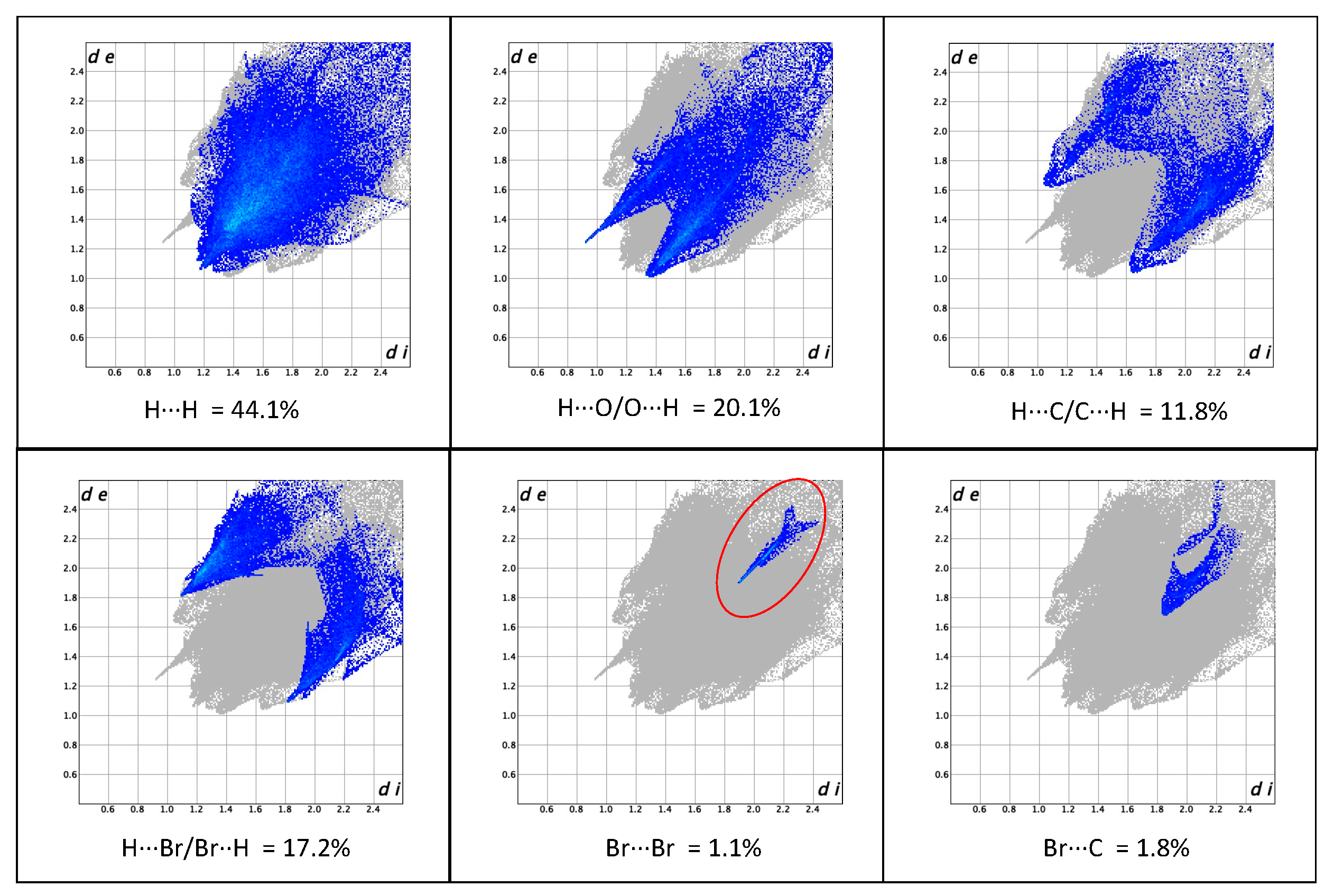
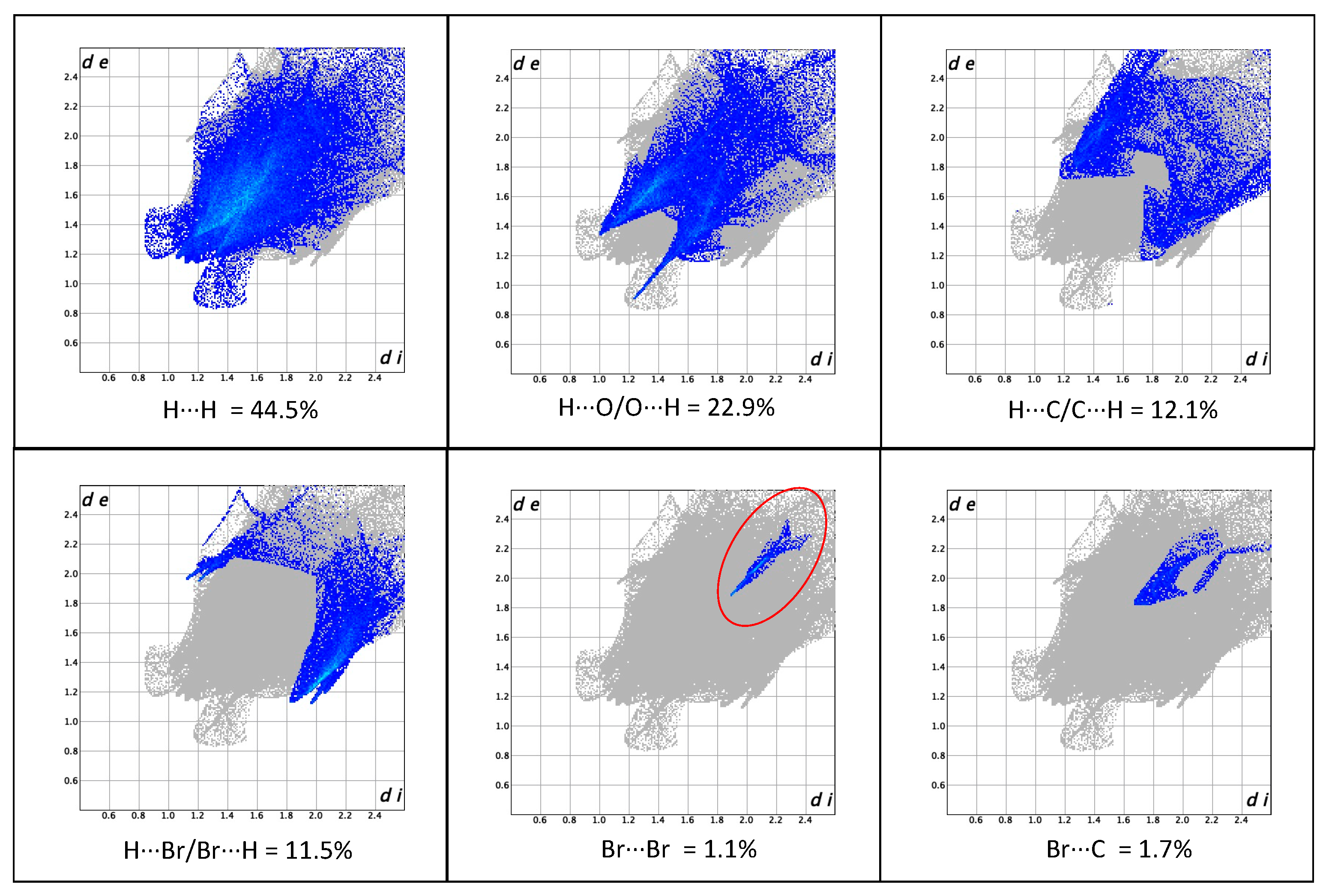
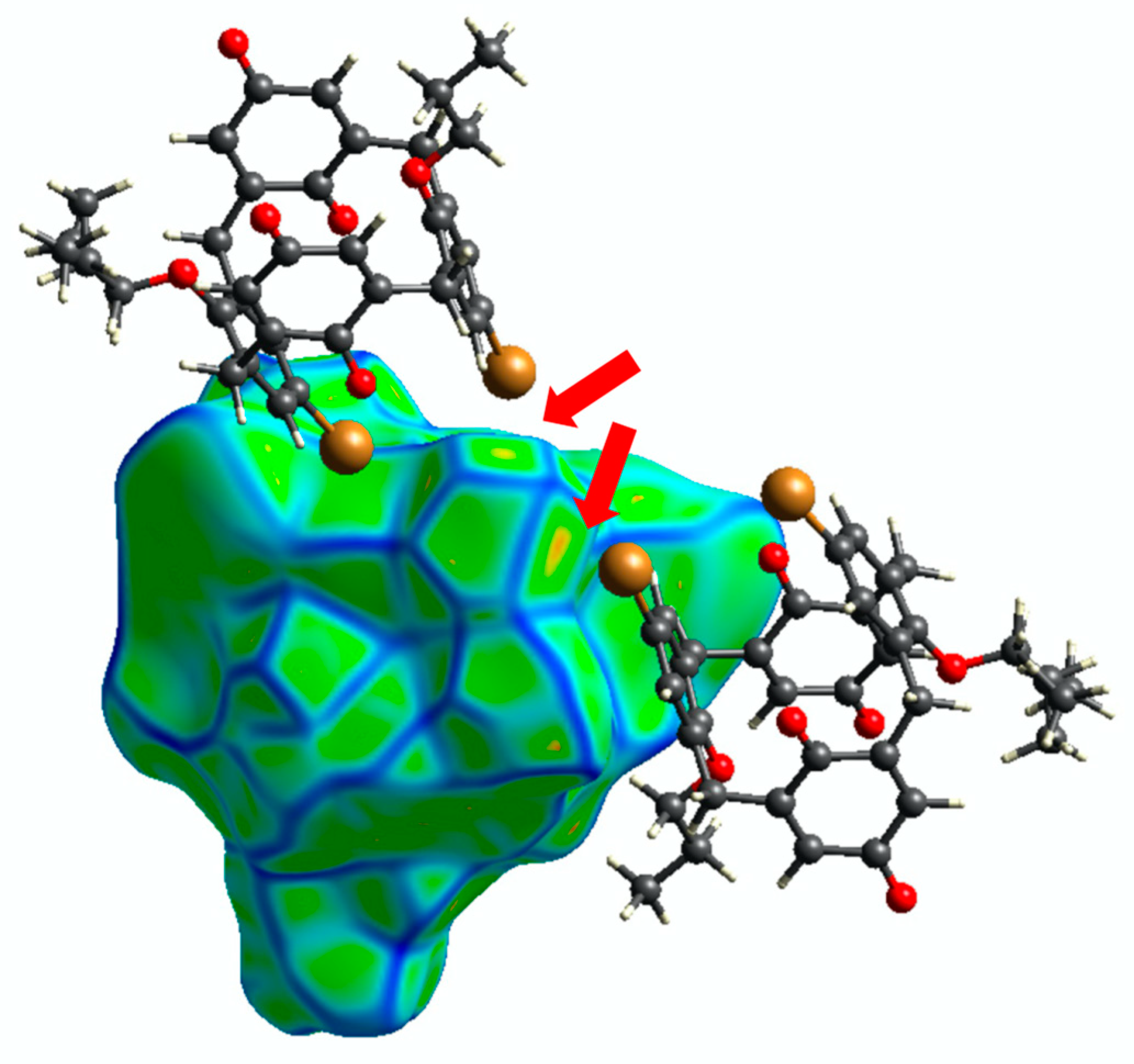
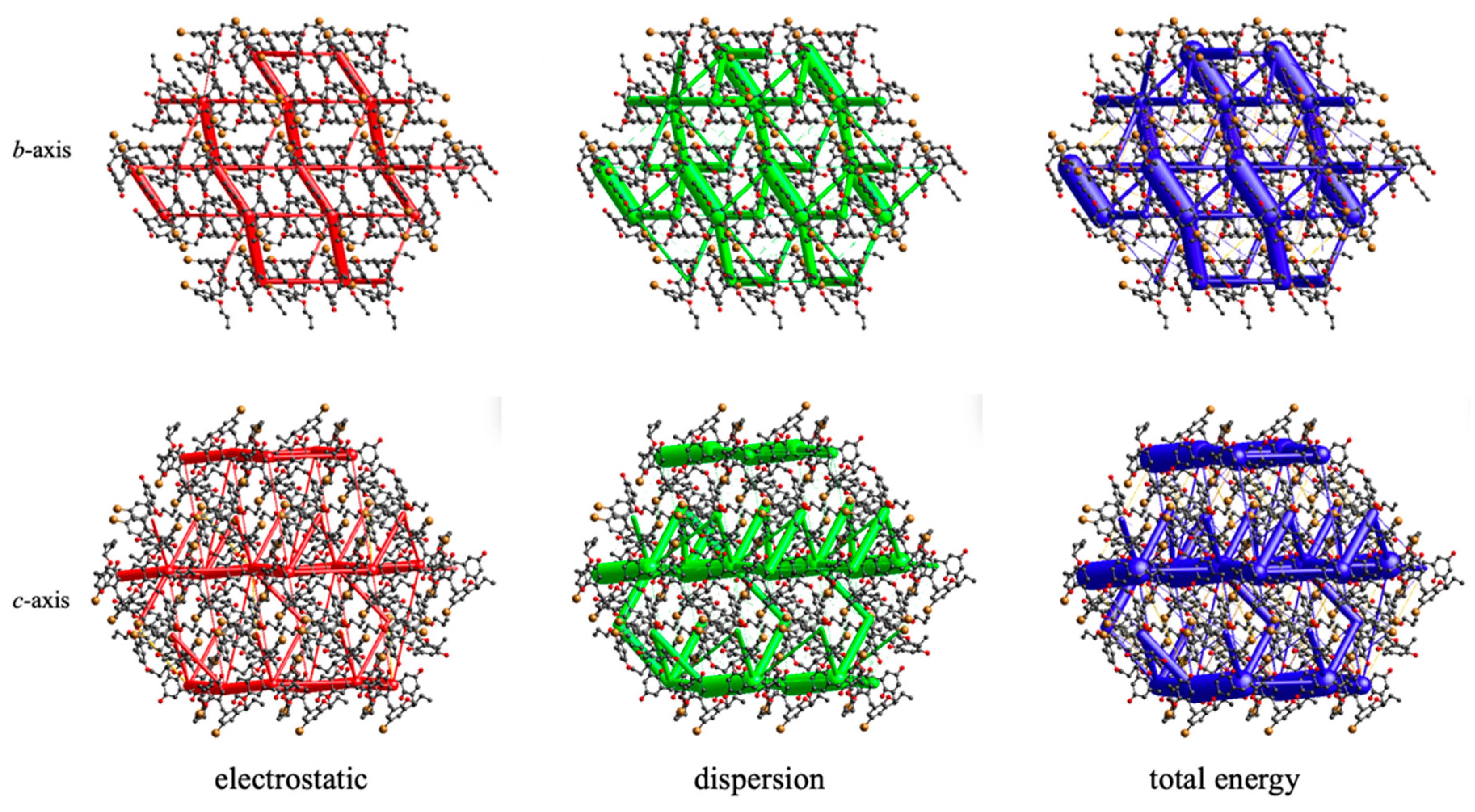
3.2. Crystal Packing and Hirshfeld Surfaces
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Suga, K.; Fujihira, M.; Morita, Y.; Agawa, T. Electrochemical study on calix[4]quinone and calix[4]hydroquinone in N,N-dimethylformamide. J. Chem. Soc. Faraday Trans. 1991, 87, 1575–1578. [Google Scholar] [CrossRef]
- Genorio, B. The synthesis of diquinone and dihydroquinone derivatives of calix[4]arene and electrochemical characterization on Au(111) surface. Acta Chim. Slov. 2016, 63, 496–508. [Google Scholar] [CrossRef][Green Version]
- Pirnat, K.; Dominko, R.; Cerc-Korosec, R.; Genorio, B.; Gaberscek, M. Electrochemically stabilised quinone based electrode composites for Li-ion batteries. J. Power Sources 2012, 199, 308–314. [Google Scholar] [CrossRef]
- Huang, W.; Zhu, Z.; Wang, L.; Wang, S.; Li, H.; Tao, Z.; Shi, J.; Guan, L.; Chen, J. Quasi-solid-state rechargeable lithium-ion batteries with a calix[4]quinone cathode and gel polymer electrolyte. Angew. Chem. Int. Ed. 2013, 52, 9162–9166. [Google Scholar] [CrossRef] [PubMed]
- Kang, S.K.; Lee, O.S.; Chang, S.K.; Chung, D.S.; Kim, H.; Chung, T.D. Calcium ion-Calixquinone complexes adsorbed on a silver electrode. J. Phys. Chem. C 2009, 113, 19981–19985. [Google Scholar] [CrossRef]
- Kim, Y.R.; Kim, R.S.; Kang, S.K.; Choi, M.G.; Kim, H.Y.; Cho, D.; Lee, J.Y.; Chang, S.K.; Chung, T.D. Modulation of quinone PCET reaction by Ca2+ ion captured by calix[4]quinone in water. J. Am. Chem. Soc. 2013, 135, 18957–18967. [Google Scholar] [CrossRef]
- Lee, M.-D.; Yang, K.-M.; Tsoo, C.-Y.; Shu, C.-M.; Lin, L.-G. Calix[4]quinone. Part 1: Synthesis of 5-hydroxycalix[4]arene by calix[4]quinone monoketal route. Tetrahedron 2001, 57, 8095–8099. [Google Scholar] [CrossRef]
- Gutsche, C.D.; Bauer, L.J. Calixarenes. 13. The conformational properties of calix[4]arenes, calix[6]arenes, calix[8]arenes, and oxacalixarenes. J. Am. Chem. Soc. 1985, 107, 6052–6059. [Google Scholar] [CrossRef]
- Gutsche, C.D.; Levine, J.A. Calixarenes. 6. Synthesis of a functionalizable calix[4]arene in a conformationally rigid cone conformation. J. Am. Chem. Soc. 1982, 104, 2652–2653. [Google Scholar] [CrossRef]
- Gutsche, C.D. Embroidering the baskets: Modifying the upper and lower rims of calixarenes. In Calixarenes Revisited; Gutsche, C.D., Ed.; Royal Society of Chemistry: Cambridge, UK, 1998; pp. 79–145. [Google Scholar]
- Casnati, A.; Comelli, E.; Fabbi, M.; Bocchi, V.; Pochini, A.; Ungaro, R.; Mori, G.; Ugozzoli, F.; Lanfredi, A.M.M. Synthesis, conformations and redox properties of diametrical calix[4]arenediquinones. Recl. Des Trav. Chim. Des Pays-Bas 1993, 112, 384–392. [Google Scholar] [CrossRef]
- McKillop, A.; Perry, D.H.; Edwards, M.; Antus, S.; Farkas, L.; Nogradi, M.; Taylor, E.C. Thallium in organic synthesis. XLII. Direct oxidation of 4-substituted phenols to 4,4-disubstituted cyclohexa-2,5-dienones using thallium(III) nitrate. J. Org. Chem. 1976, 41, 282–287. [Google Scholar] [CrossRef]
- Van Loon, J.D.; Arduini, A.; Coppi, L.; Verboom, W.; Pochini, A.; Ungaro, R.; Harkema, S.; Reinhoudt, D.N. Selective functionalization of calix[4]arenes at the upper rim. J. Org. Chem. 1990, 55, 5639–5646. [Google Scholar] [CrossRef]
- Gutsche, C.D.; Pagoria, P.F. Calixarenes. 16. Functionalized calixarenes: The direct substitution route. J. Org. Chem. 1985, 50, 5795–5802. [Google Scholar] [CrossRef]
- Dolomanov, O.V.; Bourhis, L.J.; Gildea, R.J.; Howard, J.A.K.; Puschmann, H. OLEX2: A complete structure solution, refinement and analysis program. J. Appl. Crystallogr. 2009, 42, 339–341. [Google Scholar] [CrossRef]
- Sheldrick, G.M. SHELXT-Integrated space-group and crystal-structure determination. Acta Crystallogr. Sect. A Found. Crystallogr. 2015, 71, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Crystallogr. Sect. C Struct. Chem. 2015, 71, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Turner, M.J.; McKinnon, J.J.; Wolff, S.K.; Grimwood, D.J.; Spackman, P.R.; Jayatilaka, D.; Spackman, M.A. CrystalExplorer17. University of Western Australia. 2017. Available online: http://hirshfeldsurface.net (accessed on 8 January 2022).
- Trotter, J. A three-dimensional analysis of the crystal structure of p-benzoquinone. Acta Crystallogr. 1960, 13, 86–95. [Google Scholar] [CrossRef]
- Kennedy, S.R.; Main, M.U.; Pulham, C.R.; Ling, I.; Dalgarno, S.J. A self-assembled nanotube supported by halogen bonding interactions. CrystEngComm 2019, 21, 786–790. [Google Scholar] [CrossRef]
- Jaime, C.; De Mendoza, J.; Prados, P.; Nieto, P.M.; Sanchez, C. Carbon-13 NMR chemical shifts. A single rule to determine the conformation of calix[4]arenes. J. Org. Chem. 1991, 56, 3372–3376. [Google Scholar] [CrossRef]
- Eddaif, L.; Shaban, A.; Telegdi, J. Sensitive detection of heavy metals ions based on the calixarene derivatives-modified piezoelectric resonators: A review. Int. J. Environ. Anal. Chem. 2019, 99, 824–853. [Google Scholar] [CrossRef]
- Otsuka, H.; Shinkai, S. Stereochemical control of calixarenes useful as rigid and conformationally diversiform platforms for molecular design. Supramol. Sci. 1996, 3, 189–205. [Google Scholar] [CrossRef]
- Torubaev, Y.V.; Skabitsky, I.V. Crystals at a Carrefour on the Way through the Phase Space: A Middle Path. Molecules 2021, 26, 1583. [Google Scholar] [CrossRef] [PubMed]
- Desiraju, G.R.; Parthasarathy, R. The Nature of Halogen···Halogen Interactions: Are Short Halogen Contacts Due to Specific Attractive Forces or Due to Close Packing of Nonspherical Atoms? J. Am. Chem. Soc. 1989, 111, 8725–8726. [Google Scholar] [CrossRef]
- Nishio, M. The CH/π hydrogen bond in chemistry. Conformation, supramolecules, optical resolution and interactions involving carbohydrates. Phys. Chem. Chem. Phys. 2011, 13, 13873–13900. [Google Scholar] [CrossRef]
- Turner, M.J.; Thomas, S.P.; Shi, M.W.; Jayatilaka, D.; Spackman, M.A. Energy frameworks: Insights into interaction anisotropy and the mechanical properties of molecular crystals. Chem. Commun. 2015, 51, 3735–3738. [Google Scholar] [CrossRef]
- Mackenzie, C.F.; Spackman, P.R.; Jayatilaka, D.; Spackman, M.A. CrystalExplorer model energies and energy frameworks: Extension to metal coordination compounds, organic salts, solvates and open-shell systems. IUCrJ 2017, 4, 575–587. [Google Scholar] [CrossRef]
- Groom, C.R.; Bruno, I.J.; Lightfoot, M.P.; Ward, S.C. The Cambridge Structural Database. Acta Crystallogr. Sect. B Struct. Sci. Cryst. Eng. Mater. 2016, 72, 171–179. [Google Scholar] [CrossRef]
- Beer, P.D.; Gale, P.A.; Chen, Z.; Drew, M.G.B.; Heath, J.A.; Ogden, M.I.; Powell, H.R. New Ionophoric Calix[4]diquinones: Coordination Chemistry, Electrochemistry, and X-ray Crystal Structures. Inorg. Chem. 1997, 36, 5880–5893. [Google Scholar] [CrossRef]
- Meddeb-Limem, S.; Besbes-Hentati, S.; Said, H.; Malezieux, B.; Chamoreau, L.M.; Bouvet, M. Electrosynthesis and Structural Studies of 5,17-Di-tert-butyl-26,28-dimethoxycalix[4]arene-25,27-diquinone. X-ray Struct. Anal. Online 2010, 26, 29–30. [Google Scholar] [CrossRef][Green Version]
- Reddy, P.A.; Kashyap, R.P.; Gutsche, C.D.; Watson, W.H. Watson CCDC 158880: Experimental Crystal Structure Determination. 2001. Available online: https://www.ccdc.cam.ac.uk/structures/search?id=doi:10.5517/cc5bb5z&sid=DataCite (accessed on 8 January 2022). [CrossRef]
- Fischer, C.; Gruber, T.; Eissmann, D.; Seichter, W.; Weber, E. Unusual Behavior of a Calix[4]arene Featuring the Coexistence of Basic Cone and 1,2-Alternate Conformations in a Solvated Crystal. Cryst. Growth Des. 2011, 11, 1989–1994. [Google Scholar] [CrossRef]
- Lhoták, P.; Himl, M.; Stibor, I.; Sýkora, J.; Dvořáková, H.; Lang, J.; Petříčková, H. Conformational behaviour of tetramethoxythiacalix[4]arenes: Solution versus solid-state study. Tetrahedron 2003, 59, 7581–7585. [Google Scholar] [CrossRef]












Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Taylor, D.; Ling, I.; Vilela, F.; Dalgarno, S.J. Intermolecular Interactions Drive the Unusual Co-Crystallization of Different Calix[4]arene Conformations. Crystals 2022, 12, 250. https://doi.org/10.3390/cryst12020250
Taylor D, Ling I, Vilela F, Dalgarno SJ. Intermolecular Interactions Drive the Unusual Co-Crystallization of Different Calix[4]arene Conformations. Crystals. 2022; 12(2):250. https://doi.org/10.3390/cryst12020250
Chicago/Turabian StyleTaylor, Dominic, Irene Ling, Filipe Vilela, and Scott J. Dalgarno. 2022. "Intermolecular Interactions Drive the Unusual Co-Crystallization of Different Calix[4]arene Conformations" Crystals 12, no. 2: 250. https://doi.org/10.3390/cryst12020250
APA StyleTaylor, D., Ling, I., Vilela, F., & Dalgarno, S. J. (2022). Intermolecular Interactions Drive the Unusual Co-Crystallization of Different Calix[4]arene Conformations. Crystals, 12(2), 250. https://doi.org/10.3390/cryst12020250