
Carboranes as Lewis Acids: Tetrel Bonding in CB11H11 Carbonium Ylide





Abstract
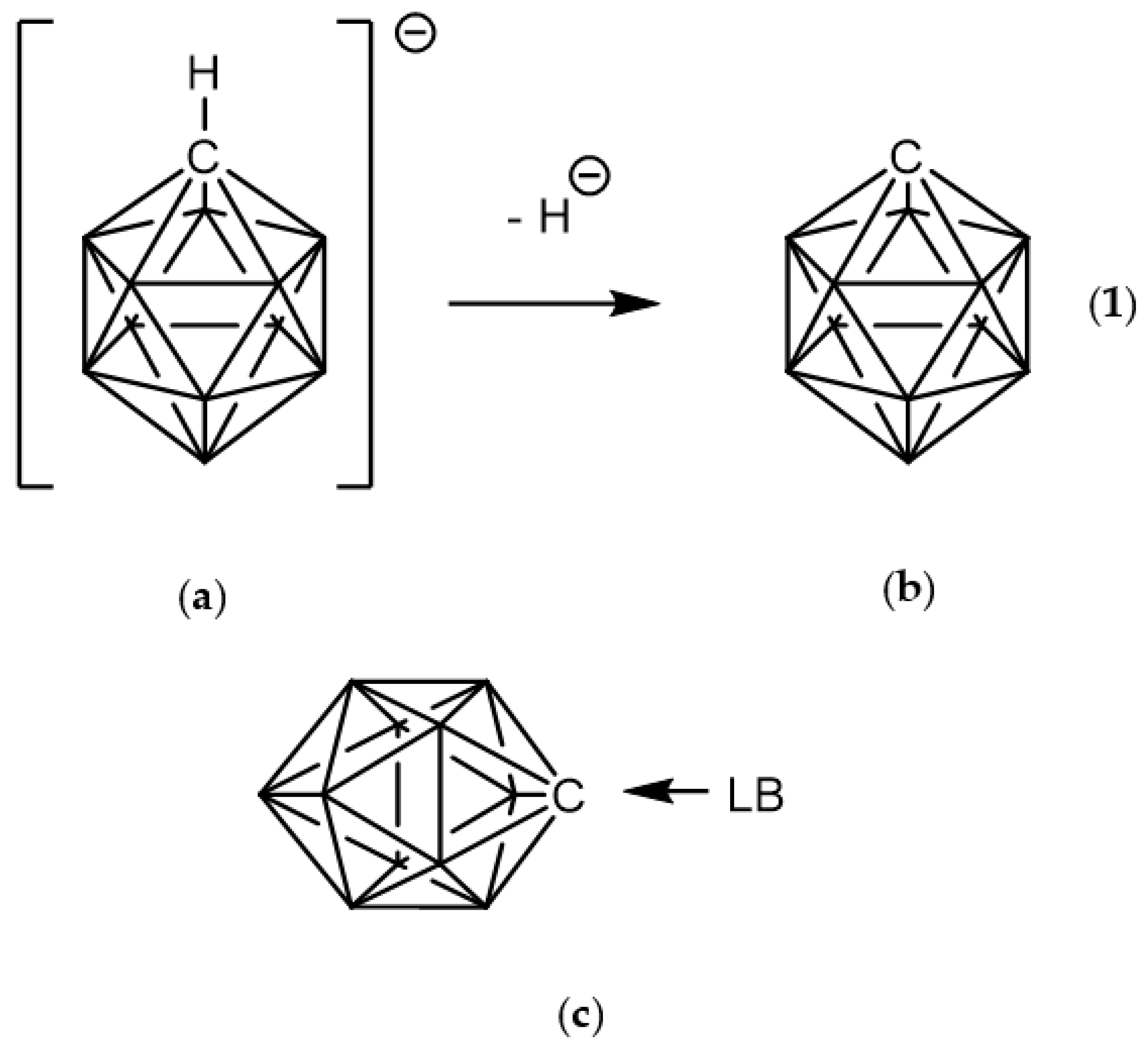
1. Introduction
2. Computational Methods
3. Results
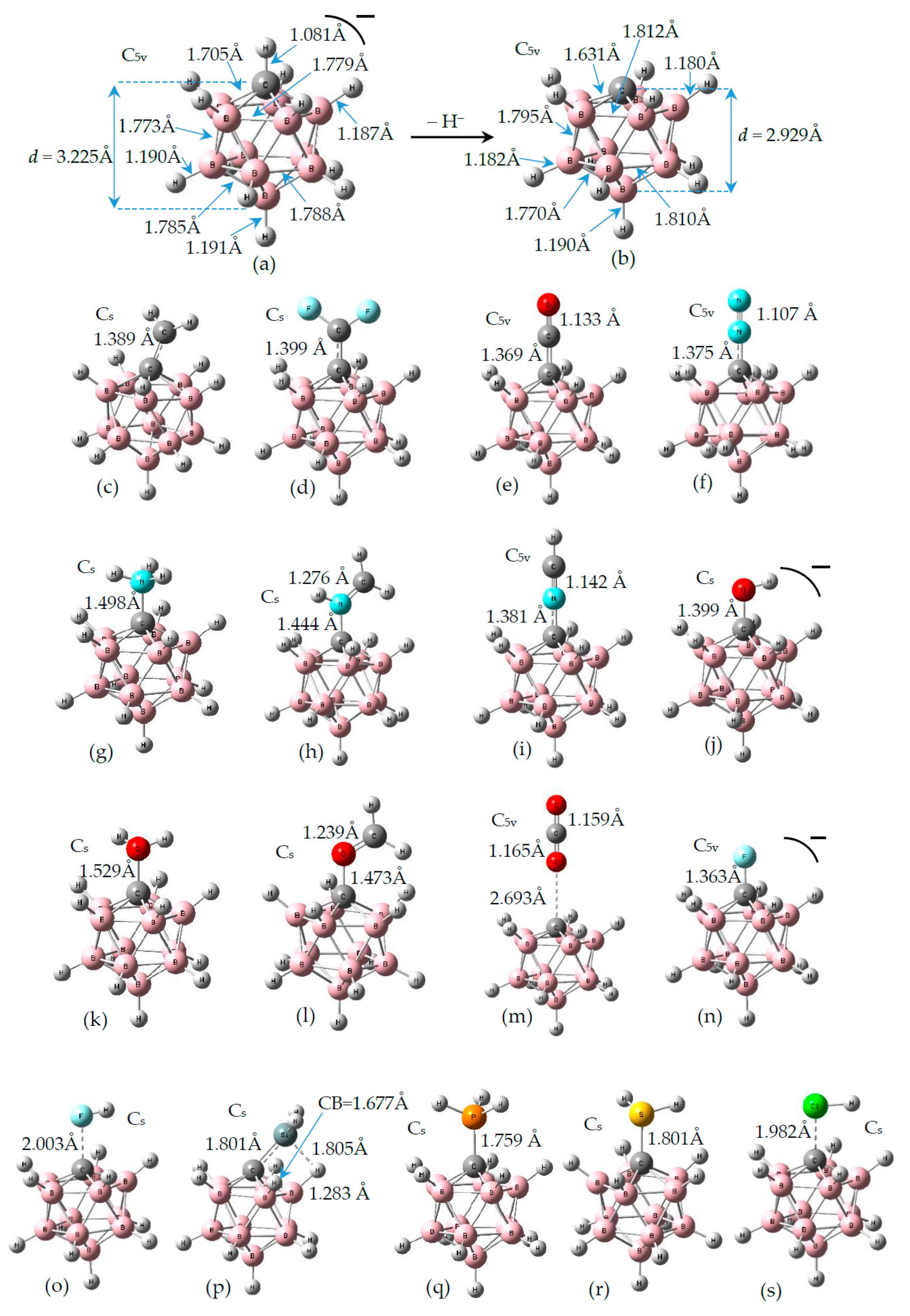
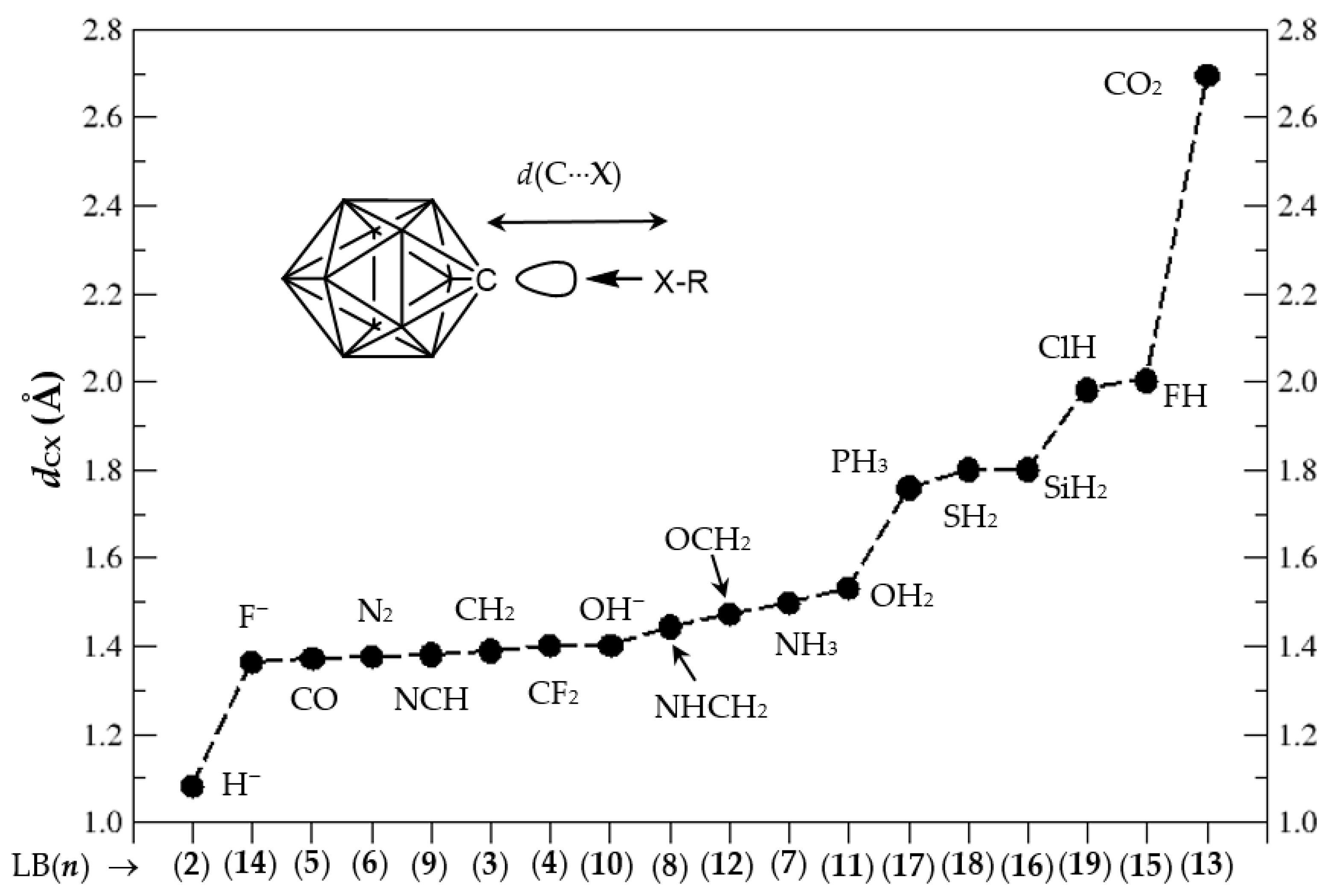
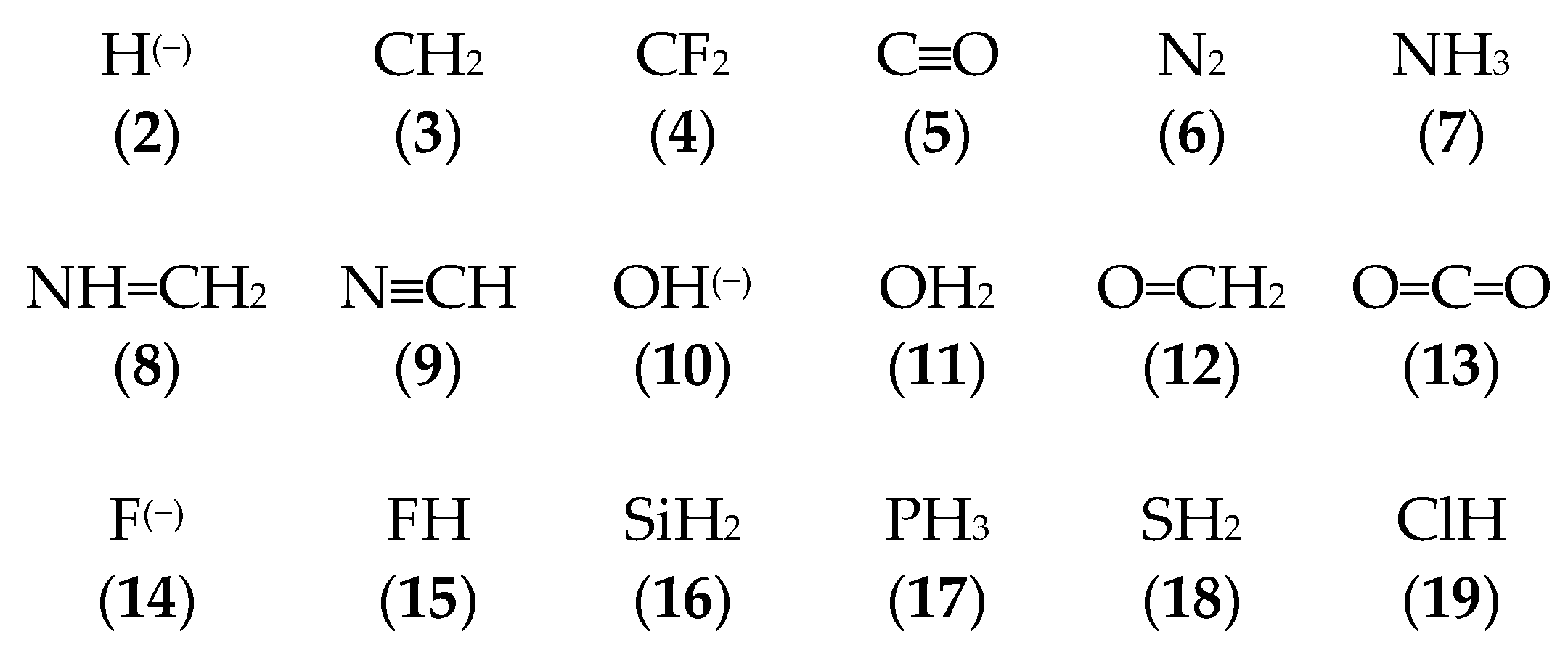
3.1. Geometries of Complexes (1:n), n = 2–19
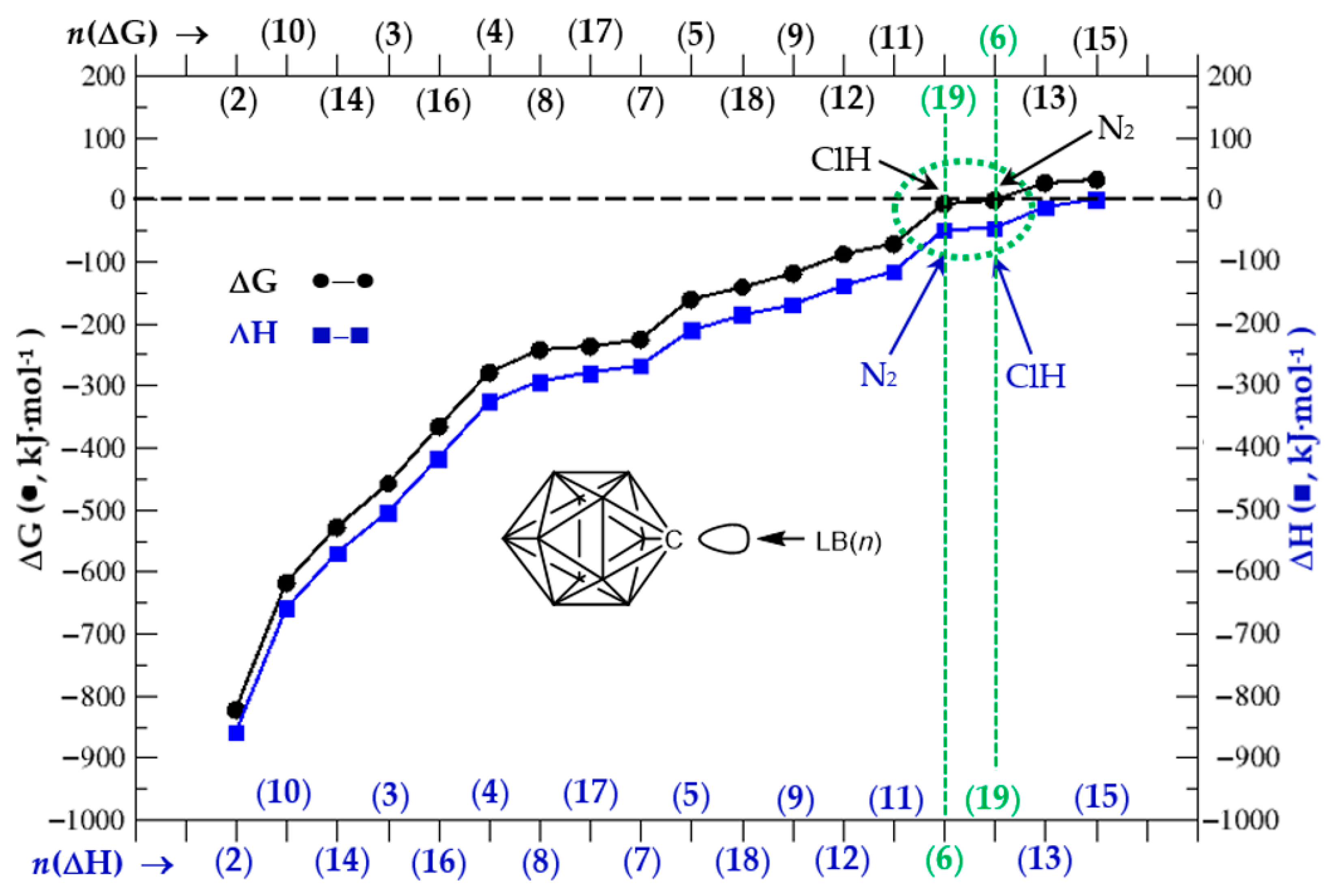
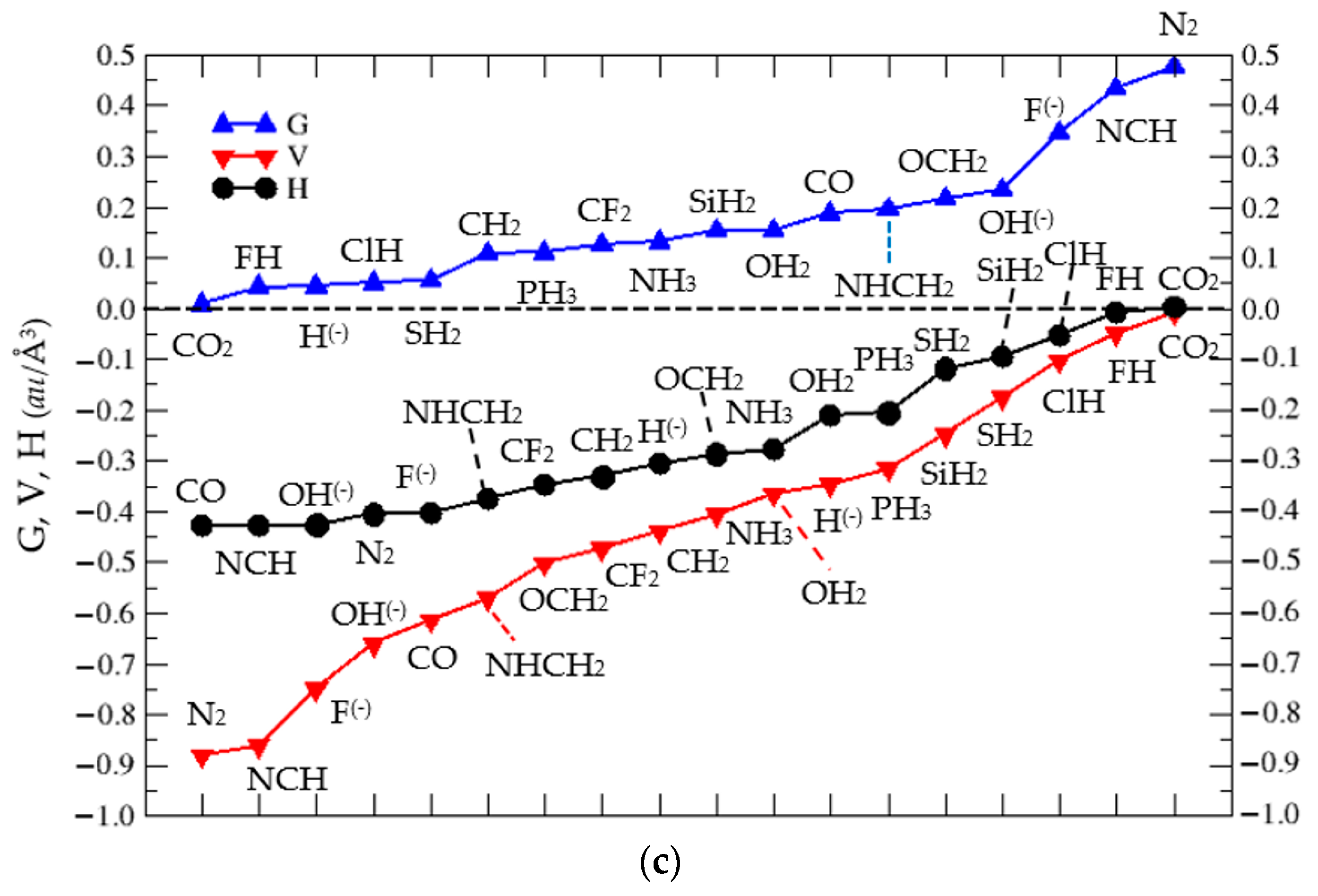
3.2. Thermochemistry of Complexes (1:n), n = 2–19
3.3. Electronic Structure of Complexes (1:n), n = 2–19
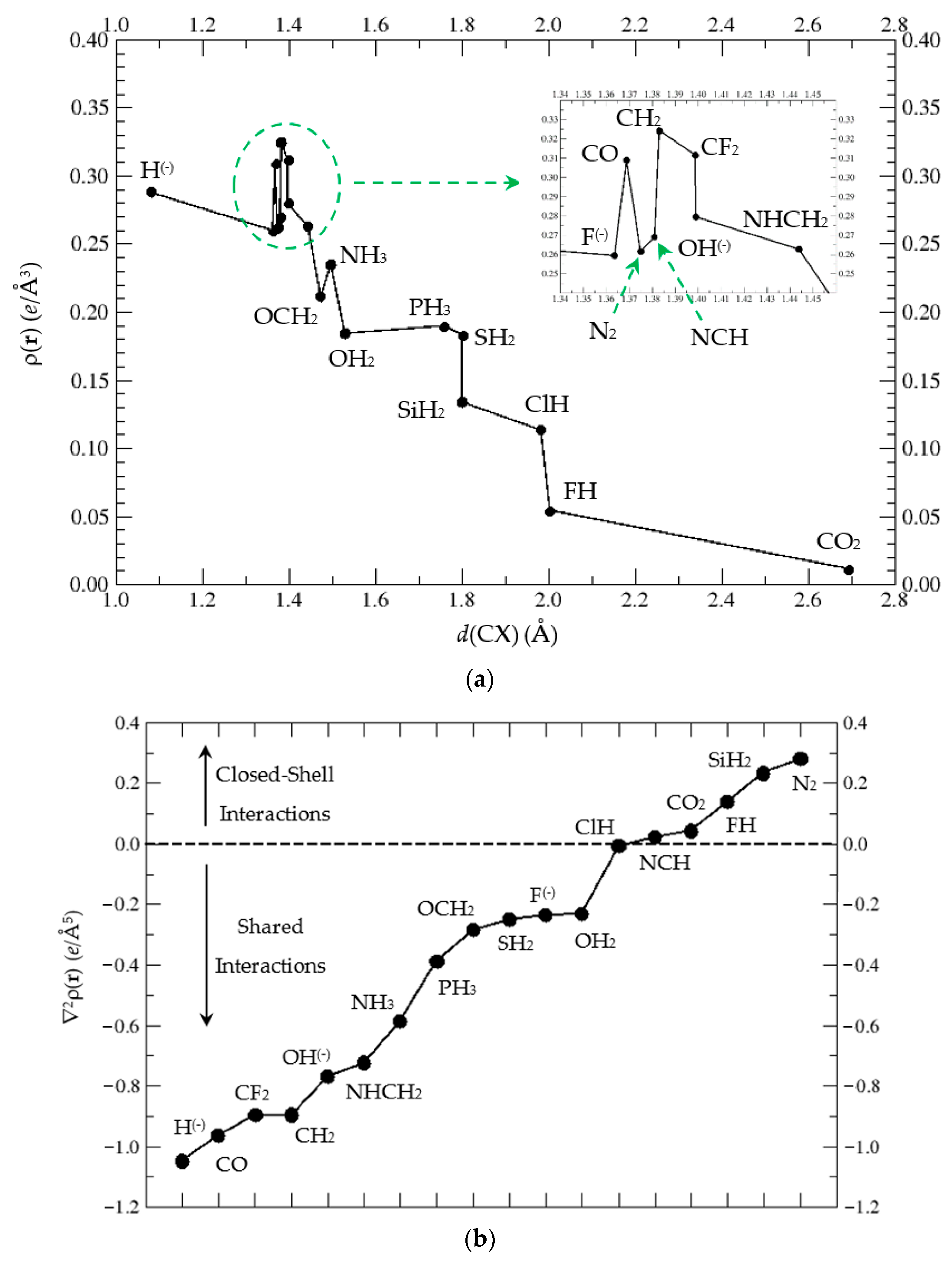
3.3.1. Atoms-in-Molecules (AIM) Topological Analysis of Complexes (1:n), n = 2–19
3.3.2. Electron Localisation Function (ELF) Analysis of Complexes (1:n), n = 2–19
- C: core basin
- V(X): monosynaptic basin, which can be associated to a lone pair
- V(X,X): disynaptic basin
- V(X,X,X): trisynaptic basin
- 5 V(B,B) = 0.42: There are 5 disynaptic basins involving two boron atoms with an average population of 0.42 electrons.
- Blue: core basin
- Red: monosynaptic basin
- Grey: polysynaptic basin
- Beige: V(X,H) basin
- Atoms: Boron (green), Carbon (black), Hydrogen (silver), Oxygen (red), and Phosphorus (tan).
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Knoth, W.H. 1-B9H9CH− and B11H11CH−. J. Am. Chem. Soc. 1967, 89, 1274–1275. [Google Scholar] [CrossRef]
- Plešek, J.; Jelínek, T.; Drdáková, E.; Heřmánek, S.; Štíbr, B. A convenient preparation of 1-CB11H12− and its C-amino derivatives. Collect. Czechoslov. Chem. Commun. 1984, 49, 1559–1562. [Google Scholar] [CrossRef]
- Franken, A.; King, B.T.; Rudolph, J.; Rao, P.; Noll, B.C.; Michl, J. Preparation of [closo-CB11H12]− by dichlorocarbene insertion into [nido-B11H14]. Collect. Czechoslov. Chem. Commun. 2001, 66, 1238–1249. [Google Scholar] [CrossRef]
- Grimes, R.N. Carboranes, 3rd ed.; Academic Press: Cambridge, MA, USA, 2016. [Google Scholar]
- Schleyer, P.v.R.; Najafian, K. Stability and three-dimensional aromaticity of closo-monocarbaborane anions, CBn-1Hn−,and closo-dicarboranes, C2Bn-2Hn. Inorg. Chem. 1998, 37, 3454–3470. [Google Scholar] [CrossRef] [PubMed]
- Michl, J. Chemistry of the three-dimensionally aromatic CB11 cage. Pure Appl. Chem. 2008, 80, 429–446. [Google Scholar] [CrossRef]
- Vyakaranam, K.; Körbe, S.; Divišová, H.; Michl, J. A new type of intermediate, C+(BCH3)11− ↔ C(BCH3)11, in a Grob fragmentation coupled with intramolecular hydride transfer. A nonclassical carbocation ylide or a carbenoid? J. Am. Chem. Soc. 2004, 126, 15795–15801. [Google Scholar] [CrossRef] [PubMed]
- Vyanakaram, K.; Havlas, Z.; Michl, J. Aromatic substitution with hypercloso C(BCH3)11: A new mechanism. J. Am. Chem. Soc. 2007, 129, 4172–4174. [Google Scholar] [CrossRef] [PubMed]
- Thomas, S.P.; Pavan, M.S.; Guru Row, T.N. Experimental evidence for ‘carbon bonding’ in the solid state from charge density analysis. Chem. Commun. 2014, 50, 49–51. [Google Scholar] [CrossRef]
- Alkorta, I.; Rozas, I.; Elguero, J. Molecular complexes between silicon derivatives and electron-rich groups. J. Phys. Chem. A 2001, 105, 743–749. [Google Scholar] [CrossRef]
- Bauzá, A.; Mooibroek, T.J.; Frontera, A. Tetrel-bonding interaction: Rediscovered supramolecular force? Angew. Chem. Int. Ed. 2013, 52, 12317–12321. [Google Scholar] [CrossRef] [PubMed]
- Grabowski, S.J. Tetrel bond–σ-hole bond as a preliminary stage of the SN2 reaction. Phys. Chem. Chem. Phys. 2014, 16, 1824–1834. [Google Scholar] [CrossRef]
- Curtiss, L.A.; Redfern, P.C.; Raghavachari, K. Gaussian-4 theory using reduced order perturbation theory. J. Chem. Phys. 2007, 127, 124105. [Google Scholar] [CrossRef] [PubMed]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 16; Revision C.01; Gaussian Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Hohenberg, P.; Kohn, W. Inhomogeneous electron gas. Phys. Rev. 1964, 136, B864–B871. [Google Scholar] [CrossRef]
- Kohn, W.; Sham, L.J. Self-consistent equations including exchange and correlation effects. Phys. Rev. 1965, 140, A1133–A1138. [Google Scholar] [CrossRef]
- Møller, C.; Plesset, M.S. Note on an approximation treatment for many-electron systems. Phys. Rev. 1934, 46, 618–622. [Google Scholar] [CrossRef]
- Bader, R.F.W. Atoms in Molecules: A Quantum Theory; Clarendon Press: Oxford, UK, 1990. [Google Scholar]
- Popelier, P.L.A. Atoms in Molecules. An Introduction; Prentice Hall: Harlow, UK, 2000. [Google Scholar]
- Keith, T.A. AIMAll; Version 19.10.12; TK Gristmill Software: Overland Park, KS, USA, 2017. [Google Scholar]
- Silvi, B.; Savin, A. Classification of chemical bonds based on topological analysis of electron localization functions. Nature 1994, 371, 683–686. [Google Scholar] [CrossRef]
- Becke, A.D.; Edgecombe, K.E. A simple measure of electron localization in atomic and molecular systems. J. Chem. Phys. 1990, 92, 5397–5403. [Google Scholar] [CrossRef]
- Noury, S.; Krokidis, X.; Fuster, F.; Silvi, B. TopMod Package; Universite Pierre et Marie Curie: Paris, France, 1997. [Google Scholar]
- Mata, I.; Alkorta, I.; Molins, E.; Espinosa, E. Universal features of the electron density distribution in hydrogen-bonding regions: A comprehensive study involving H···X (X=H, C, N, O, F, S, Cl, π) interactions. Chem. Eur. J. 2010, 16, 2442–2452. [Google Scholar] [CrossRef]
- Alkorta, I.; Solimannejad, M.; Provasi, P.F.; Elguero, J. Theoretical study of complexes and fluoride cation transfer between N2F+ and electron donors. J. Phys. Chem. A 2007, 111, 7154–7161. [Google Scholar] [CrossRef] [PubMed]
- Sánchez-Sanz, G.; Trujillo, C.; Alkorta, I.; Elguero, J. Intermolecular weak interactions in HTexH dimers (X=O, S, Se, Te): Hydrogen bonds, chalcogen-chalcogen contacts and chiral discrimination. ChemPhysChem 2012, 13, 496–503. [Google Scholar] [CrossRef] [PubMed]
- Alkorta, I.; Mata, I.; Molins, E.; Espinosa, E. Charged versus neutral hydrogen-bonded complexes: Is there a difference in the nature of the hydrogen bonds? Chem. Eur. J. 2016, 22, 9226–9234. [Google Scholar] [CrossRef] [PubMed]
- Savin, A.; Jepsen, O.; Flad, J.; Andersen, O.K.; Preuss, H.; Von Schnering, H.G. Electron localization in solid-state structures of the elements: The diamond structure. Angew. Chem. Int. Ed. 1992, 31, 187–188. [Google Scholar] [CrossRef]
- Heisenberg, W. Mehrkörperproblem und Resonanz in der Quantenmechanik. Zeit. Phys. 1985, 38, 456–471. [Google Scholar] [CrossRef]
- Dirac, P.A.M. On the theory of quantum mechanics. Proc. R. Soc. London. Ser. A Math. Phys. Sci. 1926, 112, 661–677. [Google Scholar] [CrossRef]
- Dontha, R.; Zhu, T.-C.; Shen, Y.; Wörle, M.; Hong, X.; Duttwyler, S. A 3D Analogue of phenyllithium: Solution-phase, solid-state, and computational study of the lithiacarborane [Li−CB11H11]. Angew. Chem. Int. Ed. Engl. 2019, 58, 19007–19013. [Google Scholar] [CrossRef] [PubMed]
- Groom, C.R.; Bruno, I.J.; Lightfoot, M.P.; Ward, S.C. The Cambridge structural database. Acta Crystallogr. B Struct. Sci. Cryst. Eng. Mater. 2016, B72, 171–179. [Google Scholar] [CrossRef] [PubMed]
- Pecyna, J.; Ringstrand, B.; Domagała, S.; Kaszyński, P.; Woźniak, K. Synthesis and characterization of 12-pyridinium derivatives of the [closo-1-CB11H12]− anion. Inorg. Chem. 2014, 53, 12617–12626. [Google Scholar] [CrossRef]
- Douglass, A.G.; Janousek, Z.; Kaszynski, P.; Young, V.G., Jr. Synthesis and molecular structure of 12-iodo-1-(4-pentylquinuclidin-1-yl)-1-carba-closo-dodecaborane. Inorg. Chem. 1998, 37, 6361–6365. [Google Scholar] [CrossRef]
- Finze, M.; Sprenger, J.A.P. 1-amino-2-fluoromonocarba-closo-dodecaborates: K[1-H2N-2-F-closo-1-CB11H10] and [Et4N][1-H2N-2-F-closo-1-CB11I10]. Z. Anorg. Allg. Chem. 2010, 636, 1538–1542. [Google Scholar] [CrossRef]
- Pecyna, J.; Kaszynski, P.; Ringstrand, B.; Pociecha, D.; Pakhomov, S.; Douglass, A.G.; Young, V.G., Jr. Synthesis and characterization of quinuclidinium derivatives of the [closo-1-CB11H12]− anion as potential polar components of liquid crystal materials. Inorg. Chem. 2016, 55, 4016–4025. [Google Scholar] [CrossRef]
- Maly, K.; Subrtova, V.; Petricek, V. Structure of 1-trimethylamine-1-carba-closo-dodecaborane(11). Acta Crystallogr. Sect. C Cryst. Struct. Commun. 1987, C43, 593–594. [Google Scholar] [CrossRef]
- Hailmann, M.; Herkert, L.; Himmelspach, A.; Finze, M. Difunctionalized {closo-1-CB11} clusters: 1- and 2-amino-12-ethynylcarba-closo-dodecaborates. Chem. Eur. J. 2013, 19, 15745–15758. [Google Scholar] [CrossRef] [PubMed]
- Morris, J.H.; Peters, G.S.; Spicer, M.D. 1-Me2NH-2-CH2Cl-closo-l-CB11H10. An unusual product from the insertion reaction of Me2NBCl2 with Li2[7-Me3N-nido-7-CB10H10]. J. Organomet. Chem. 1995, 494, 195–198. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
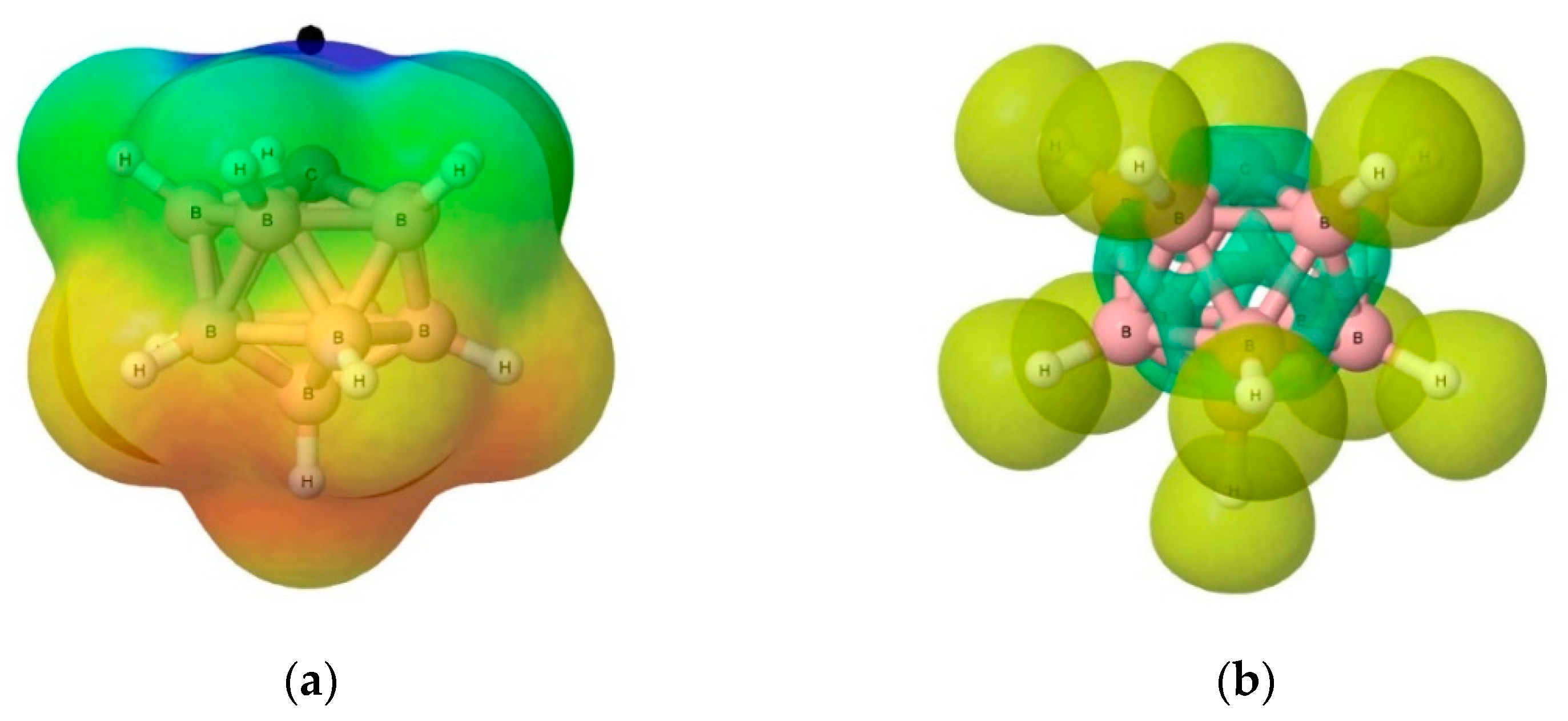{kind=link}
{kind=link}
{kind=link}
{kind=link}
| n = 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 | |
|---|---|---|---|---|---|---|---|---|---|
| LB | H(−) | CH2 | CF2 | C≡O | N2 | NH3 | NH=CH2 | N≡CH | OH(−) |
| ΔH | −858.6 | −504.9 | −326.2 | −211.1 | −50.5 | −268.6 | −294.6 | −170.6 | −659.3 |
| ΔG | −822.3 | −457.7 | −279.7 | −162.9 | −2.8 | −226.8 | −242.7 | −120.8 | −618.0 |
| 11 | 12 | 13 | 14 | 15 | 16 | 17 | 18 | 19 | |
| LB | OH2 | O=CH2 | O=C=O | F(−) | FH | SiH2 | PH3 | SH2 | ClH |
| ΔH | −116.2 | −139.8 | −14.3 | −571.5 | −0.7 | −418.0 | −280.9 | −186.1 | −47.6 |
| ΔG | −72.1 | −87.9 | 25.7 | −529.7 | 31.3 | −366.8 | −238.1 | −140.9 | −8.9 |
| H(−) | N≡N | PH3 | CO2 |
 11 C(B) = 2.07 1 C(C) = 2.11 11 V(B,H) = 2.05 10 V(B,B,B) = 0.81 15 V(B,B) = 0.76 5 V(C,B) = 1.01 1 V(C,H) = 2.05 |  11 C(B) = 2.06 1 C(C) = 2.06 2 C(N) = 2.07 11 V(B,H) = 2.05 1 V(N,N) = 4.01 5 V(B,B) = 0.46 5 V(B,B,B) = 2.43 5 V(B,B,B) = 0.97 5 V(C,B) = 1.02 1 V(C,N2) = 2.62 1 V(N1) = 3.51 |  11 C(B) = 2.07 1 C(C) = 2.07 1 C(P) = 9.89 11 V(B,H) = 2.05 3 V(P,H) = 2.05 5 V(C,B) = 1.01 1 V(C,P) = 2.21 5 V(B,B) = 0.51 5 V(B,B,B) = 2.44 5 V(B,B,B) = 0.91 |  11 C(B) = 2.07 2 C(C) = 2.07 2 C(O) = 2.13 11 V(B,H) = 2.03 1 V(C2,O2) = 2.26 1 V(C2,O1) = 2.51 5 V(C,B) = 1.20 15 V(B,B) = 0.54 7 V(B,B,B) = 1.08 3 V(C,B,B) = 1.08 1 V(O2) = 5.64 1 V(O1) = 3.44 1 V(O1) = 1.82 |
| n | LB | V(C, X) | n | LB | V(C, X) | |
|---|---|---|---|---|---|---|
| 2 | H(−) | 2.05 | 11 | OH2 | 1.56 | |
| 3 | CH2 | 2.53 | 12 | O=CH2 | 1.65 | |
| 4 | CF2 | 2.71 | 13 | O=C=O | - | |
| 5 | C≡O | 2.78 | 14 | F(−) | 0.99 | |
| 6 | N≡N | 2.62 | 15 | FH | - | |
| 7 | NH3 | 1.76 | 16 | SiH2 | 2.40 | |
| 8 | NH=CH2 | 2.02 | 17 | PH3 | 2.21 | |
| 9 | N≡CH | 2.34 | 18 | SH2 | 1.85 | |
| 10 | OH(−) | 1.32 | 19 | ClH | 1.11 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ferrer, M.; Alkorta, I.; Elguero, J.; Oliva-Enrich, J.M. Carboranes as Lewis Acids: Tetrel Bonding in CB11H11 Carbonium Ylide. Crystals 2021, 11, 391. https://doi.org/10.3390/cryst11040391
Ferrer M, Alkorta I, Elguero J, Oliva-Enrich JM. Carboranes as Lewis Acids: Tetrel Bonding in CB11H11 Carbonium Ylide. Crystals. 2021; 11(4):391. https://doi.org/10.3390/cryst11040391
Chicago/Turabian StyleFerrer, Maxime, Ibon Alkorta, José Elguero, and Josep M. Oliva-Enrich. 2021. "Carboranes as Lewis Acids: Tetrel Bonding in CB11H11 Carbonium Ylide" Crystals 11, no. 4: 391. https://doi.org/10.3390/cryst11040391
APA StyleFerrer, M., Alkorta, I., Elguero, J., & Oliva-Enrich, J. M. (2021). Carboranes as Lewis Acids: Tetrel Bonding in CB11H11 Carbonium Ylide. Crystals, 11(4), 391. https://doi.org/10.3390/cryst11040391