Electrochemical Characterization of CVD-Grown Graphene for Designing Electrode/Biomolecule Interfaces


Abstract
:1. Introduction
2. Materials and Methods
2.1. Reagents and Materials
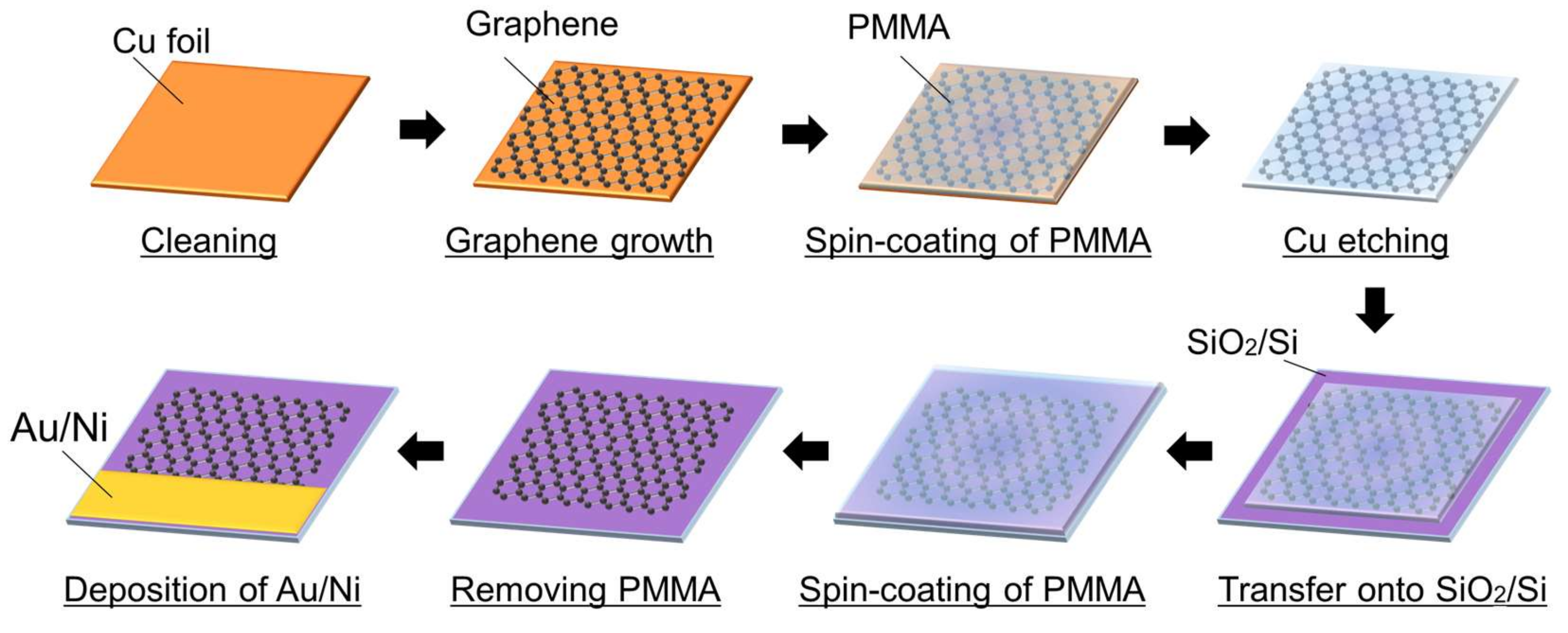
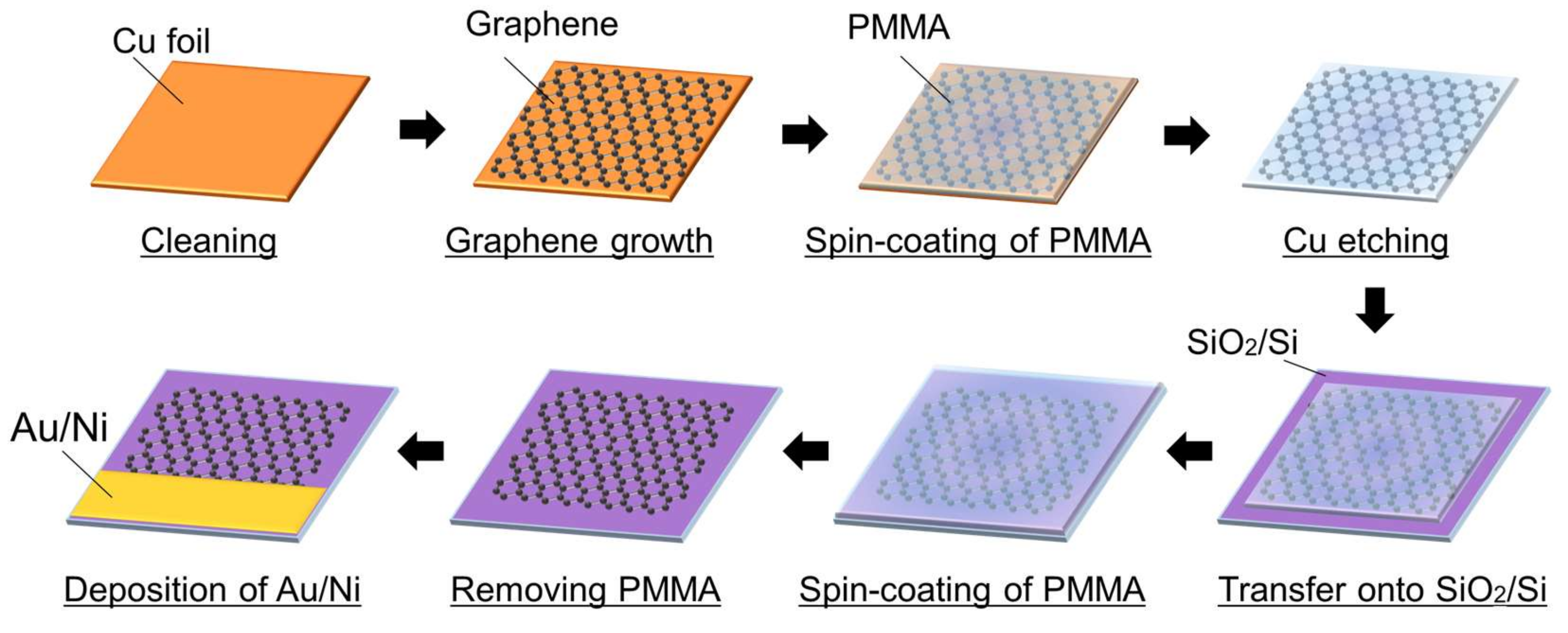
2.2. Fabrication of Graphene Electrode
2.3. Characterizaiton of Graphene Electrode
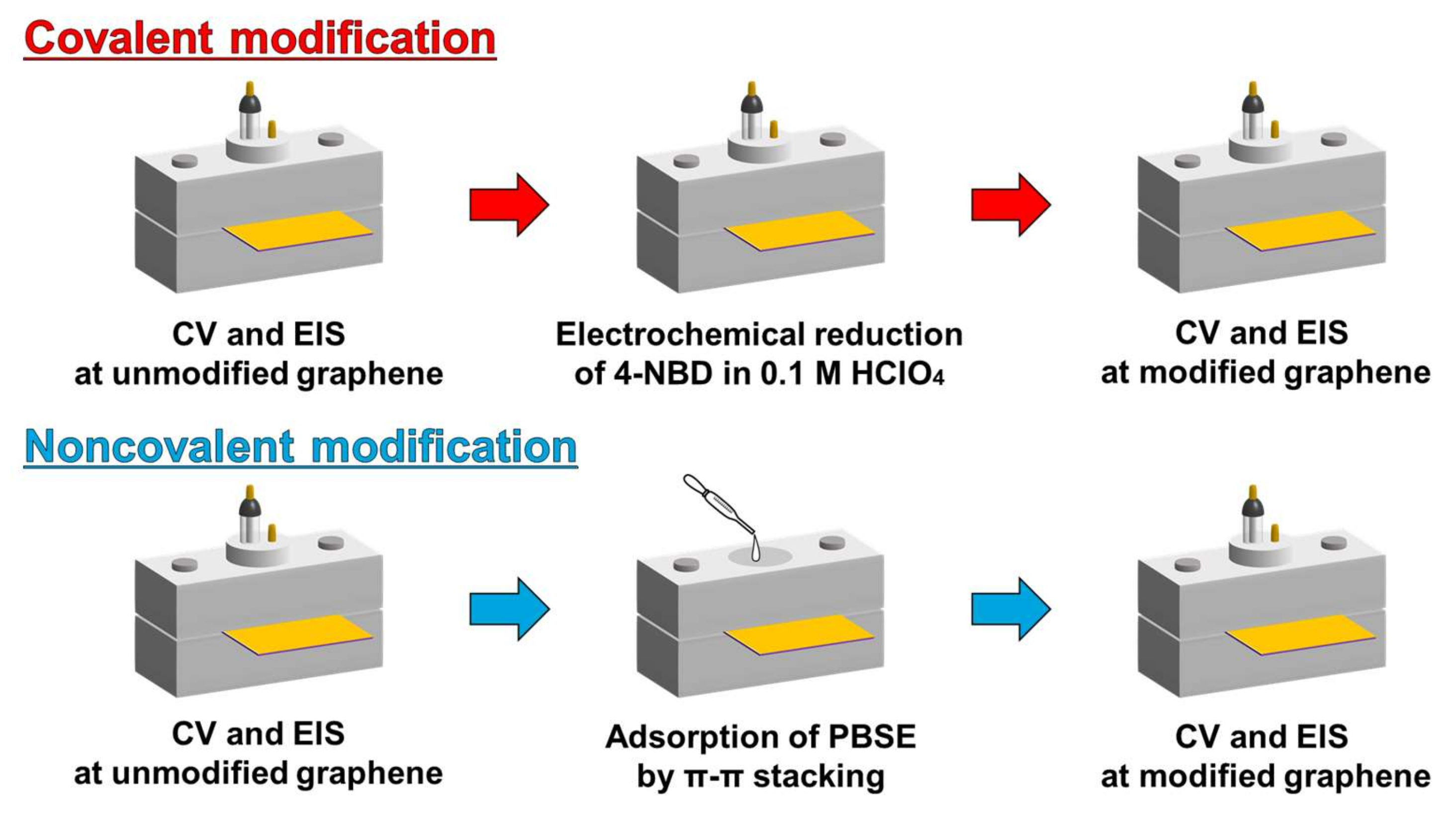
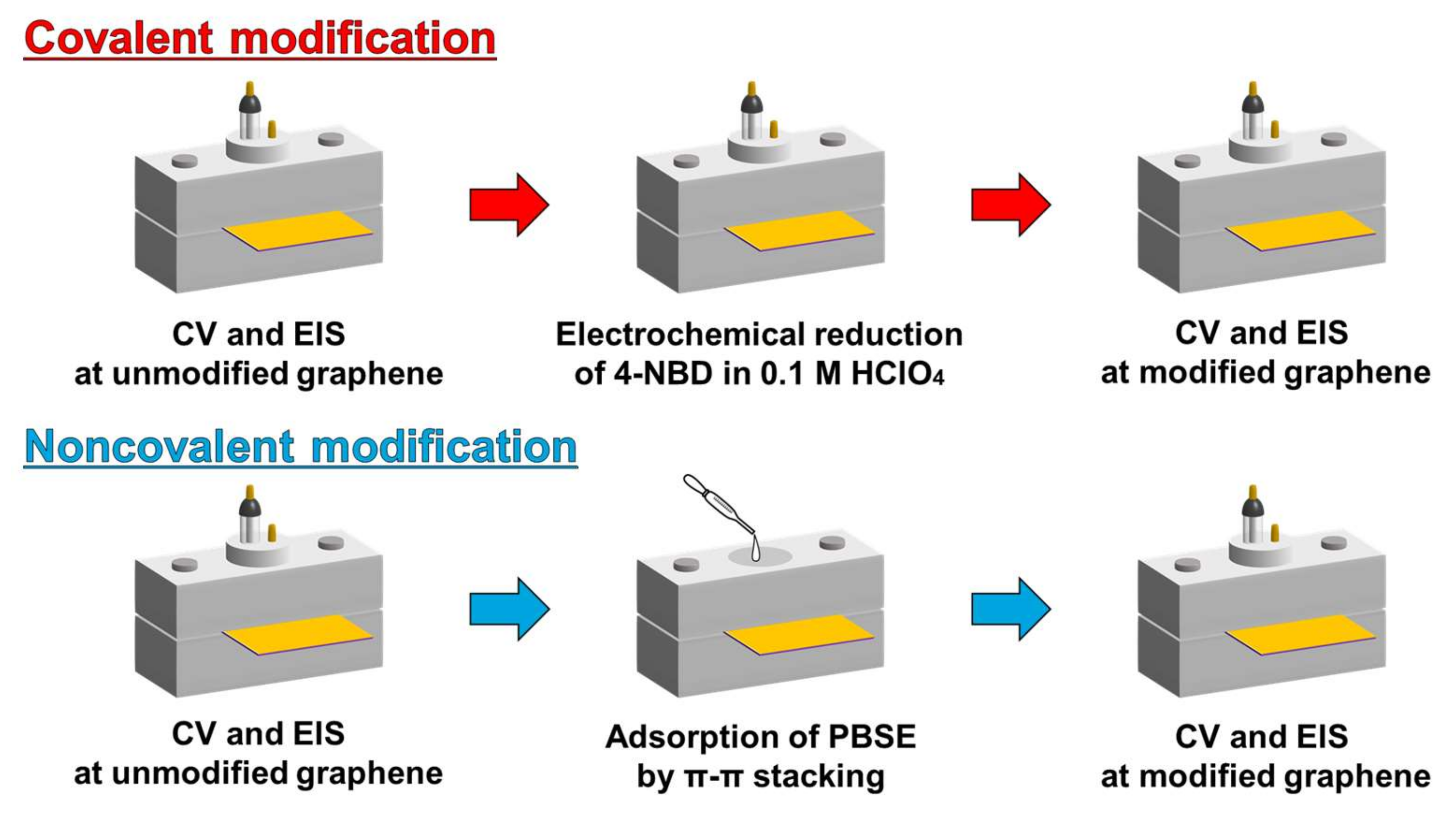
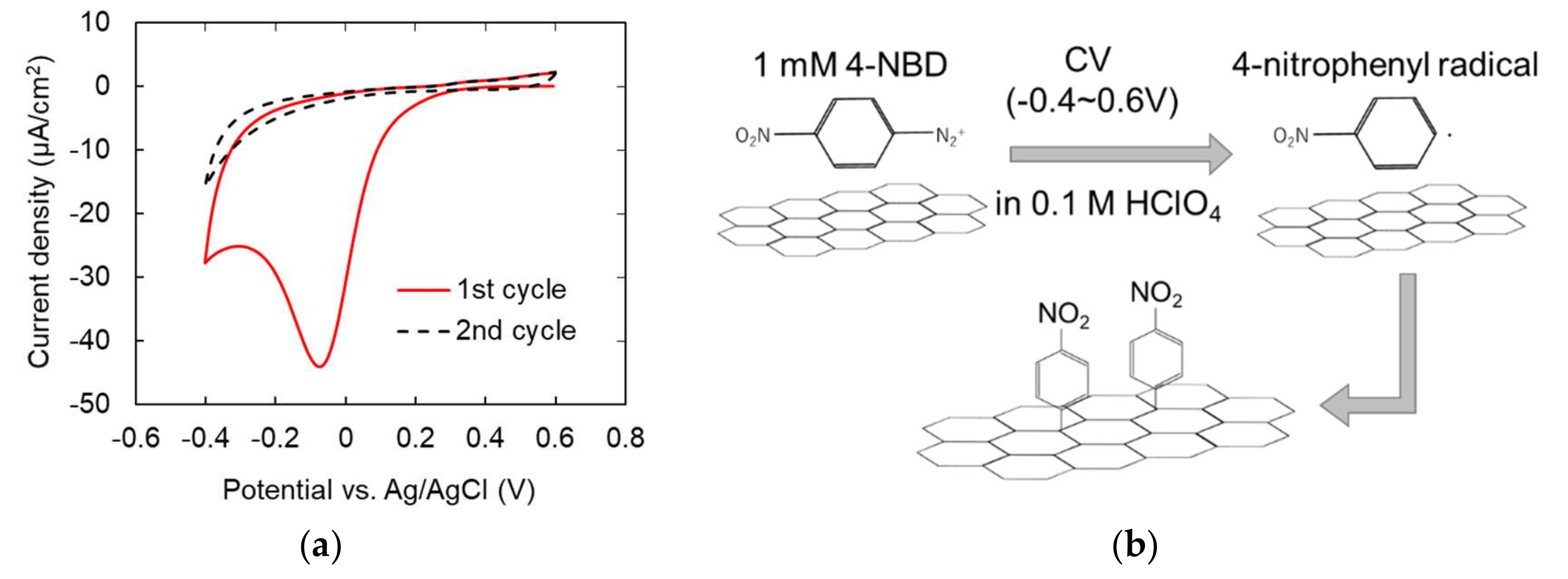
2.4. Surface Modification of Gaphene Eectrode
3. Results and Discussion
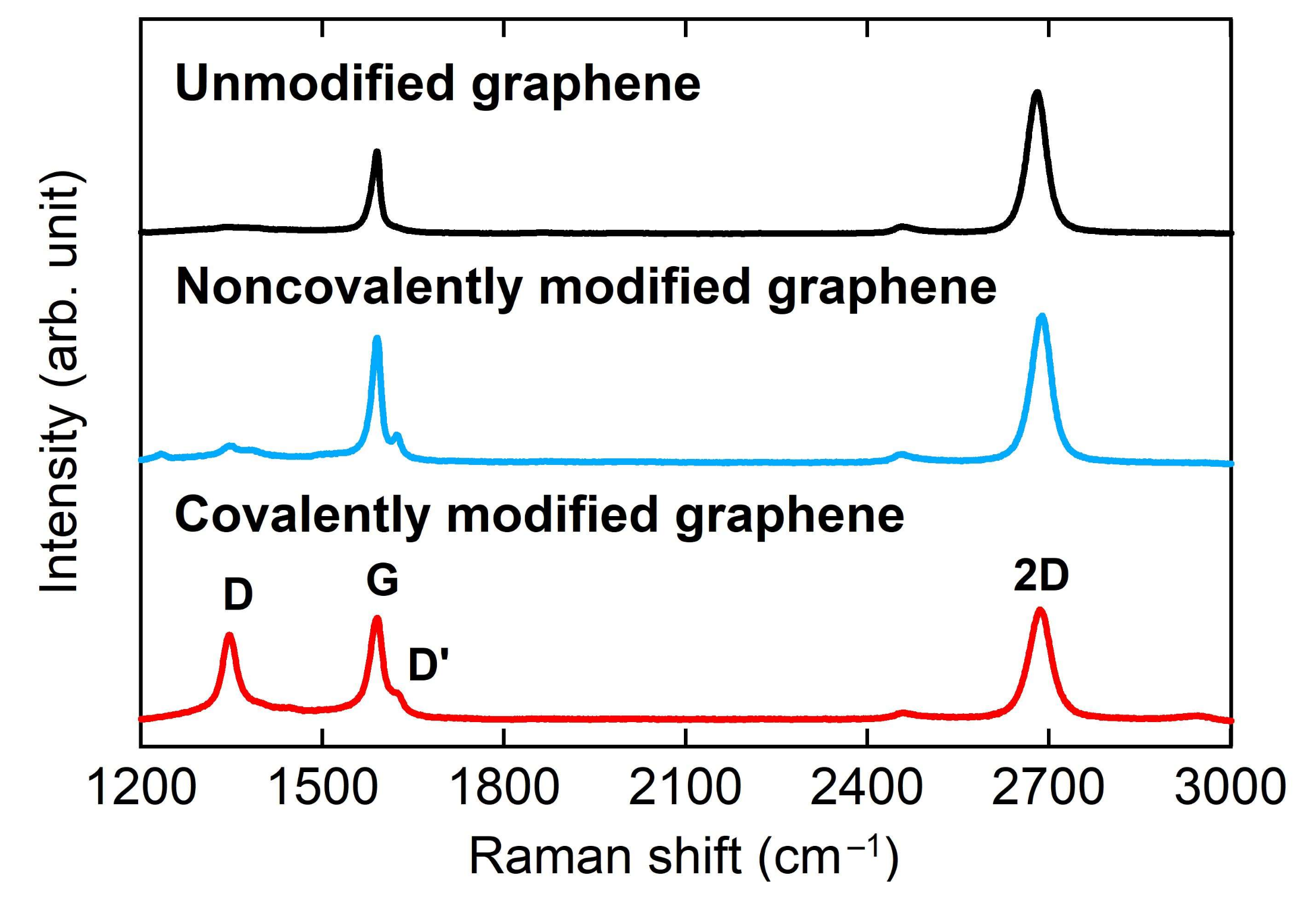
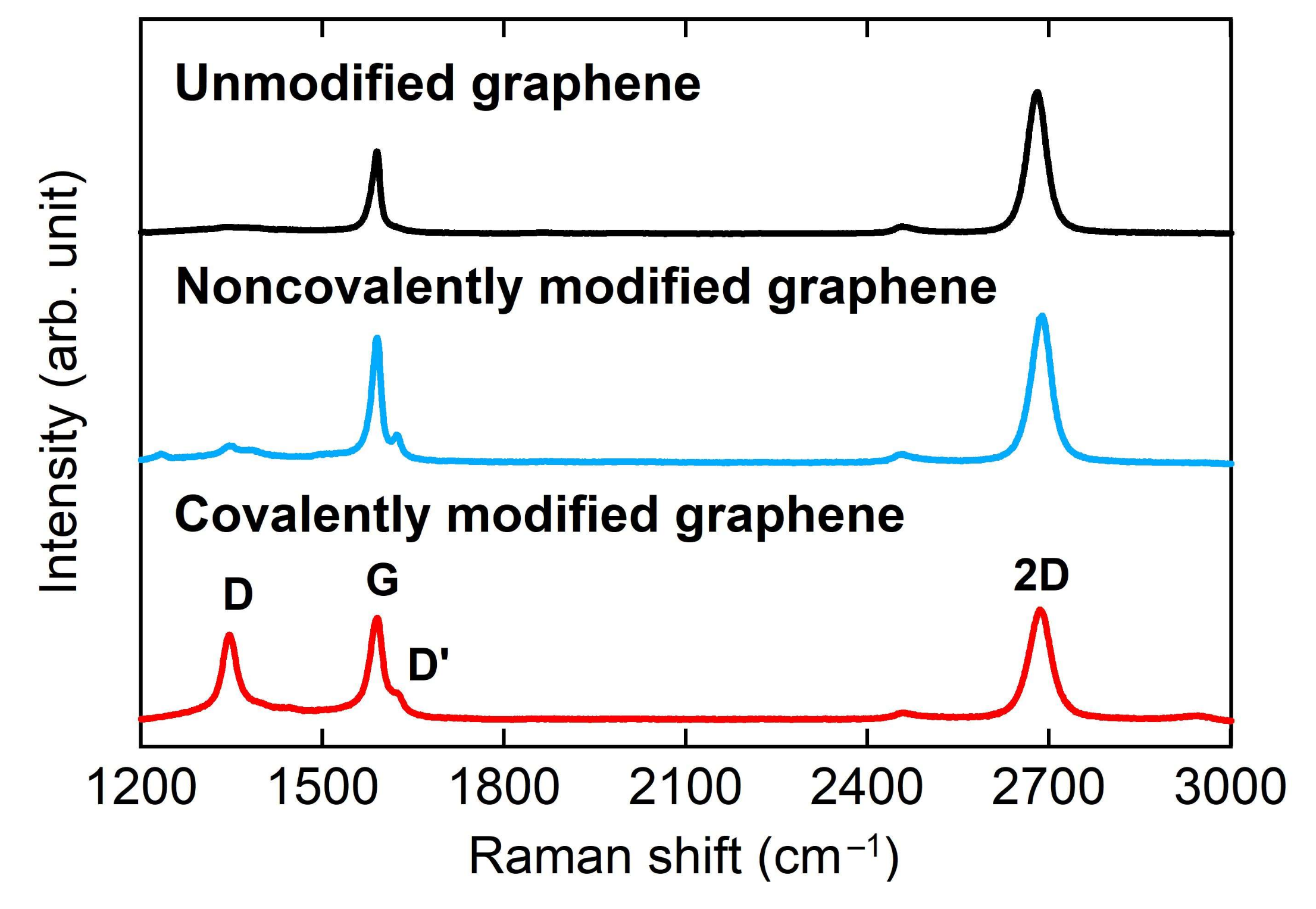
3.1. Characterizaiton of Graphene Electrode
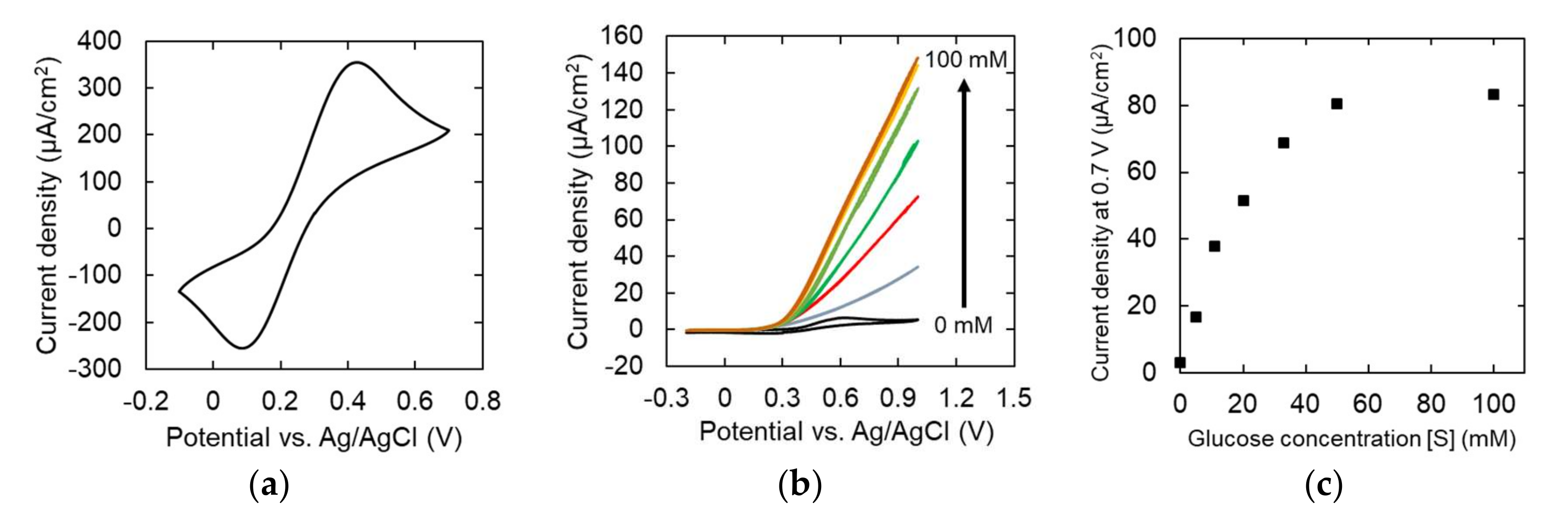
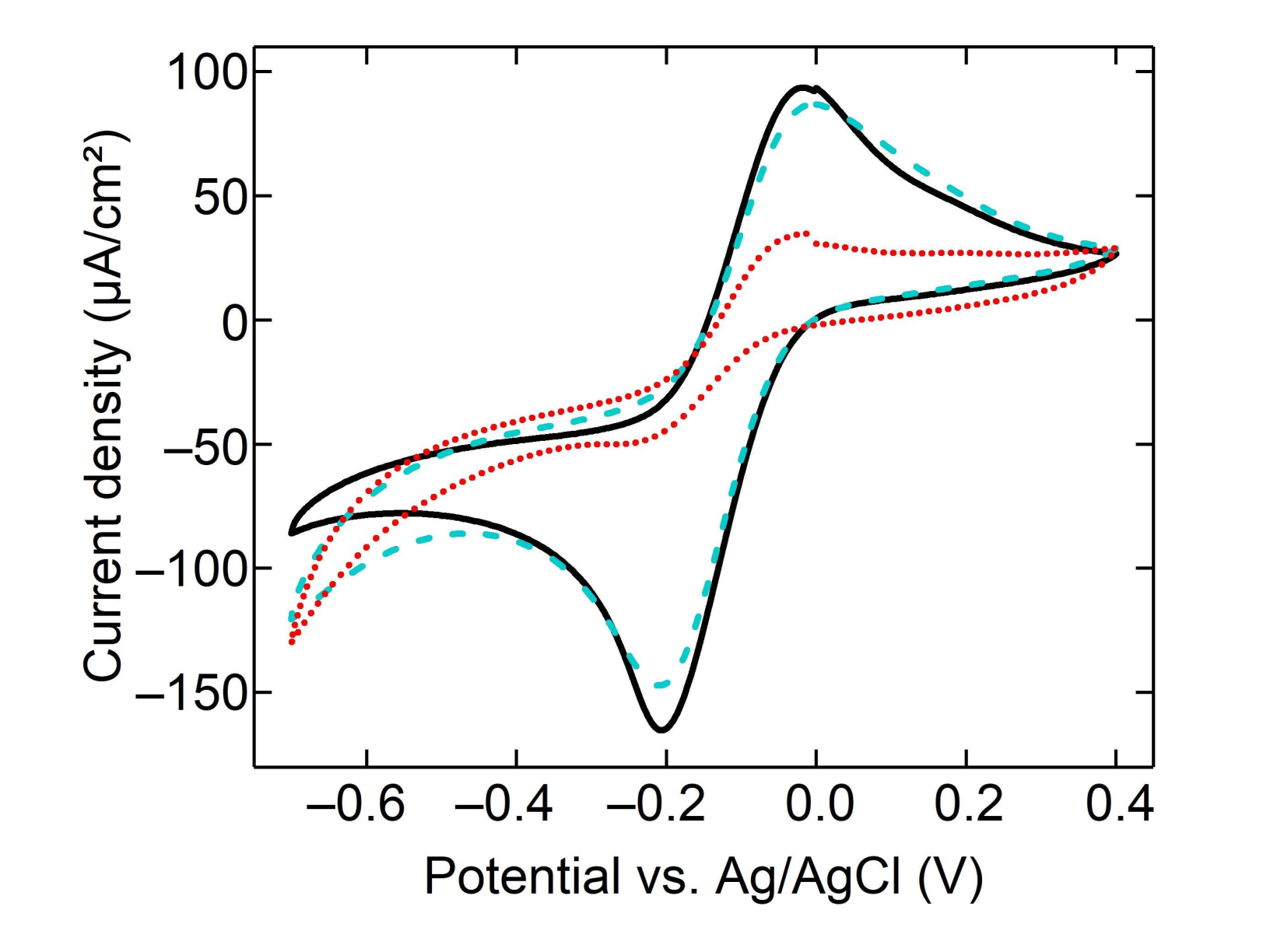
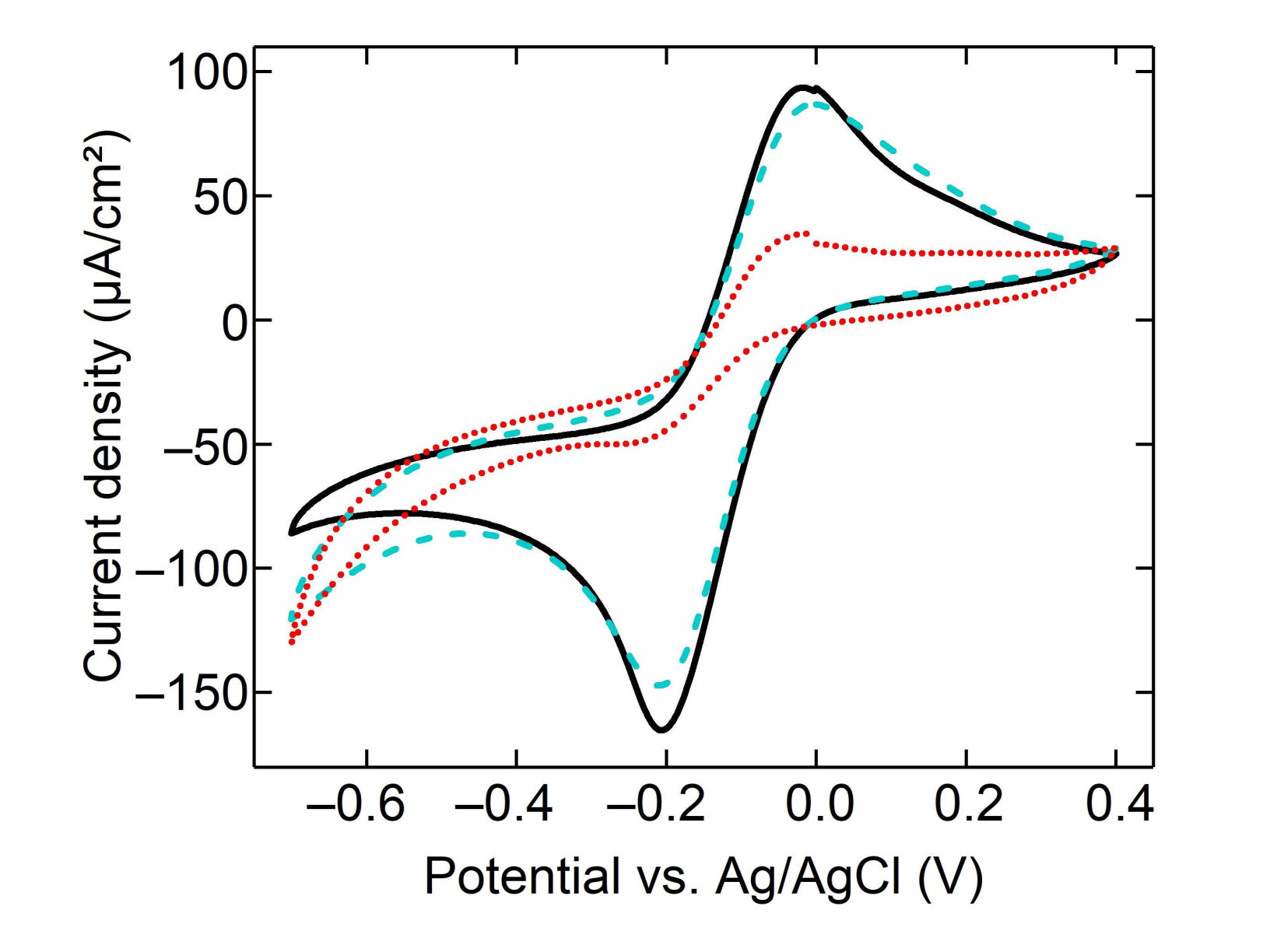
3.2. Electrochemical Behavior of Monolayer Gaphene Eectrode
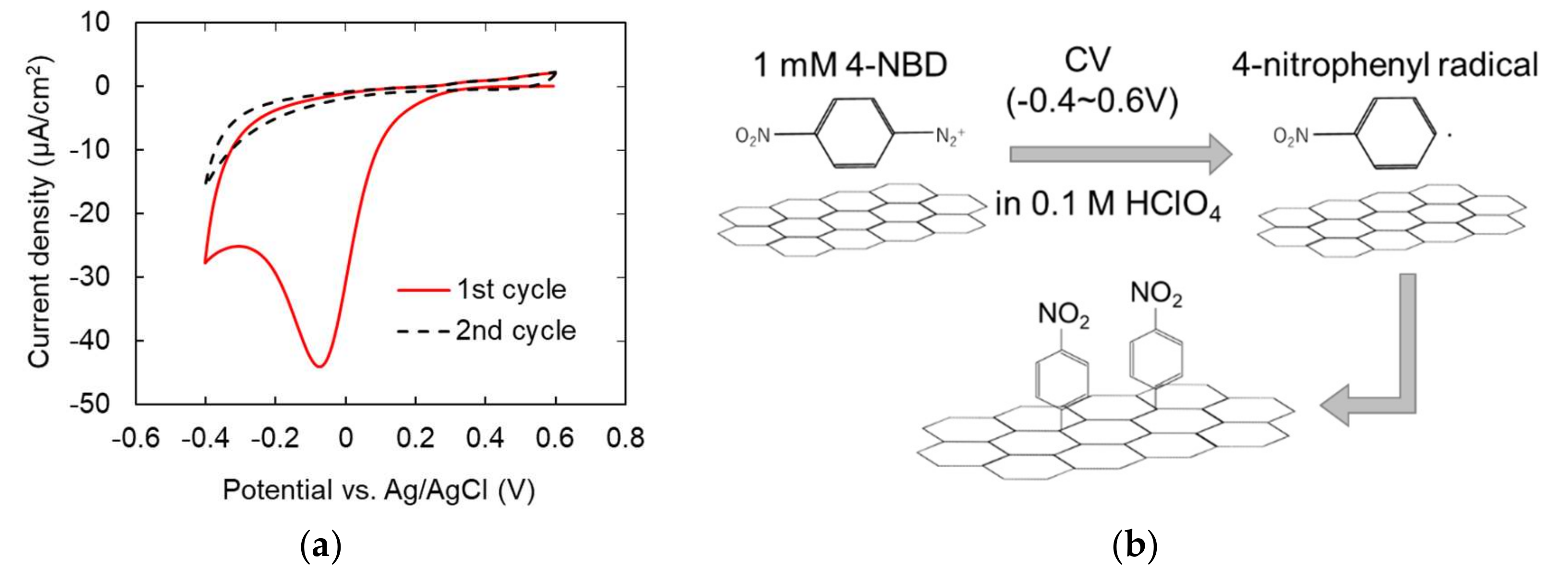
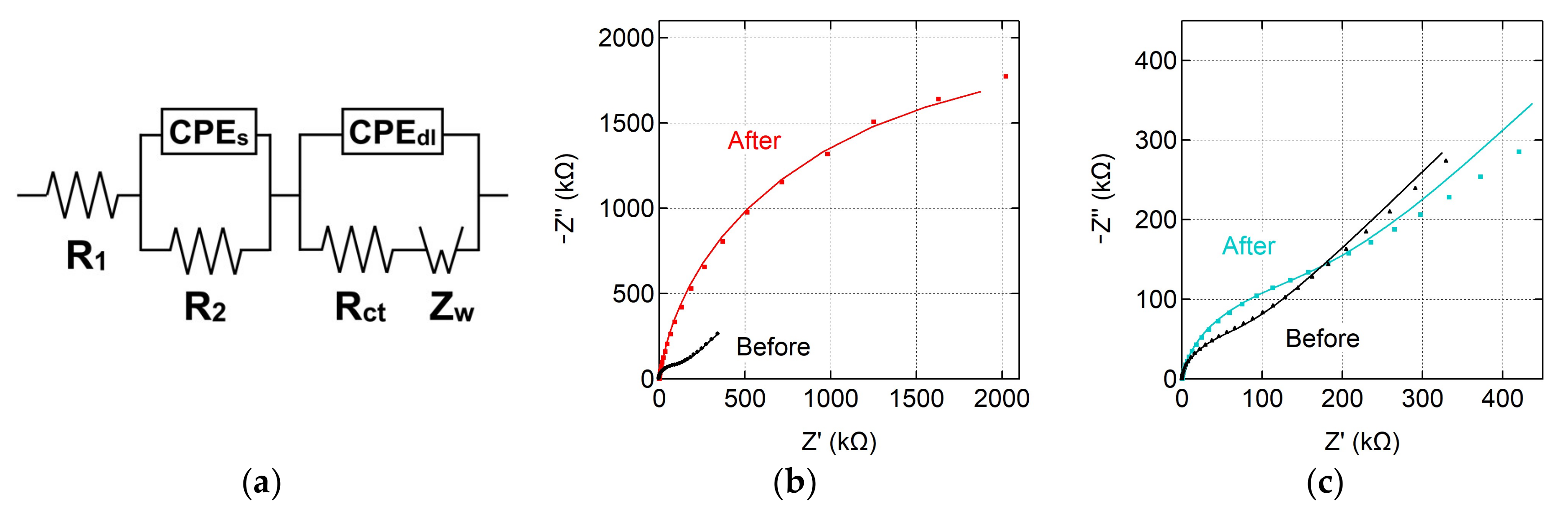
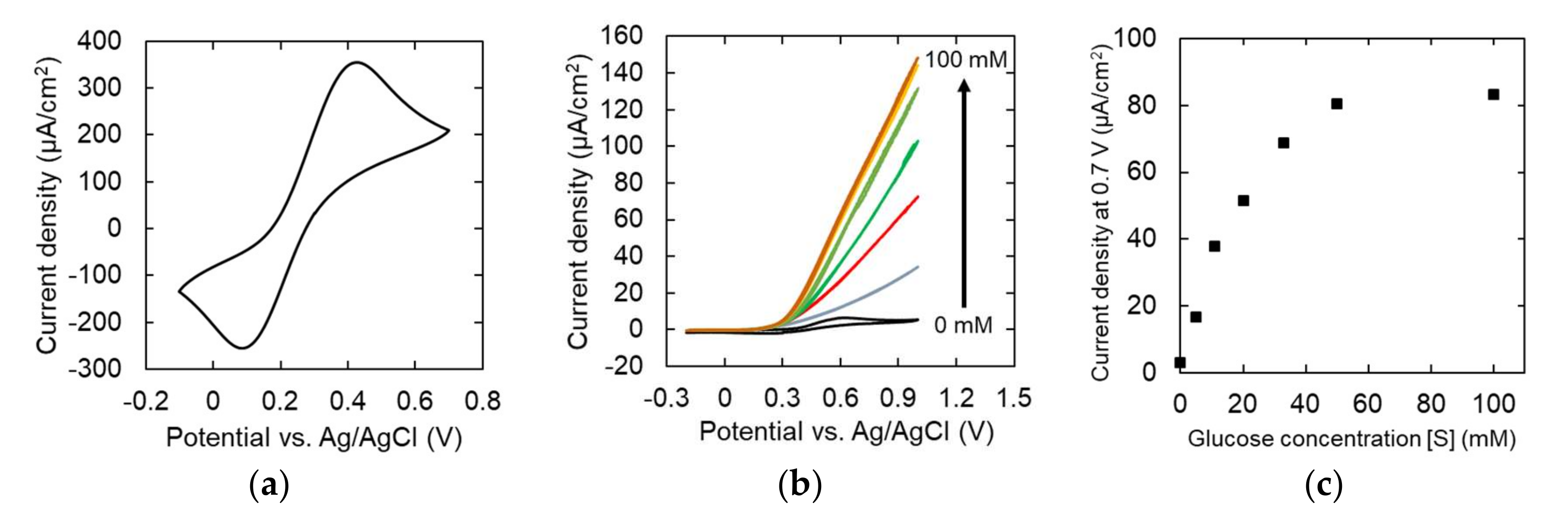
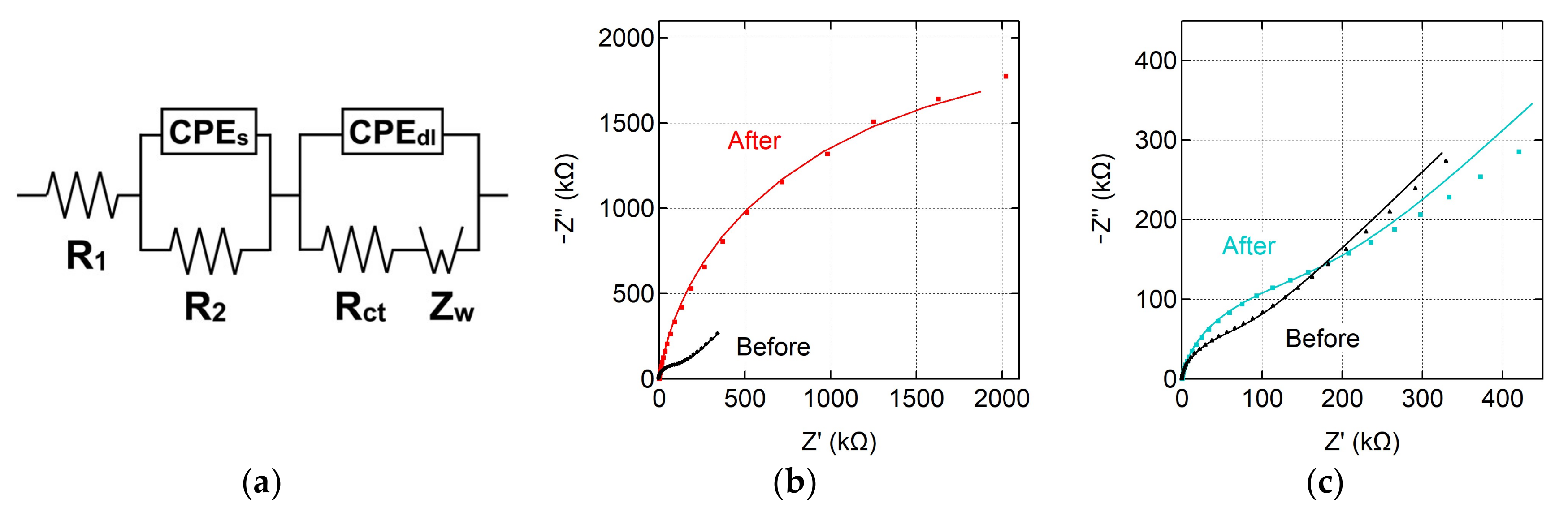
3.3. Characterization of Modified Graphene Electrodes
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Larminie, J.; Dicks, A. Fuel Cell Systems Explained; John Wiley & Sons, Ltd.: England, UK, 2003; ISBN 9781118878330. [Google Scholar]
- Cao, L. Immobilised enzymes: Science or art? Curr. Opin. Chem. Biol. 2005, 9, 217–226. [Google Scholar] [CrossRef]
- Boland, S.; Jenkins, P.; Kavanagh, P.; Leech, D. Biocatalytic fuel cells: A comparison of surface pre-treatments for anchoring biocatalytic redox films on electrode surfaces. J. Electroanal. Chem. 2009, 626, 111–115. [Google Scholar] [CrossRef]
- Cracknell, J.A.; Vincent, K.A.; Armstrong, F.A. Enzymes as working or inspirational electrocatalysts for fuel cells and electrolysis. Chem. Rev. 2008, 108, 2439–2461. [Google Scholar] [CrossRef] [PubMed]
- Barton, S.C.; Gallaway, J.; Atanassov, P. Enzymatic biofuel cells for implantable and microscale devices. Chem. Rev. 2004, 104, 4867–4886. [Google Scholar] [CrossRef] [PubMed]
- Gallaway, J.W.; Calabrese Barton, S.A. Effect of redox polymer synthesis on the performance of a mediated laccase oxygen cathode. J. Electroanal. Chem. 2009, 626, 149–155. [Google Scholar] [CrossRef]
- Shim, J.; Kim, G.Y.; Moon, S.H. Covalent co-immobilization of glucose oxidase and ferrocenedicarboxylic acid for an enzymatic biofuel cell. J. Electroanal. Chem. 2011, 653, 14–20. [Google Scholar] [CrossRef]
- Halámková, L.; Halámek, J.; Bocharova, V.; Szczupak, A.; Alfonta, L.; Katz, E. Implanted biofuel cell operating in a living snail. J. Am. Chem. Soc. 2012, 134, 5040–5043. [Google Scholar] [CrossRef]
- Hoshi, K.; Muramatsu, K.; Sumi, H.; Nishioka, Y. Graphene-coated carbon fiber cloth for flexible electrodes of glucose fuel cells. Jpn. J. Appl. Phys. 2016, 55, 02BE05. [Google Scholar] [CrossRef]
- Liu, C.; Alwarappan, S.; Chen, Z.; Kong, X.; Li, C.Z. Membraneless enzymatic biofuel cells based on graphene nanosheets. Biosens. Bioelectron. 2010, 25, 1829–1833. [Google Scholar] [CrossRef]
- Zebda, A.; Gondran, C.; Le Goff, A.; Holzinger, M.; Cinquin, P.; Cosnier, S. Mediatorless high-power glucose biofuel cells based on compressed carbon nanotube-enzyme electrodes. Nat. Commun. 2011, 2, 370. [Google Scholar] [CrossRef] [Green Version]
- Holzinger, M.; Le Goff, A.; Cosnier, S. Carbon nanotube/enzyme biofuel cells. Electrochim. Acta 2012, 82, 179–190. [Google Scholar] [CrossRef]
- Das, B.; Voggu, R.; Rout, C.S.; Rao, C.N.R. Changes in the electronic structure and properties of graphene induced by molecular charge-transfer. Chem. Commun. 2008, 44, 5155–5157. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, C.; Du, D.; Lin, Y. Graphene-like 2D nanomaterial-based biointerfaces for biosensing applications. Biosens. Bioelectron. 2017, 89, 43–55. [Google Scholar] [CrossRef]
- Reina, A.; Jia, X.; Ho, J.; Nezich, D.; Son, H.; Bulovic, V.; Dresselhaus, M.S.; Jing, K. Large area, few-layer graphene films on arbitrary substrates by chemical vapor deposition. Nano Lett. 2009, 9, 30–35. [Google Scholar] [CrossRef] [PubMed]
- Geim AK, N.K. The rise of graphene. Chem. Rev. 2017, 114, E9793–E9801. [Google Scholar]
- Ogawa, Y.; Komatsu, K.; Kawahara, K.; Tsuji, M.; Tsukagoshi, K.; Ago, H. Structure and transport properties of the interface between CVD-grown graphene domains. Nanoscale 2014, 6, 7288–7294. [Google Scholar] [CrossRef]
- Novoselov, K.S. Electric Field Effect in Atomically Thin Carbon Films. Science 2004, 306, 666–669. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kang, J.; Shin, D.; Bae, S.; Hong, B.H. Graphene transfer: Key for applications. Nanoscale 2012, 4, 5527–5537. [Google Scholar] [CrossRef]
- Li, X.; Zhu, Y.; Cai, W.; Borysiak, M.; Han, B.; Chen, D.; Piner, R.D.; Colombo, L.; Ruoff, R.S. Transfer of Large-Area Graphene Films for High-Performance Transparent Conductive Electrodes. Nano Lett. 2009, 9, 4359–4363. [Google Scholar] [CrossRef]
- Kosuga, S.; Suga, K.; Suga, R.; Watanabe, T.; Hashimoto, O.; Koh, S. Radiation properties of graphene-based optically transparent dipole antenna. Microw. Opt. Technol. Lett. 2018, 60, 2992–2998. [Google Scholar] [CrossRef]
- Greenwood, J.; Phan, T.H.; Fujita, Y.; Li, Z.; Ivasenko, O.; Vanderlinden, W.; Van Gorp, H.; Frederickx, W.; Lu, G.; Tahara, K.; et al. Covalent modification of graphene and graphite using diazonium chemistry: Tunable grafting and nanomanipulation. ACS Nano 2015, 9, 5520–5535. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, R.J.; Zhang, Y.; Wang, D.; Dai, H. Noncovalent Sidewall Functionalization of Single-Walled Carbon Nanotubes for Protein Immobilization. J. Am. Chem. Soc. 2001, 123, 3838–3839. [Google Scholar] [CrossRef] [PubMed]
- Ebrish, M.A.; Olson, E.J.; Koester, S.J. Effect of noncovalent basal plane functionalization on the quantum capacitance in graphene. ACS Appl. Mater. Interfaces 2014, 6, 10296–10303. [Google Scholar] [CrossRef]
- Blake, P.; Hill, E.W.; Castro Neto, A.H.; Novoselov, K.S.; Jiang, D.; Yang, R.; Booth, T.J.; Geim, A.K. Making graphene visible. Appl. Phys. Lett. 2007, 91, 063124. [Google Scholar] [CrossRef] [Green Version]
- Ferrari, A.C.; Basko, D.M. Raman spectroscopy as a versatile tool for studying the properties of graphene. Nat. Nanotechnol. 2013, 8, 235–246. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rabti, A.; Raouafi, N.; Merkoçi, A. Bio(Sensing) devices based on ferrocene–functionalized graphene and carbon nanotubes. Carbon N. Y. 2016, 108, 481–514. [Google Scholar] [CrossRef]
- Kirkman, P.M.; Güell, A.G.; Cuharuc, A.S.; Unwin, P.R. Spatial and temporal control of the diazonium modification of sp 2 carbon surfaces. J. Am. Chem. Soc. 2014, 136, 36–39. [Google Scholar] [CrossRef]
- Baranton, S.; Bélanger, D. Electrochemical derivatization of carbon surface by reduction of in situ generated diazonium cations. J. Phys. Chem. B 2005, 109, 24401–24410. [Google Scholar] [CrossRef]
- Ferrari, A.C.; Meyer, J.C.; Scardaci, V.; Casiraghi, C.; Lazzeri, M.; Mauri, F.; Piscanec, S.; Jiang, D.; Novoselov, K.S.; Roth, S.; et al. Raman spectrum of graphene and graphene layers. Phys. Rev. Lett. 2006, 97, 187401. [Google Scholar] [CrossRef] [Green Version]
- Malard, L.M.; Pimenta, M.A.; Dresselhaus, G.; Dresselhaus, M.S. Raman spectroscopy in graphene. Phys. Rep. 2009, 473, 51–87. [Google Scholar] [CrossRef]
- Wu, G.; Tang, X.; Meyyappan, M.; Lai, K.W.C. Doping effects of surface functionalization on graphene with aromatic molecule and organic solvents. Appl. Surf. Sci. 2017, 425, 713–721. [Google Scholar] [CrossRef]
- Sharma, R.; Baik, J.H.; Perera, C.J.; Strano, M.S. Anomalously large reactivity of single graphene layers and edges toward electron transfer chemistries. Nano Lett. 2010, 10, 398–405. [Google Scholar] [CrossRef] [PubMed]
- Nicholson, R.S. Theory and Application of Cyclic Voltammetry for Measurement of Electrode Reaction Kinetics. Anal. Chem. 1965, 37, 1351–1355. [Google Scholar] [CrossRef]
- Lavagnini, I.; Antiochia, R.; Magno, F. An extended method for the practical evaluation of the standard rate constant from cyclic voltammetric data. Electroanalysis 2004, 16, 505–506. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | R₁ (Ω) | R₂ (Ω) | CPEs | CPEdl | Rct (kΩ) | Zw (µΩ∙s½) | χ2 | ||
|---|---|---|---|---|---|---|---|---|---|
| Q₁ (µF∙s(n−1)) | n₁ | Q₂ (µF∙s(n−1)) | n₂ | ||||||
| Before covalent modification | 100 | 780 | 1.25 | 0.69 | 1.90 | 0.99 | 104 | 3.47 | 0.003 |
| After covalent modification | 107 | 800 | 4.64 | 0.61 | 3.18 | 0.92 | 2753 | 1.13 | 0.025 |
| Before noncovalent modification | 231 | 490 | 1.45 | 0.75 | 2.00 | 0.97 | 67 | 3.18 | 0.010 |
| After noncovalent modification | 271 | 387 | 0.50 | 0.85 | 2.69 | 0.93 | 158 | 2.66 | 0.044 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Miki, K.; Watanabe, T.; Koh, S. Electrochemical Characterization of CVD-Grown Graphene for Designing Electrode/Biomolecule Interfaces. Crystals 2020, 10, 241. https://doi.org/10.3390/cryst10040241
Miki K, Watanabe T, Koh S. Electrochemical Characterization of CVD-Grown Graphene for Designing Electrode/Biomolecule Interfaces. Crystals. 2020; 10(4):241. https://doi.org/10.3390/cryst10040241
Chicago/Turabian StyleMiki, Keishu, Takeshi Watanabe, and Shinji Koh. 2020. "Electrochemical Characterization of CVD-Grown Graphene for Designing Electrode/Biomolecule Interfaces" Crystals 10, no. 4: 241. https://doi.org/10.3390/cryst10040241
APA StyleMiki, K., Watanabe, T., & Koh, S. (2020). Electrochemical Characterization of CVD-Grown Graphene for Designing Electrode/Biomolecule Interfaces. Crystals, 10(4), 241. https://doi.org/10.3390/cryst10040241