Structural Refinement and Density Functional Theory Study of Synthetic Ge-Akaganéite (β-FeOOH)


Abstract
1. Introduction
2. Materials and Methods
2.1. Experimental Synthesis and Characterization
2.2. X-Ray Diffraction and Rietveld Refinement
2.3. Computational Approach
3. Results and Discussion
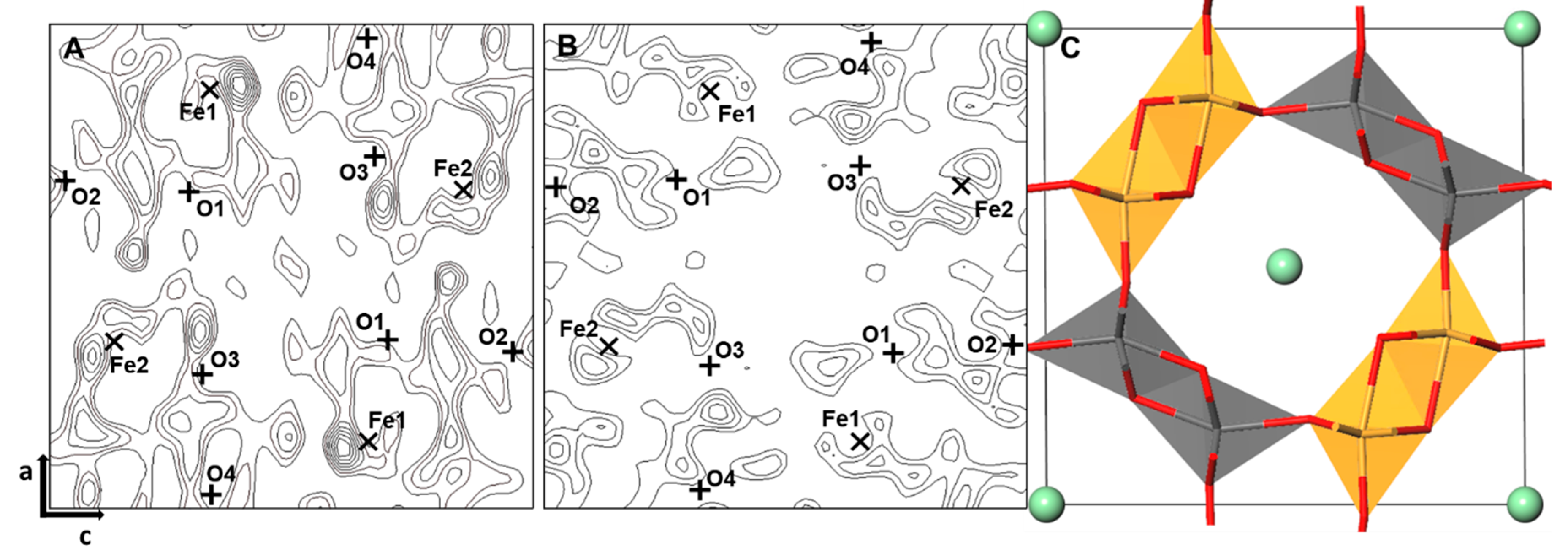
3.1. Finding the Ge Sites Using DELF Contour Map Analysis
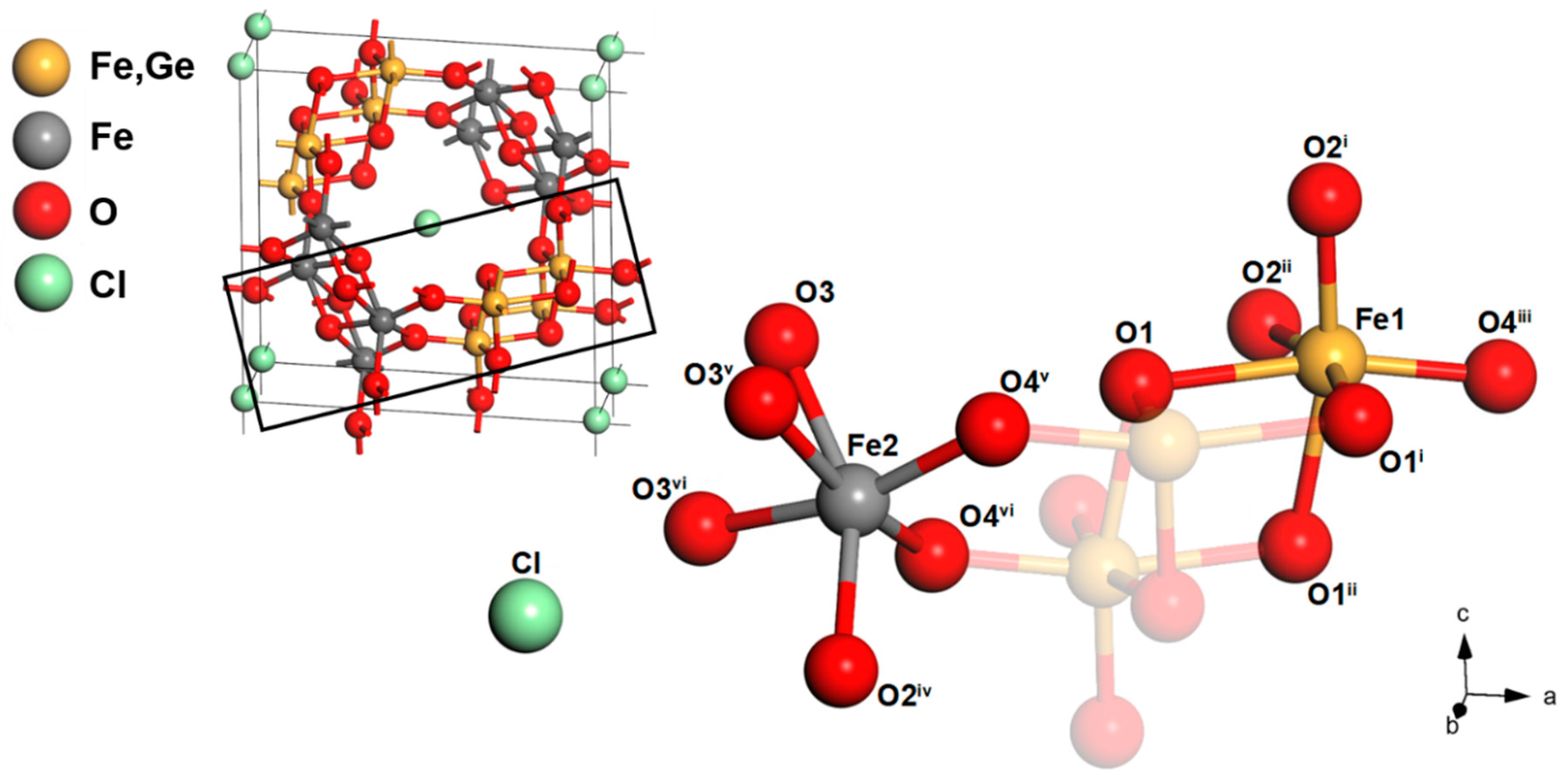
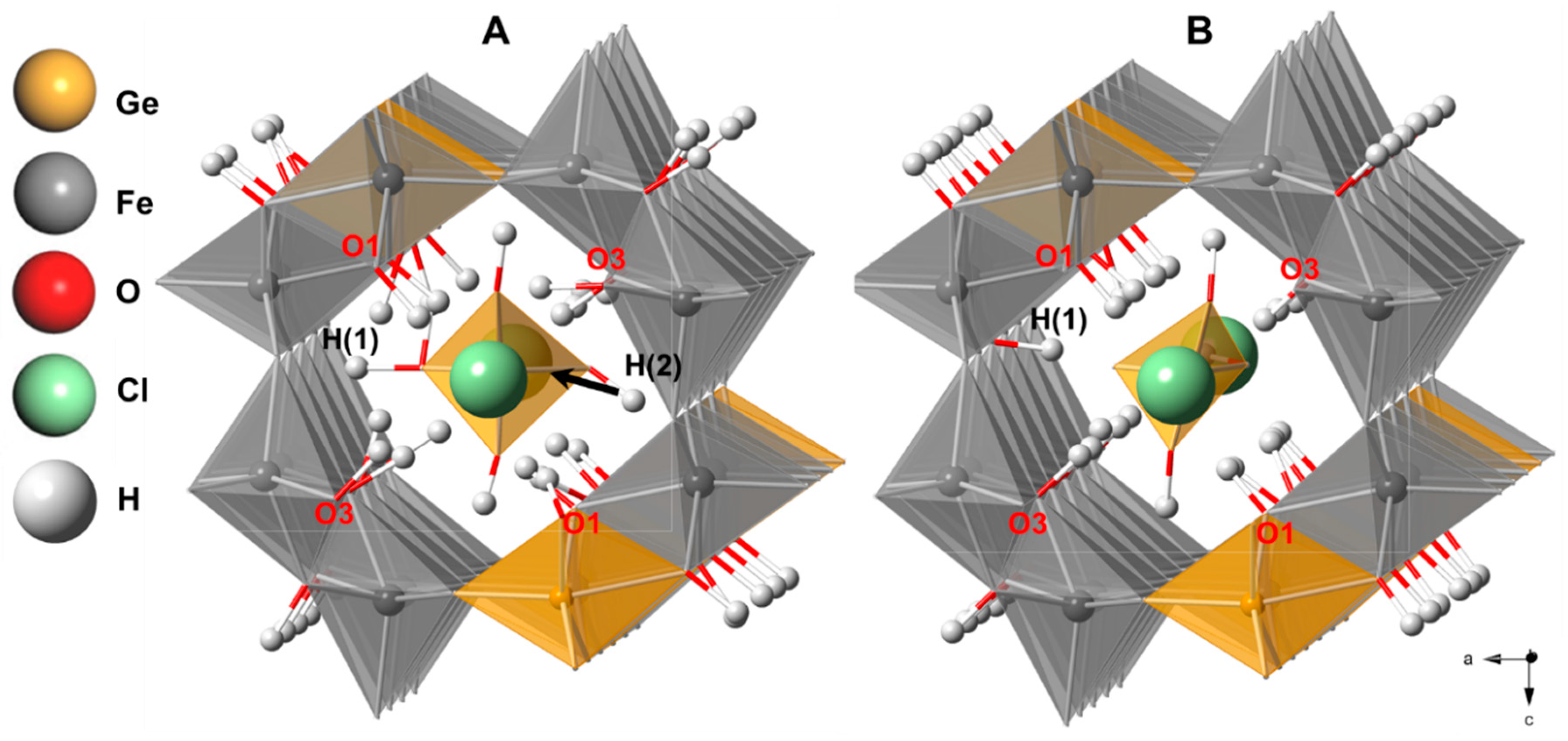
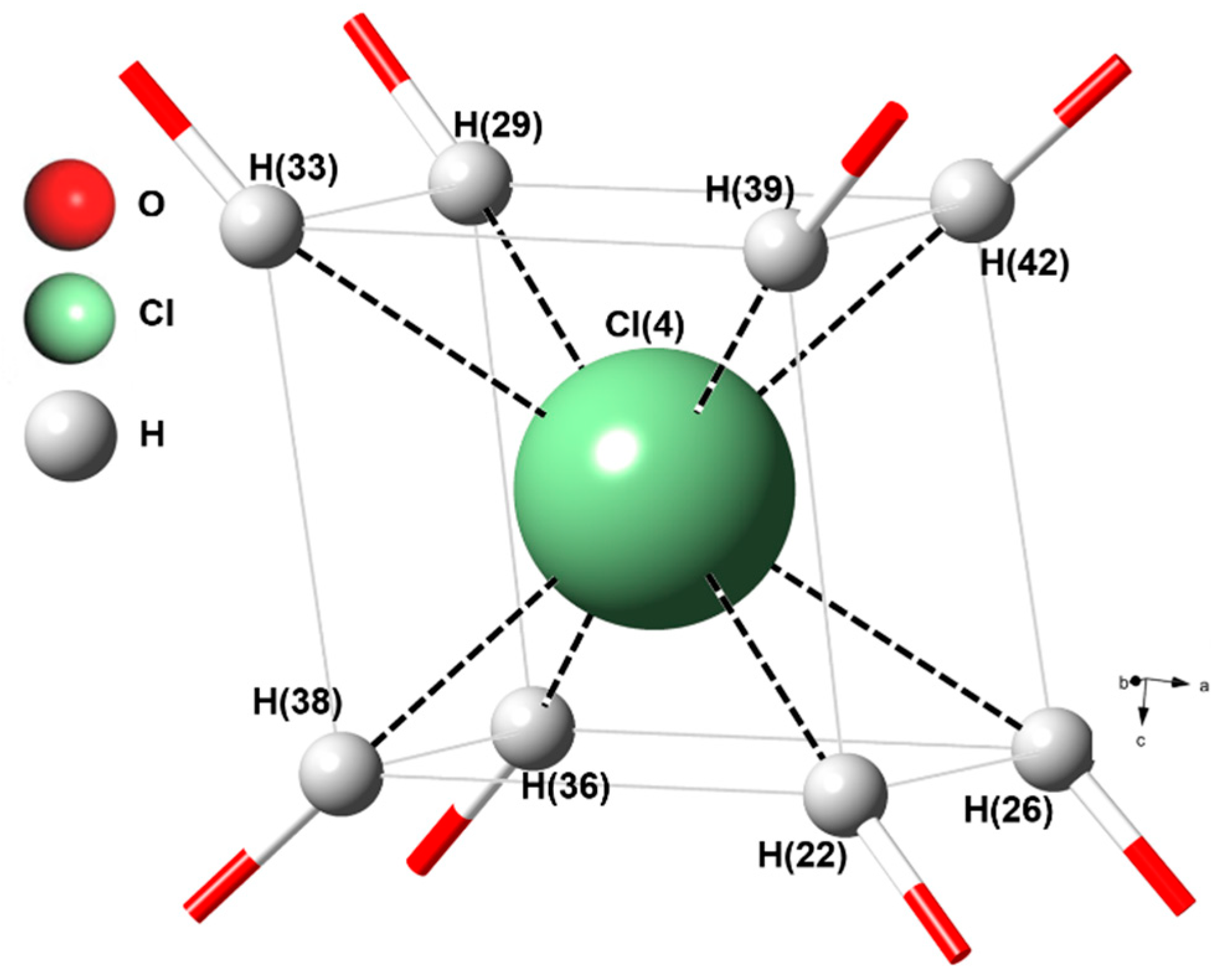
3.2. Structure of Ge-Akaganéite
3.3. Model Verification Using the Enthalpy of Formation
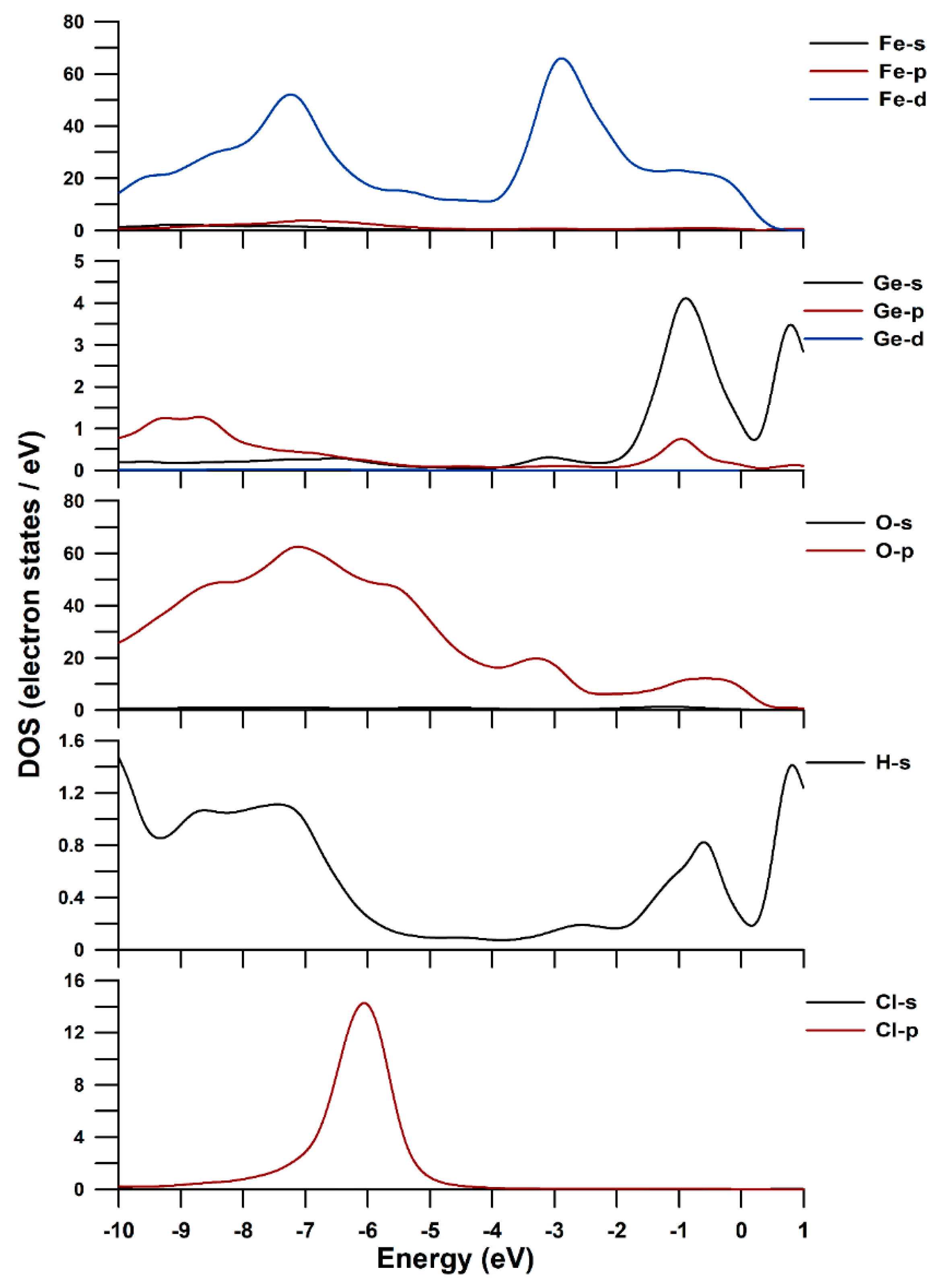
3.4. DFT-D Calculation and DOS of Ge-Akaganéite
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Holm, N.G.; Dowler, M.J.; Wadsten, T.; Arrhenius, G. β-FeOOH · Cln (akaganéite) and Fe1−xO (wüstite) in hot brine from the atlantis ii deep (red sea) and the uptake of amino acids by synthetic β-FeOOH · Cln. Geochim. Cosmochim. Acta 1983, 47, 1465–1470. [Google Scholar] [CrossRef]
- Buchwald, V.F.; Clarke, J.; Roy, S. Corrosion of Fe-Ni alloys by Cl-containing akaganeite (β-FeOOH): The antarctic meteorite case. Am. Mineral. 1989, 74, 656–667. [Google Scholar]
- Post, J.E.; Buchwald, V.F. Crystal-structure refinement of akaganeite. Am. Mineral. 1991, 76, 272–277. [Google Scholar]
- Post, J.E.; Heaney, P.J.; Dreele, R.B.V.; Hanson, J.C. Neutron and temperature-resolved synchrotron X-ray Powder Diffraction study of akaganéite. Am. Mineral. 2003, 88, 782–788. [Google Scholar] [CrossRef]
- Ståhl, K.; Nielsen, K.; Jiang, J.; Lebech, B.; Hanson, J.C.; Norby, P.; Van Lanschot, J. On the akaganéite crystal structure, phase transformations and possible role in post-excavational corrosion of iron artifacts. Corros. Sci. 2003, 45, 2563–2575. [Google Scholar] [CrossRef]
- Piao, Y.; Kim, J.; Na, H.B.; Kim, D.; Baek, J.S.; Ko, M.K.; Lee, J.H.; Shokouhimehr, M.; Hyeon, T. Wrap-bake-peel process for nanostructural transformation from β-FeOOH nanorods to biocompatible iron oxide nanocapsules. Nat. Mater. 2008, 7, 242. [Google Scholar] [CrossRef]
- Chaudhari, N.K.; Yu, J.S. Size control synthesis of uniform β-FeOOH to high coercive field porous magnetic α-Fe2O3 nanorods. J. Phys. Chem. C 2008, 112, 19957–19962. [Google Scholar] [CrossRef]
- Peterson, K.M.; Heaney, P.J.; Post, J.E. A kinetic analysis of the transformation from akaganeite to hematite: An in situ time-resolved X-ray diffraction study. Chem. Geol. 2016, 444, 27–36. [Google Scholar] [CrossRef]
- Harijan, D.K.L.; Chandra, V. Akaganeite nanorods decorated graphene oxide sheets for removal and recovery of aqueous phosphate. J. Water Proc. Eng. 2017, 19, 120–125. [Google Scholar] [CrossRef]
- Zhang, Y.X.; Jia, Y. A facile solution approach for the synthesis of akaganéite (β-FeOOH) nanorods and their ion-exchange mechanism toward As(V) ions. Appl. Surf. Sci. 2014, 290, 102–106. [Google Scholar] [CrossRef]
- Cho, M.K.; Jo, J.H.; Choi, J.U.; Kim, J.; Yashiro, H.; Yuan, S.; Shi, L.; Sun, Y.K.; Myung, S.T. Tunnel-type β-FeOOH cathode material for high rate sodium storage via a new conversion reaction. Nano Energy 2017, 41, 687–696. [Google Scholar] [CrossRef]
- Wu, Q.; Zhao, R.; Liu, W.; Zhang, X.; Shen, X.; Li, W.; Diao, G.; Chen, M. In-depth nanocrystallization enhanced Li-ions batteries performance with nitrogen-doped carbon coated Fe3O4 yolk− shell nanocapsules. J. Power Sources 2017, 344, 74–84. [Google Scholar] [CrossRef]
- Wang, D.; Song, C.; Zhao, Y.; Yang, M. Synthesis and characterization of monodisperse iron oxides microspheres. J. Phys. Chem. C 2008, 112, 12710–12715. [Google Scholar] [CrossRef]
- Jolivet, J.P.; Chanéac, C.; Tronc, E. Iron oxide chemistry. From molecular clusters to extended solid networks. Chem. Commun. 2004, 481–483. [Google Scholar]
- Wang, X.; Chen, X.; Gao, L.; Zheng, H.; Ji, M.; Tang, C.; Shen, T.; Zhang, Z. Synthesis of β-FeOOH and α-Fe2O3 nanorods and electrochemical properties of β-FeOOH. J. Mater. Chem. 2004, 14, 905–907. [Google Scholar] [CrossRef]
- Song, Y.; Bac, B.H.; Lee, Y.B.; Kim, M.H.; Kang, I.M. Highly ordered Ge-incorporated akaganeite (β-FeOOH): A tunnel-type nanorod. CrystEngComm 2011, 13, 287–292. [Google Scholar] [CrossRef]
- Toby, B.H. Expgui, a graphical user interface for gsas. J. Appl. Crystallogr. 2001, 34, 210–213. [Google Scholar] [CrossRef]
- Larson, A.C.; Von Dreele, R.B. General Structure Analysis System (GSAS); Los Alamos National Laboratory Report; LAUR: USA, 2004; pp. 86–748. Available online: https://11bm.xray.aps.anl.gov/documents/GSASManual.pdf (accessed on 25 March 2020).
- Shannon, R.D. Revised effective ionic radii and systematic studies of interatomic distances in halides and chalcogenides. Acta Crystallogr. Sect. A Cryst. Phys. Diffr. Theor. Gen. Crystallogr. 1976, 32, 751–767. [Google Scholar] [CrossRef]
- Segall, M.; Lindan, P.J.; Probert, M.A.; Pickard, C.; Hasnip, P.J.; Clark, S.; Payne, M. First-principles simulation: Ideas, illustrations and the castep code. J. Phys. Condens. Matter 2002, 14, 2717. [Google Scholar] [CrossRef]
- Clark, S.J.; Segall, M.D.; Pickard, C.J.; Hasnip, P.J.; Probert, M.I.; Refson, K.; Payne, M.C. First principles methods using castep. Z. Kristallogr.-Cryst. Mater. 2005, 220, 567–570. [Google Scholar] [CrossRef]
- Hohenberg, P.; Kohn, W. Inhomogeneous electron gas. Phys. Rev. 1964, 136, 864–871. [Google Scholar] [CrossRef]
- Kohn, W.; Sham, L.J. Self-consistent equations including exchange and correlation effects. Phys. Rev. 1965, 140, A1133. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 1996, 77, 3865. [Google Scholar] [CrossRef] [PubMed]
- Kaltak, M.; Fernández-Serra, M.; Hybertsen, M.S. Charge localization and ordering in A2Mn8O16 hollandite group oxides: Impact of density functional theory approaches. Phys. Rev. Mater. 2017, 1, 075401. [Google Scholar] [CrossRef]
- Xu, Y.; Wen, Y.; Grote, R.; Amoroso, J.; Nickles, L.S.; Brinkman, K.S. A-site compositional effects in Ga-doped hollandite materials of the form BaxCsyGa2x + yTi8 − 2x − yO16: Implications for Cs immobilization in crystalline ceramic waste forms. Sci. Rep. 2016, 6, 27412. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Feygenson, M.; Page, K.; Nickles, L.S.; Brinkman, K.S. Structural evolution in hollandite solid solutions across the a-site compositional range from Ba1.33Ga2.66Ti5.34O16 to Cs1.33Ga1.33Ti6.67O16. J. Am. Ceram. Soc. 2016, 99, 4100–4106. [Google Scholar] [CrossRef]
- Tkatchenko, A.; Scheffler, M. Accurate molecular van der waals interactions from ground-state electron density and free-atom reference data. Phys. Rev. Lett. 2009, 102, 073005. [Google Scholar] [CrossRef]
- Pfrommer, B.G.; Côté, M.; Louie, S.G.; Cohen, M.L. Relaxation of crystals with the quasi-newton method. J. Comput. Phy. 1997, 131, 233–240. [Google Scholar] [CrossRef]
- Monkhorst, H.J.; Pack, J.D. Special points for brillouin-zone integrations. Phys. Rev. B 1976, 13, 5188–5192. [Google Scholar] [CrossRef]
- Sturge, M. The jahn-teller effect in solids. In Solid State Physics; Elsevier: Amsterdam, The Netherlands, 1968; Volume 20, pp. 91–211. [Google Scholar]
- Yamashita, Y.; Ueda, K. Spin-driven Jahn-Teller Distortion in a pyrochlore system. Phys. Rev. Lett. 2000, 85, 4960. [Google Scholar] [CrossRef]
- Tunega, D. Theoretical study of properties of goethite (α-FeOOH) at ambient and high-pressure conditions. J. Phys. Chem. C 2012, 116, 6703–6713. [Google Scholar] [CrossRef]
- Yang, L.M.; Ravindran, P.; Vajeeston, P.; Tilset, M. Ab initio investigations on the crystal structure, formation enthalpy, electronic structure, chemical bonding, and optical properties of experimentally synthesized isoreticular metal–organic framework-10 and its analogues: M-irmof-10 (m = Zn, Cd, Be, Mg, Ca, Sr and Ba). RSC Adv. 2012, 2, 1618–1631. [Google Scholar]
- Yeung, H.H.M.; Kosa, M.; Parrinello, M.; Forster, P.M.; Cheetham, A.K. Structural diversity and energetics in anhydrous lithium tartrates: Experimental and computational studies of novel chiral polymorphs and their racemic and meso analogues. Cryst. Growth Des. 2010, 11, 221–230. [Google Scholar] [CrossRef]
- Gronert, S. Theoretical studies of proton transfers. 1. The potential energy surfaces of the identity reactions of the first-and second-row non-metal hydrides with their conjugate bases. J. Am. Chem. Soc. 1993, 115, 10258–10266. [Google Scholar] [CrossRef]
- Weckler, B.; Lutz, H. Lattice vibration spectra. Part xcv. Infrared spectroscopic studies on the iron oxide hydroxides goethite (α), akaganéite (β), lepidocrocite (γ), and feroxyhite (δ). Eur. J. Solid State Inorg. Chem. 1998, 35, 531–544. [Google Scholar] [CrossRef]
- Grabowski, S.J. Ab initio calculations on conventional and unconventional hydrogen bonds—Study of the hydrogen bond strength. J. Phys. Chem. A 2001, 105, 10739–10746. [Google Scholar] [CrossRef]
- Desiraju, G.R. Hydrogen bridges in crystal engineering: Interactions without borders. Acc. Chem. Res. 2002, 35, 565–573. [Google Scholar] [CrossRef]
- Huggins, M.L. Hydrogen bridges in ice and liquid water. J. Phys. Chem. 1936, 40, 723–731. [Google Scholar] [CrossRef]
- Rollmann, G.; Rohrbach, A.; Entel, P.; Hafner, J. First-principles calculation of the structure and magnetic phases of hematite. Phys. Rev. B 2004, 69, 165107. [Google Scholar] [CrossRef]
- Escamilla-Roa, E.; Huertas, F.J.; Hernández-Laguna, A.; Sainz-Díaz, C.I. A DFT study of the adsorption of glycine in the interlayer space of montmorillonite. Phys. Chem. Chem. Phys. 2017, 19, 14961–14971. [Google Scholar] [CrossRef]
- Grimme, S. Semiempirical GGA-type density functional constructed with a long-range dispersion correction. J. Comput. Chem. 2006, 27, 1787–1799. [Google Scholar] [CrossRef] [PubMed]
- Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H. A consistent and accurate Ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys. 2010, 132, 154104. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Zhao, J.; Li, F.; Chen, Z. Appropriate description of intermolecular interactions in the methane hydrates: An assessment of DFT methods. J. Comput. Chem. 2013, 34, 121–131. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Space Group | Akaganéite – X-Ray * I2/m | Ge-Akaganéite – X-Ray I2/m | |||||
|---|---|---|---|---|---|---|---|
| Unit Cell | |||||||
| a (Å) | 10.5876 (5) | 10.4441 (8) | |||||
| b (Å) | 3.03357 (8) | 3.01781 (15) | |||||
| c (Å) | 10.5277 (6) | 10.5015 (6) | |||||
| β (⁰) | 90.14 (2) | 90.339 (11) | |||||
| V (Å3) | 338.13 (2) | 330.99 (5) | |||||
| Refinement Parameters | |||||||
| No. of data points | 2173 | 5937 | |||||
| Diffraction range (dÅ) | 1.0–12 | 0.86–11 | |||||
| No. of variables | 65 | 27 | |||||
| R (F2) | 0.015 | 0.11396 | |||||
| Rwp | 0.016 | 0.068 | |||||
| χ2 | 2.56 | 2.890 | |||||
| Atomic Coordinates and Isotropic Displacements for Ge-Akaganéite | |||||||
| Atom | x | y | z | Site Occupancy | Uiso (Å2) ** | ||
| Fe1 | 0.8581 | 0 | 0.3416 | 0.8 | 0.0173 | ||
| Fe2 | 0.3470 | 0 | 0.1456 | 1.0 | 0.0173 | ||
| O1 | 0.652 | 0 | 0.298 | 1.0 | 0.002 | ||
| O2 | 0.674 | 0 | 0.049 | 1.0 | 0.002 | ||
| O3 | 0.279 | 0 | 0.323 | 1.0 | 0.002 | ||
| O4 | 0.036 | 0 | 0.336 | 1.0 | 0.002 | ||
| Cl | 0 | 0.071 | 0 | 0.201 | 0.10 | ||
| Ge1 | 0.8581 | 0 | 0.3416 | 0.2 | 0.0173 | ||
| Ge(OH)4 *** | 0 | 0.071 | 0 | 0.051 | 0.17 | ||
| Fe1–O1 | 2.190 (14) | O1–Fe1–O4 iii | 166.461 |
| Fe1–O1 i | 2.110 (10) | O1 i–Fe1–O2 ii | 165.153 |
| Fe1–O1 ii | 2.110 (10) | O1 ii–Fe1–O2 i | 165.153 |
| Fe1–O2 i | 1.926 (10) | - | - |
| Fe1–O2 ii | 1.926 (10) | - | - |
| Fe1–O4 iii | 1.868 (13) | O2 iv–Fe2–O3 | 153.305 |
| Fe2–O2 iv | 2.057 (17) | O3 v–Fe2–O4 vi | 165.176 |
| Fe2–O3 | 2.000 (16) | O3 vi–Fe2–O4 v | 165.176 |
| Fe2–O3 v | 2.035 (9) | - | - |
| Fe2–O3 vi | 2.035 (9) | - | - |
| Fe2–O4 v | 1.944 (8) | - | - |
| Fe2–O4 vi | 1.944 (8) | - | - |
| Number of Tunnel Tetrahedra N in the 1 × 5 × 1 Supercell | |||||
|---|---|---|---|---|---|
| Model Number (N value) | I (0) | II (1) | III (2) | IV (3) | V (4) |
| No. of Elements in Supercell | |||||
| H | 39 | 44 | 49 | 54 | 59 |
| O | 80 | 84 | 88 | 92 | 96 |
| Cl | 4 | 4 | 4 | 4 | 4 |
| Fe | 35 | 36 | 37 | 38 | 39 |
| Ge | 5 | 5 | 5 | 5 | 5 |
| No. of Cl–H * | 31 | 32 | 33 | 31 | 32 |
| No. of Cl–O ** | 0 | 0 | 1 | 6 | 9 |
| Formation enthalpy differences (kcal/mol) † | 10.382 | 0 | 0.021 | 5.551 | 81.927 |
| Bond | Bond Length (Å) | Bond | Bond Length (Å) |
|---|---|---|---|
| Ge–O(T1) | 1.639 | - | - |
| Ge–O(T2) | 1.660 | - | - |
| Ge–O(T3) | 1.678 | - | - |
| Ge–O(T4) | 1.946 | - | - |
| O(T1)–H(4) | 1.056 | O(T3)–H(44) | 1.207 |
| O(T1)–H(13) | 1.533 | O(T3)–H(1) | 1.283 |
| O(T1)–H(43) | 1.731 | O(T3)–H(35) | 1.332 |
| O(T2)–H(3) | 1.033 | O(T4)–H(2) | 0.975 |
| O(T2)–H(20) | 1.403 | O(T4)–H(18) | 1.372 |
| O(T2)–H(34) | 1.622 | O(T4)–H(12) | 1.408 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chung, D.; Park, C.; Choi, W.; Song, Y. Structural Refinement and Density Functional Theory Study of Synthetic Ge-Akaganéite (β-FeOOH). Crystals 2020, 10, 239. https://doi.org/10.3390/cryst10040239
Chung D, Park C, Choi W, Song Y. Structural Refinement and Density Functional Theory Study of Synthetic Ge-Akaganéite (β-FeOOH). Crystals. 2020; 10(4):239. https://doi.org/10.3390/cryst10040239
Chicago/Turabian StyleChung, Donghoon, Changyun Park, Woohyun Choi, and Yungoo Song. 2020. "Structural Refinement and Density Functional Theory Study of Synthetic Ge-Akaganéite (β-FeOOH)" Crystals 10, no. 4: 239. https://doi.org/10.3390/cryst10040239
APA StyleChung, D., Park, C., Choi, W., & Song, Y. (2020). Structural Refinement and Density Functional Theory Study of Synthetic Ge-Akaganéite (β-FeOOH). Crystals, 10(4), 239. https://doi.org/10.3390/cryst10040239