Deuteration Effects on the Transport Properties of (TMTTF)2X Salts



Abstract
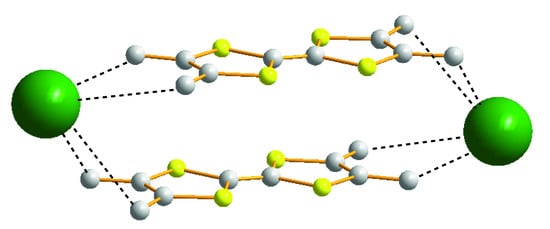
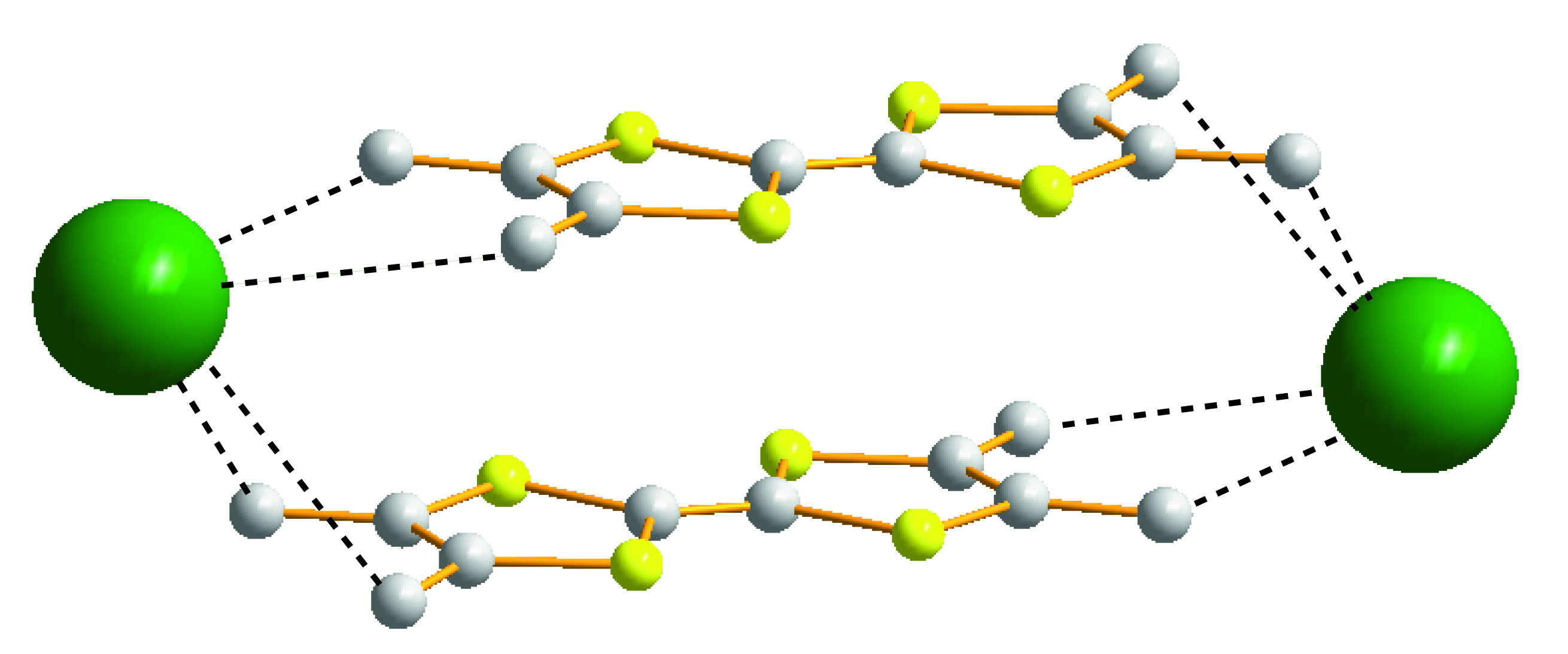
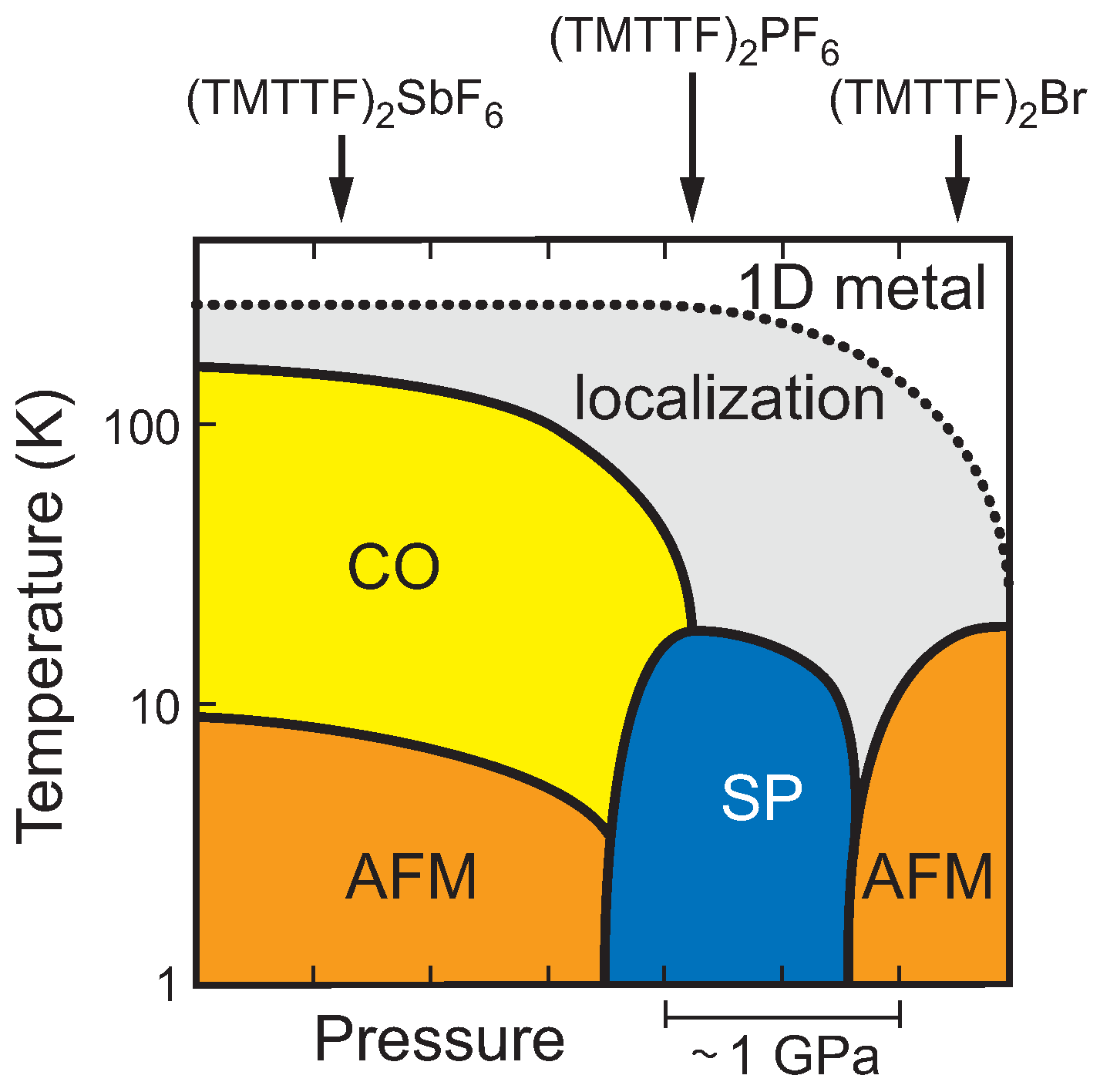
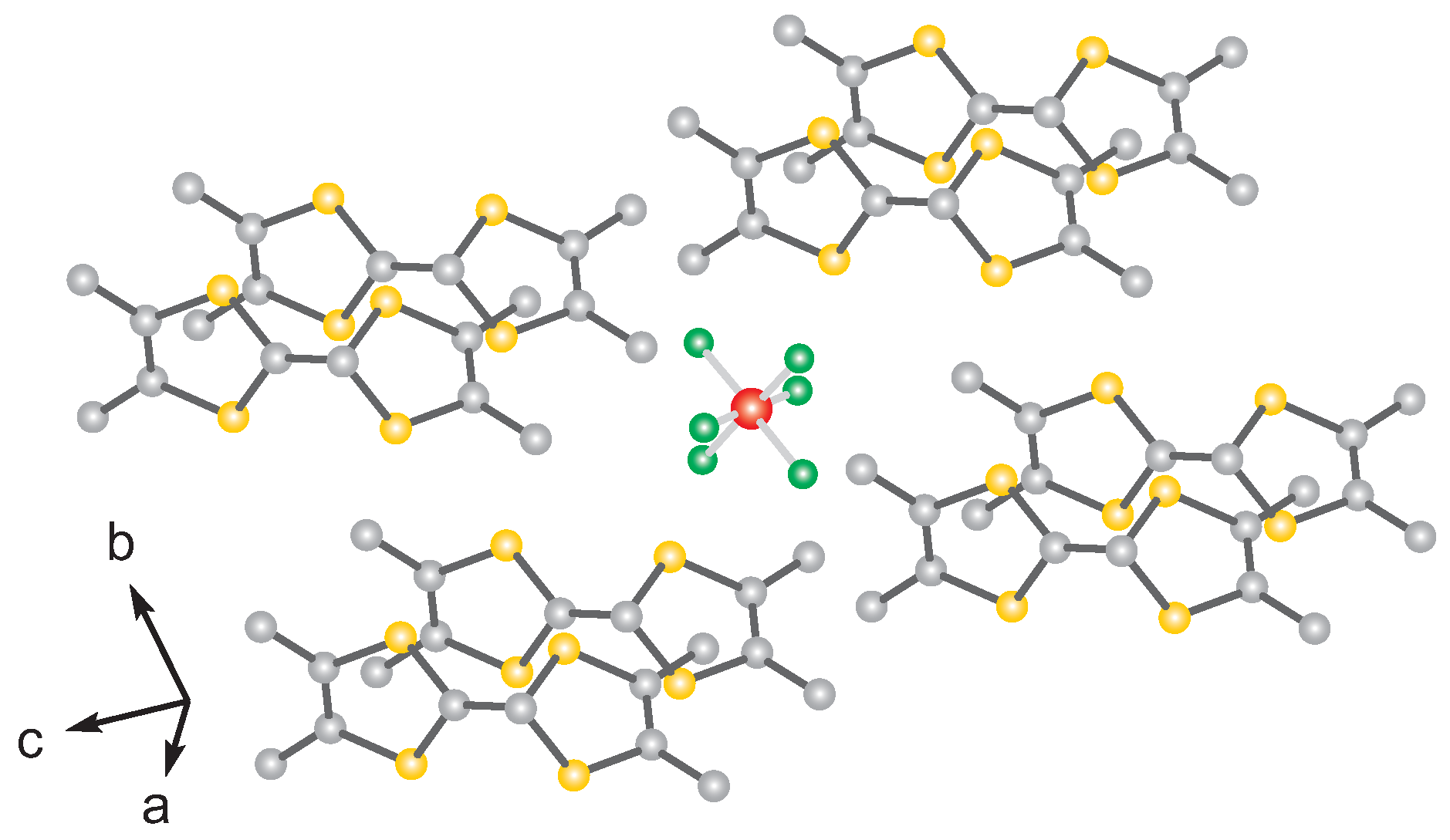
1. Introduction
2. Experimental Details
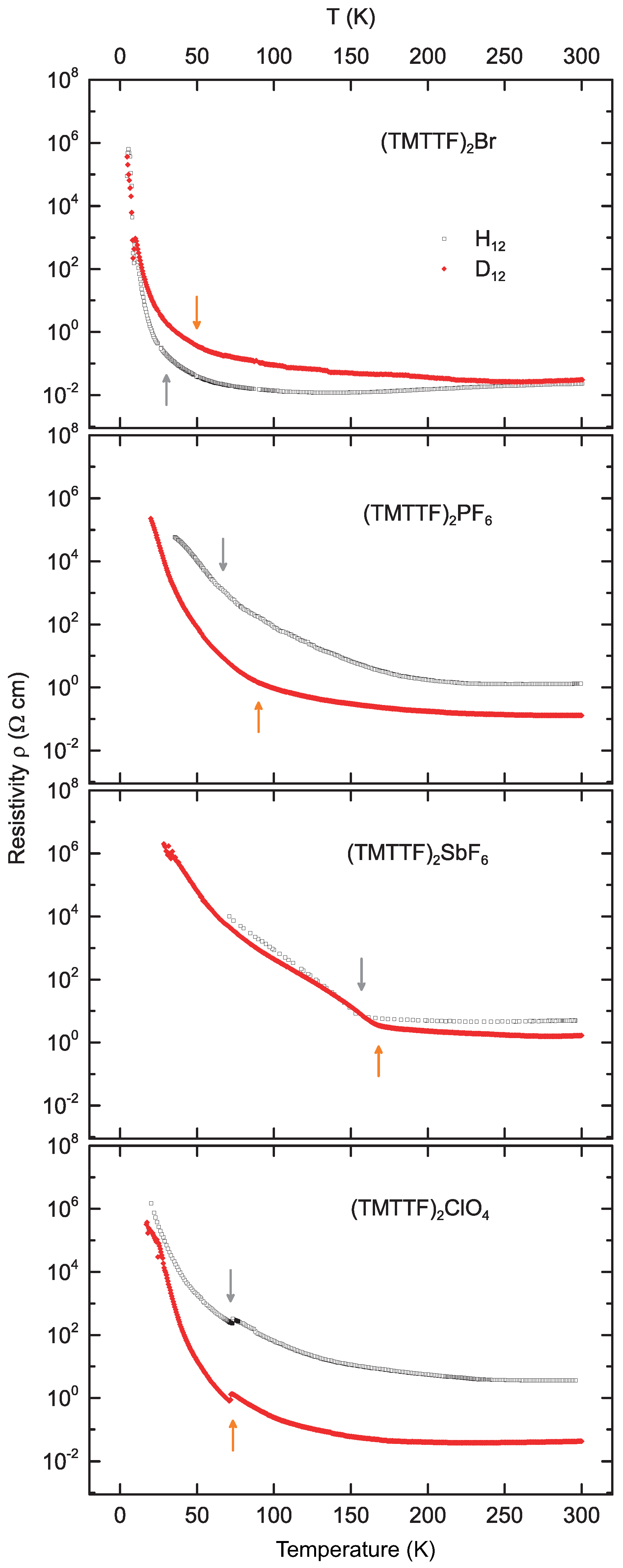
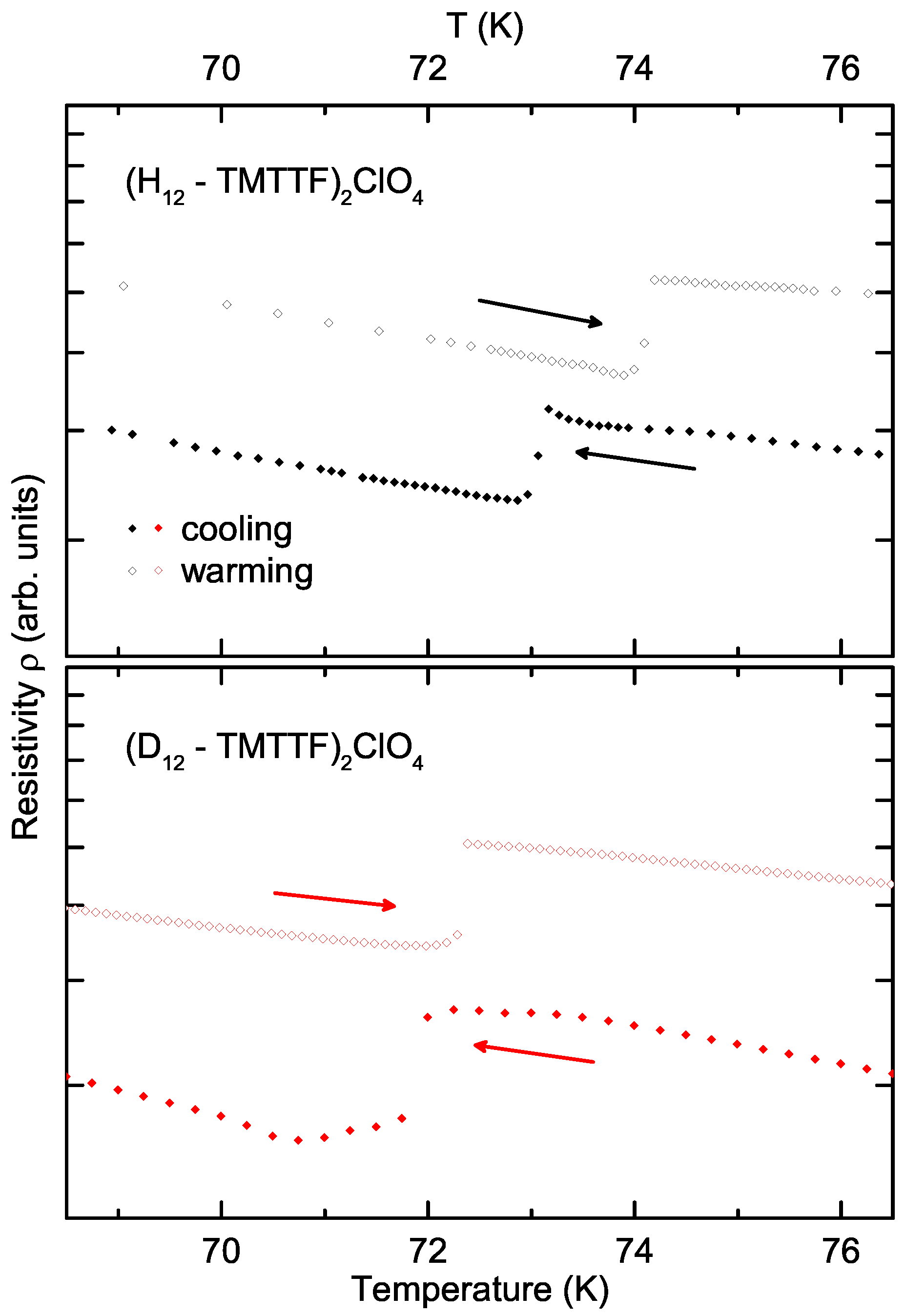
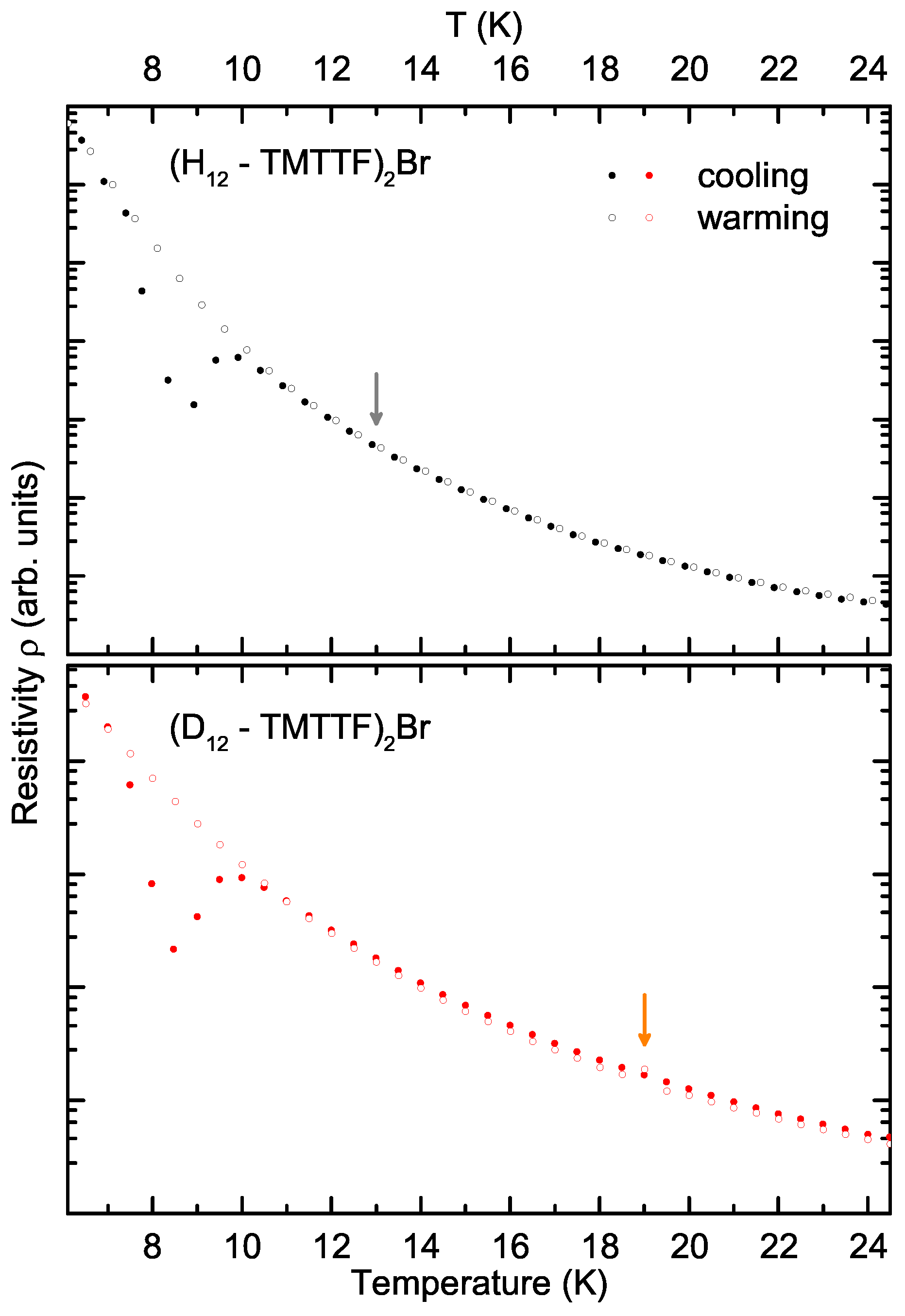
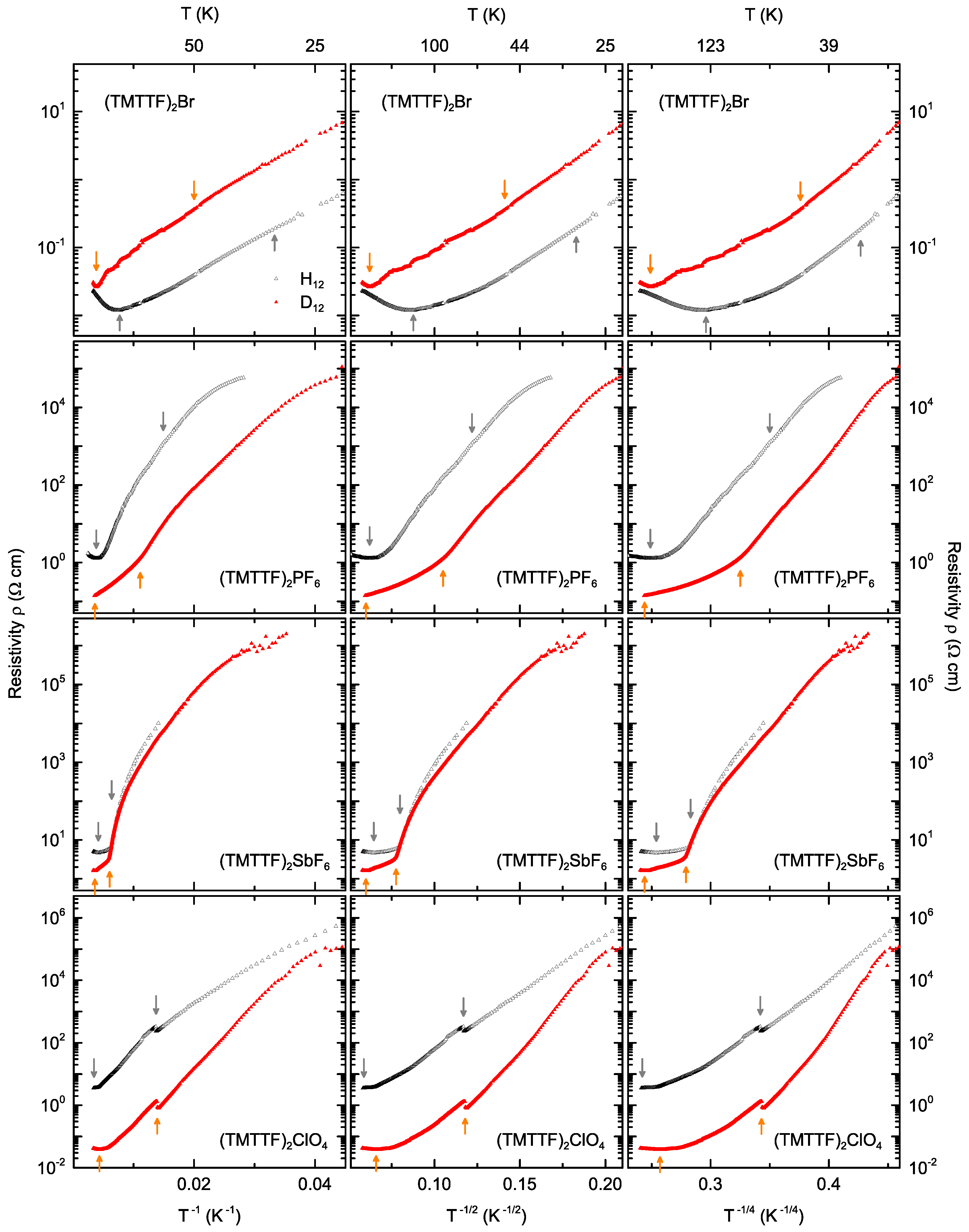
3. Results and Discussion
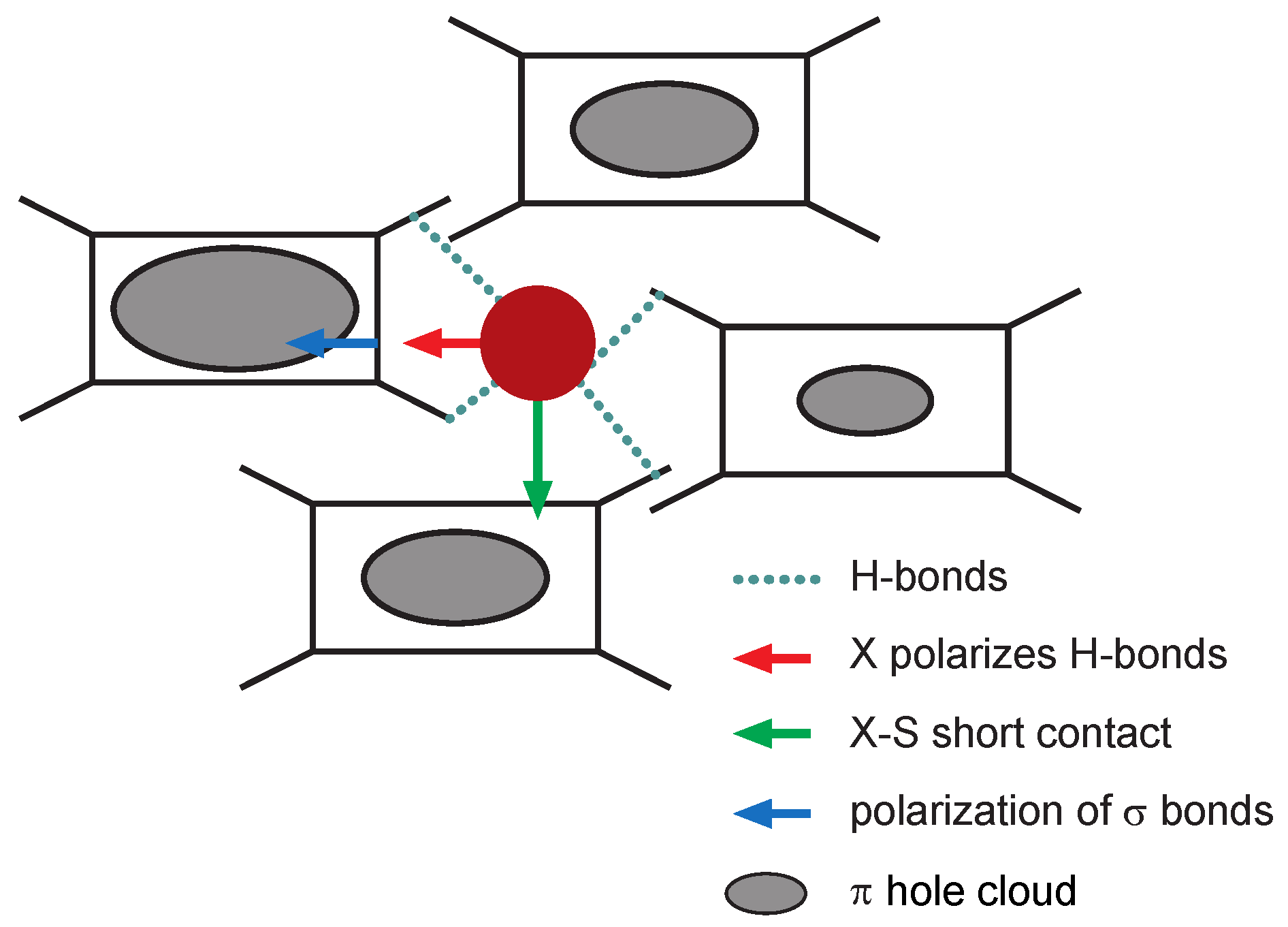
4. Analysis of the Transport Mechanisms
4.1. Charge Transport Mechanisms
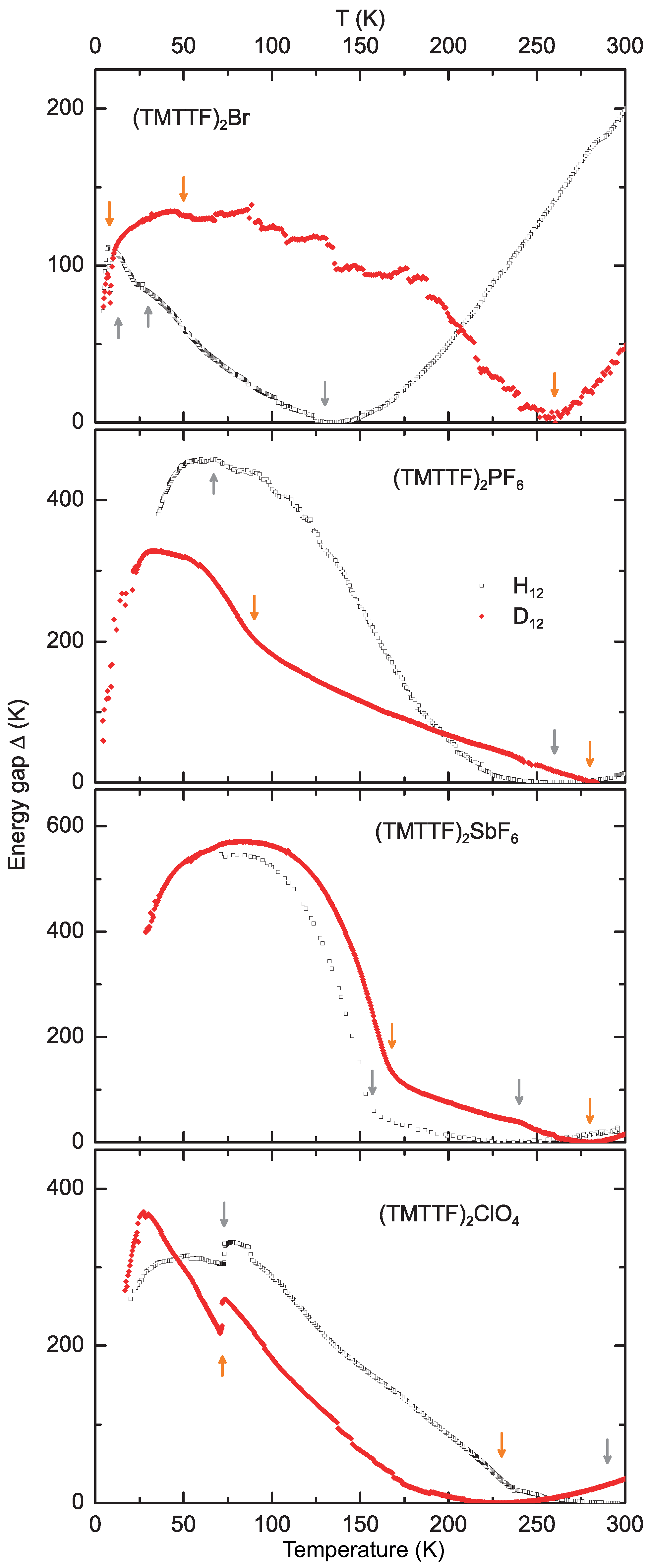
4.2. Energy Gap
5. Summary
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Appendix A

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| #Linear Fit# over Entire# Temp Range | Best# Linear# Fit | Linear Fit# over Entire# Temp Range | Best# Linear# Fit | Linear Fit# over Entire# Temp Range | Best# Linear# Fit | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| (TMTTF)X | 300 K- | or | or or or 4 K | |||||||||||
| H–Br | actual temperature range (K) | 300–130 | 130–30 | 30–13 | ||||||||||
| best fit temp range (K) | 300–180 | 130–60 | 60–30 | 24–13 | ||||||||||
| slope | −89.595 | −106.554 | 48.078 | 38.048 | 52.804 | 54.757 | 57.149 | |||||||
| y-intercept | −1.352 | −1.282 | −2.349 | −2.233 | −2.457 | −2.598 | −2.741 | |||||||
| best fit temp range (K) | 300–181 | 130–61 | 60–27 | 24–13 | ||||||||||
| slope | −11.206 | −14.082 | 12.462 | 8.118 | 16.354 | 24.579 | 26.979 | |||||||
| y-intercept | −1.005 | −0.819 | −3.126 | −2.662 | −3.711 | −5.317 | −5.899 | |||||||
| best fit temp range (K) | 300–167 | 130–63 | 49–27 | 22–10 | ||||||||||
| slope | −6.044 | −7.076 | 8.819 | 5.167 | 13.597 | 25.026 | 29.727 | |||||||
| y-intercept | −0.193 | 0.069 | −4.671 | −3.482 | −6.529 | −11.669 | −13.983 | |||||||
| D–Br | actual temperature range (K) | 300–260 | 260–50 | 50–8 | ||||||||||
| best fit temp range (K) | 295–260 | 157–174 | 173–89 | 89–48 | 50–25 | 25–10 | ||||||||
| slope | −127.055 | −119.421 | 71.181 | 141.631 | 74.232 | 54.111 | 40.052 | 52.862 | 42.037 | |||||
| y-intercept | −1.096 | −1.124 | −1.789 | −2.146 | −1.789 | −1.537 | −1.050 | −1.461 | −1.002 | |||||
| best fit temp range (K) | 295–260 | 255–54 | 50–25 | 25–10 | ||||||||||
| slope | −15.423 | −14.362 | 14.053 | 13.994 | 18.514 | 17.777 | 21.234 | |||||||
| y-intercept | −0.628 | −0.692 | −2.443 | −2.438 | −3.047 | −2.942 | −3.640 | |||||||
| best fit temp range (K) | 295–261 | 257–177 | 186–67 | 67–48 | 50–25 | 25–10 | ||||||||
| slope | −7.535 | −6.995 | 8.638 | 10.224 | 8.547 | 11.621 | 17.357 | 14.510 | 21.126 | |||||
| y-intercept | 0.292 | 0.159 | −3.751 | −4.155 | −3.730 | −4.816 | −7.047 | −5.896 | −8.875 | |||||
| H–PF | actual temperature range (K) | 300–260 | 260–67 | 67–19 (data ends at 35) | ||||||||||
| best fit temp range (K) | 300–260 | 200–107 | 109–67 | 100–51 | ||||||||||
| slope | −34.808 | (not linear) | 299.215 | 371.644 | 235.220 | 132.518 | 211.301 | |||||||
| y-intercept | 0.240 | −1.166 | −1.650 | −0.419 | 1.274 | −0.124 | ||||||||
| best fit temp range (K) | 300–260 | 181–65 | 124–47 | |||||||||||
| slope | −4.125 | (not linear) | 53.106 | 56.448 | 38.392 | 50.922 | ||||||||
| y-intercept | 0.363 | −3.432 | −3.758 | −1.483 | −3.168 | |||||||||
| best fit temp range (K) | 300–260 | 188–44 | 67–35 | |||||||||||
| slope | −1.974 | (not linear) | 31.610 | 34.964 | 29.290 | (see previous range) | ||||||||
| y-intercept | 0.598 | −8.103 | −9.142 | −7.059 | (see previous range) | |||||||||
| D–PF | actual temperature range (K) | 300–280 | 280–90 | 90–13 (good data up to 20) | ||||||||||
| best fit temp range (K) | 300–280 | 239–119 | 157–90 | 59–30 | ||||||||||
| slope | 4.722 | (too small) | 124.022 | 115.341 | 139.517 | 148.637 | 151.236 | |||||||
| y-intercept | −0.869 | −1.325 | −1.282 | −1.458 | −1.221 | −1.113 | ||||||||
| best fit temp range (K) | 300–280 | 287–161 | 163–101 | 87–26 | ||||||||||
| slope | 0.532 | (too small) | 20.038 | 14.253 | 23.332 | 46.486 | 47.952 | |||||||
| y-intercept | −0.883 | −2.115 | −1.710 | −2.426 | −4.693 | −4.879 | ||||||||
| best fit temp range (K) | 300–280 | 280–155 | 163–103 | 89–23 | ||||||||||
| slope | 0.268 | (too small) | 11.213 | 7.761 | 13.707 | 36.144 | 36.353 | |||||||
| y-intercept | −0.917 | −3.675 | −2.764 | −4.438 | −11.642 | −11.717 | ||||||||
| H–SbF | actual temperature range (K) | 300–240 | 240–157 | 157–8 (good data ends at 70) | ||||||||||
| best fit temp range (K) | 296–251 | 228–165 (not linear) | 110–70 (not linear) | |||||||||||
| slope | −47.048 | −54.097 | 70.625 | 66.416 | 393.592 | 273.853 | ||||||||
| y-intercept | 0.859 | 0.884 | 0.351 | 0.367 | −1.177 | 0.196 | ||||||||
| best fit temp range (K) | 296–249 | 219–165 (not linear) | 157–114 | 112–70 | ||||||||||
| slope | −5.746 | −6.532 | 10.123 | 10.124 | 78.505 | 121.771 | 59.678 | |||||||
| y-intercept | 1.034 | 1.081 | −0.011 | −0.018 | −5.042 | −8.825 | −3.043 | |||||||
| best fit temp range (K) | 297–252 | 234–182 | 199–157 | 157–114 | 116–71 | |||||||||
| slope | −2.734 | −3.209 | 5.413 | 3.589 | 7.915 | 49.339 | 71.707 | 38.894 | ||||||
| y-intercept | 1.359 | 1.475 | −0.734 | −0.256 | −1.421 | −12.770 | −19.378 | −9.373 | ||||||
| D–SbF | actual temperature range (K) | 300–280 | 280–166 | 166–8 (good data to 27) | ||||||||||
| best fit temp range (K) | 300–286 | 231–181 | 111–64 (not linear) | |||||||||||
| slope | −101.615 | −126.631 | 134.304 | 128.026 | 203.142 | 247.267 | ||||||||
| y-intercept | 0.560 | 0.644 | −0.296 | −0.274 | 0.336 | 0.176 | ||||||||
| best fit temp range (K) | 300–287 | 231–184 | 112–44 | |||||||||||
| slope | −11.269 | −14.926 | 18.243 | 17.713 | 52.762 | 52.129 | ||||||||
| y-intercept | 0.871 | 1.084 | −0.912 | −0.886 | −2.845 | −2.557 | ||||||||
| best fit temp range (K) | 300–286 | 229–183 | 126–38 | |||||||||||
| slope | −5.666 | −7.150 | 9.511 | 9.482 | 37.198 | 36.190 | ||||||||
| y-intercept | 1.582 | 1.940 | −2.150 | −2.154 | −9.280 | −8.818 | ||||||||
| H–ClO | actual temperature range (K) | 300–290 | 290–73 | 74–72 | 73–4 (good data ends at 20) | |||||||||
| best fit temp range (K) | 300–290 | 232–73.2 | 87–73.2 | 73.17–72.87 | 72.5–52 | 53–29 | ||||||||
| slope | 4.567 | (too small) | 204.468 | 209.608 | 147.605 | −2694.026 | 116.853 | 147.693 | 119.150 | |||||
| y-intercept | 0.536 | −0.264 | −0.316 | 0.495 | 39.318 | 0.838 | 0.334 | 0.903 | ||||||
| best fit temp range (K) | 300–290 | 233–131 | 130–88 | 87–73.0 | 73.17–72.82 | 72.6–54 | 72–24 | |||||||
| slope | 0.533 | (too small) | 35.389 | 28.927 | 43.917 | 31.979 | −630.607 | 37.248 | 36.931 | 37.704 | ||||
| y-intercept | 0.520 | −1.723 | −1.304 | −2.591 | −1.233 | 73.220 | −2.002 | −1.970 | −2.063 | |||||
| best fit temp range (K) | 300–290 | 237–136 | 138–89 | 87–73.1 | 135–73.1 | 73.17–72.87 | 72.8–53 | 53–22 | 72.6–22 | |||||
| slope | 0.257 | (too small) | 21.392 | 15.578 | 26.857 | 22.312 | 27.831 | −308.947 | 29.329 | 26.114 | 30.649 | 29.310 | ||
| y-intercept | 0.489 | −4.929 | −3.397 | −6.691 | −5.118 | −6.988 | 108.098 | −7.715 | −6.583 | −8.268 | −7.709 | |||
| D–ClO | actual temperature range (K) | 300–230 | 230–73 | 73–70 | 73–4 (good data to 18) | |||||||||
| best fit temp range (K) | 300–230 | 138–71.1 | 90–72.4 | 72.23–70.74 | 70.6–56 | 70.4–29 | ||||||||
| slope | −44.010 | (not linear) | 172.713 | 197.157 | 193.418 | −655.223 | 167.089 | 220.575 | 208.745 | |||||
| y-intercept | −1.234 | −2.309 | −2.569 | −2.514 | 0.137 | −2.198 | −3.206 | −3.014 | ||||||
| best fit temp range (K) | 300–230 | 137–96 | 96–72.2 | 72.26–70.73 | 70.8–57 | 56–30 | ||||||||
| slope | −5.544 | (not linear) | 30.793 | 33.654 | 43.408 | −185.305 | 56.477 | 55.196 | 64.775 | |||||
| y-intercept | −1.060 | −3.641 | −3.989 | −4.948 | 21.896 | −6.765 | −6.656 | −7.990 | ||||||
| best fit temp range (K) | 300–230 | 137–96 | 95–72.1 | 72.24–70.74 | 70.6–56 | 70.9–43 | 46–25 | |||||||
| slope | −2.778 | (not linear) | 18.215 | 20.594 | 28.769 | −106.880 | 45.719 | 39.238 | 41.638 | 55.276 | ||||
| y-intercept | −0.712 | −6.314 | −7.137 | −9.713 | 36.728 | −15.919 | −13.627 | −14.473 | −19.754 | |||||
References and Notes
- Lebed, A. (Ed.) The Physics of Organic Superconductors and Conductors; Springer: Berlin, Germany, 2008. [Google Scholar]
- Dressel, M. Spin-charge separation in quasi one-dimensional organic conductors. Naturwissenschaften 2003, 90, 337–344. [Google Scholar] [CrossRef] [PubMed]
- Dressel, M. Ordering phenomena in quasi-one-dimensional organic conductors. Naturwissenschaften 2007, 94, 527–541. [Google Scholar] [CrossRef] [PubMed]
- Dressel, M.; Dumm, M.; Knoblauch, T.; Masino, M. Comprehensive optical investigations of charge order in organic chain compounds (TMTTF)2X. Crystals 2012, 2, 528–578. [Google Scholar] [CrossRef]
- Nad, F.; Monceau, P.; Fabre, J.M. Low frequency dielectric permittivity of quasi-one-dimensional conductor (TMTTF)2Br. Eur. Phys. J. B 1998, 3, 301–306. [Google Scholar] [CrossRef]
- Coulon, C.; Lalet, G.; Pouget, J.P.; Foury-Leylekian, P.; Mouradpour, A.; Fabre, J.M. Anisotropic conductivity and charge ordering in (TMTTF)2X salts probed by ESR. Phys. Rev. B 2007, 76, 085126. [Google Scholar] [CrossRef]
- Köhler, B.; Rose, E.; Dumm, M.; Untereiner, G.; Dressel, M. Comprehensive transport study of anisotropy and ordering phenomena in quasi-one-dimensional (TMTTF)2X salts (X = PF6, AsF6, SbF6, BF4, ClO4, ReO4). Phys. Rev. B 2011, 84, 035124. [Google Scholar] [CrossRef]
- Rose, E.; Dressel, M. Coupling between molecular chains and anions in (TMTTF)2X salts. Phys. B Condens. Matter 2012, 407, 1787–1792. [Google Scholar] [CrossRef]
- Rösslhuber, R.; Rose, E.; Ivek, T.; Pustogow, A.; Breier, T.; Geiger, M.; Schrem, K.; Untereiner, G.; Dressel, M. Structural and Electronic Properties of (TMTTF)2X Salts with Tetrahedral Anions. Crystals 2018, 8, 121. [Google Scholar] [CrossRef]
- Pustogow, A.; Peterseim, T.; Kolatschek, S.; Engel, L.; Dressel, M. Electronic correlations versus lattice interactions: Interplay of charge and anion orders in (TMTTF)2X. Phys. Rev. B 2016, 94, 195125. [Google Scholar] [CrossRef]
- Pouget, J.P. Structural Aspects of the Bechgaard and Fabre Salts: An Update. Crystals 2012, 2, 466–520. [Google Scholar] [CrossRef]
- Pouget, J.P.; Alemany, P.; Canadell, E. Donor-anion interactions in quarter-filled low-dimensional organic conductors. Mater. Horiz. 2018, 5, 590–640. [Google Scholar] [CrossRef]
- Pouget, J.P.; Foury-Leylekian, P.; Bolloc’h, D.; Hennion, B.; Ravy, S.; Coulon, C.; Cardoso, V.; Moradpour, A. Neutron-scattering evidence for a spin-peierls ground state in (TMTTF)2PF6. J. Low Temp. Phys. 2006, 142, 147–152. [Google Scholar] [CrossRef]
- Asada, M.; Nakamura, T. Magnetic resonance investigation for a possible antiferromagnetic subphase in (TMTTF)2Br. Phys. Rev. B 2017, 96, 125120. [Google Scholar] [CrossRef]
- Furukawa, K.; Hara, T.; Nakamura, T. Deuteration effect and possible origin of the charge-ordering transition of (TMTTF)2X. J. Phys. Soc. Jpn. 2005, 74, 3288. [Google Scholar] [CrossRef]
- Nad, F.; Monceau, P.; Nakamura, T.; Furukawa, K. The effect of deuteration on the transition into a charge ordered state of (TMTTF)2X salts. J. Phys. Condens. Matter 2005, 17, L399–L406. [Google Scholar] [CrossRef]
- Dumm, M.; Loidl, A.; Fravel, B.W.; Starkey, K.P.; Montgomery, L.K.; Dressel, M. Electron spin resonance studies on the organic linear-chain compounds (TMTCF)2X(C = S, Se; X = PF6, AsF6, ClO4, Br). Phys. Rev. B 2000, 61, 511–521. [Google Scholar] [CrossRef]
- Salameh, B.; Yasin, S.; Dumm, M.; Untereiner, G.; Montgomery, L.; Dressel, M. Spin dynamics of the organic linear chain compounds (TMTTF)2X (X = SbF6, AsF6, BF4, ReO4, and SCN). Phys. Rev. B 2011, 83, 205126. [Google Scholar] [CrossRef]
- We cannot guarantee that the experiments have been conducted under exactly the same conditions. Cooling rates as well as heat transfer rates may have differed between the measurements on the deuterated and protonated crystals, which may influence the hysteresis of this transition.
- Pouget, J.P.; Ravy, S. Structural aspects of the Bechgaard salts and related compounds. J. Phys. I 1996, 6, 1501–1525. [Google Scholar] [CrossRef]
- Dumm, M.; Loidl, A.; Alavi, B.; Starkey, K.P.; Montgomery, L.K.; Dressel, M. Comprehensive ESR-Study of the Antiferromagnetic Ground States in the One-Dimensional Spin Systems (TMTSF)2PF6, (TMTSF)2AsF6, and (TMTTF)2Br. Phys. Rev. B 2000, 62, 6512–6520. [Google Scholar] [CrossRef]
- Tomić, S.; Biskup, N.; Dolamski Babić, S.; Maki, K. Commensurate Spin-Density Wave State in (TMTTF)2Br: Single-Particle and Collective Charge Dynamics. Europhys. Lett. 1994, 26, 295–301. [Google Scholar] [CrossRef]
- Baranovski, S. Charge Transport in Disordered Solids with Applications in Electronics; John Wiley & Sons: West Sussex, UK, 2006. [Google Scholar]
- Mott, N.F.; Davies, E. Electronic Processes in Non-Crystalline Materials, 2nd ed.; Clarendon Press: Oxford, UK, 1979. [Google Scholar]
- Efros, A.L.; Shklovskii, B.I. Coulomb gap and low temperature conductivity of disordered systems. J. Phys. C 1975, 8, L49. [Google Scholar] [CrossRef]
- Shklovskii, B.I.; Efros, A.L. Electronic Properties of Doped Semiconductors; Springer: Berlin, Germany, 1984. [Google Scholar]
- Khondaker, S.I.; Shlimak, I.S.; Nicholls, J.T.; Pepper, M.; Ritchie, D.A. Crossover phenomenon for two-dimensional hopping conductivity and density-of-states near the Fermi level. Solid State Commun. 1999, 109, 751. [Google Scholar] [CrossRef]
- Strictly speaking γ = 2 is also obtained in the case of one-dimensional transport (d = 1). The thorough study of anisotropic transport reveals that at low-temperatures one-dimensional models do not apply despite the appreciable anisotropy.
- Dressel, M.; Petukhov, K.; Salameh, B.; Zornoza, P.; Giamarchi, T. Scaling Behavior of the Longitudinal and Transverse Transport in Quasi One-Dimensional Organic Conductors. Phys. Rev. B 2005, 71, 075104. [Google Scholar] [CrossRef]
- Joung, D.; Khondaker, S.I. Efros-Shklovskii variable range hopping in reduced graphene oxide sheets of varying carbon sp2 fraction. Phys. Rev. B 2012, 86, 235423. [Google Scholar] [CrossRef]








| Anions | Transition Temperatures (K) | |||||
|---|---|---|---|---|---|---|
| X | ||||||
| Br | H | 130 | 30 | 13 | ||
| D | 260 | 50 | 19 | |||
| PF | H | 260 | 67 | 19 | ||
| D | 280 | 90 | 13 | |||
| SbF | H | 240 | 157 | 6 | ||
| D | 280 | 168 | 8 | |||
| ClO | H | 290 | 73.1 | |||
| D | 230 | 71.9 | ||||
| Anions | Change in Transition Temperature (K) | ||||
|---|---|---|---|---|---|
| X | |||||
| Br | −130 | −20 | −6 | ||
| PF | −20 | −23 | 6 | ||
| SbF | −40 | −11 | −2 | ||
| ClO | 60 | 1.2 | |||
| Anions X | (cm) | |
|---|---|---|
| Br | H | 0.01184 |
| D | 0.02606 | |
| PF | H | 1.286 |
| D | 0.1401 | |
| SbF | H | 4.638 |
| D | 1.585 | |
| ClO | H | 3.557 |
| D | 0.03818 |
| Anions X | (K) | (K) | (K) | (K) | |
|---|---|---|---|---|---|
| Br | H | 90 | 110 | 140 | |
| D | 130 | 135 | 190 | ||
| PF | H | 460 | 460 | 650 | |
| D | 200 | 330 | 390 | ||
| SbF | H | 60 | 545 | 550 | |
| D | 140 | 570 | 585 | ||
| ClO | H | 330 | 315 | 450 | |
| D | 260 | 370 | 450 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rohwer, A.; Dressel, M.; Nakamura, T. Deuteration Effects on the Transport Properties of (TMTTF)2X Salts. Crystals 2020, 10, 1085. https://doi.org/10.3390/cryst10121085
Rohwer A, Dressel M, Nakamura T. Deuteration Effects on the Transport Properties of (TMTTF)2X Salts. Crystals. 2020; 10(12):1085. https://doi.org/10.3390/cryst10121085
Chicago/Turabian StyleRohwer, Andrea, Martin Dressel, and Toshikazu Nakamura. 2020. "Deuteration Effects on the Transport Properties of (TMTTF)2X Salts" Crystals 10, no. 12: 1085. https://doi.org/10.3390/cryst10121085
APA StyleRohwer, A., Dressel, M., & Nakamura, T. (2020). Deuteration Effects on the Transport Properties of (TMTTF)2X Salts. Crystals, 10(12), 1085. https://doi.org/10.3390/cryst10121085