Pseudokinases: From Allosteric Regulation of Catalytic Domains and the Formation of Macromolecular Assemblies to Emerging Drug Targets

 ,
,  and
and 
Abstract
:1. Introduction
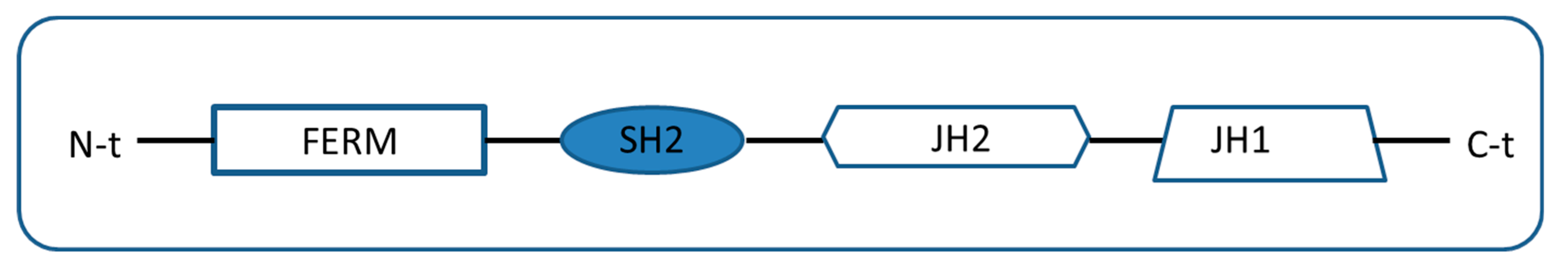
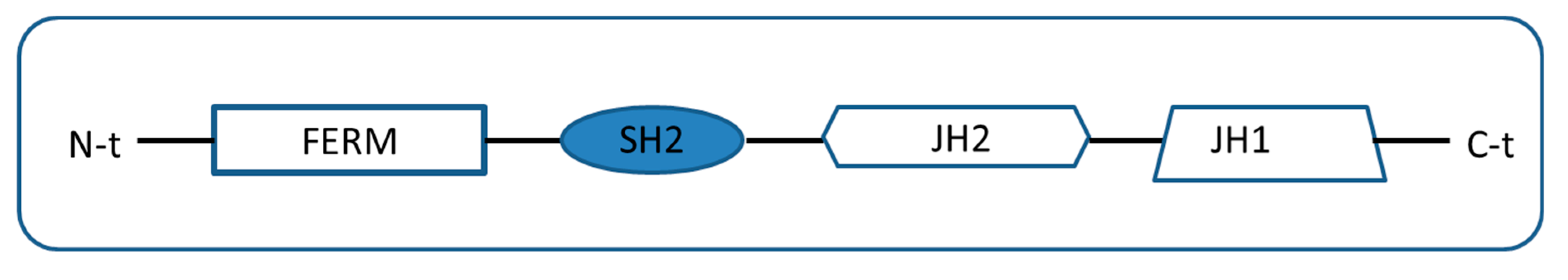
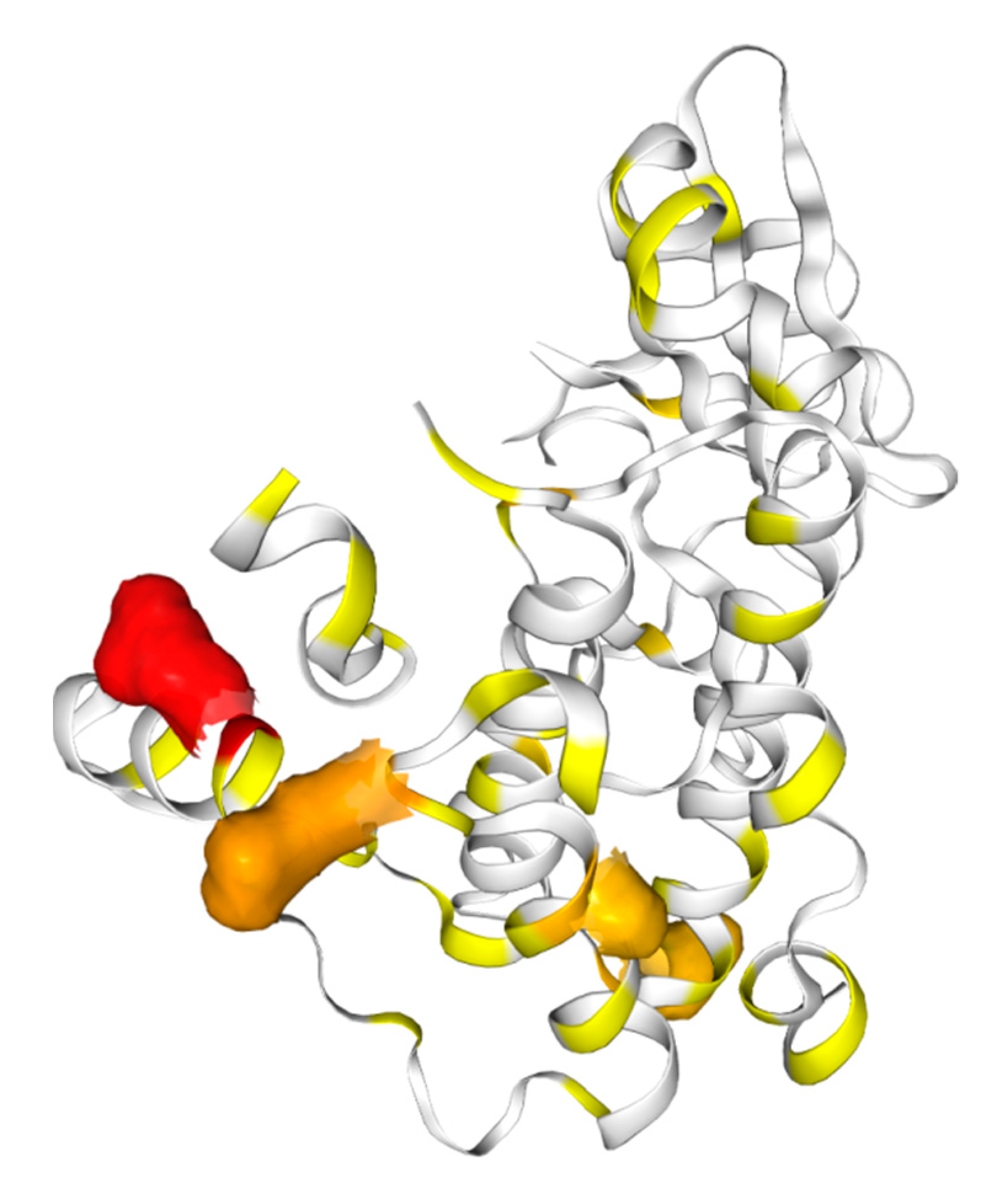
2. Types of Pseudokinases Proteins
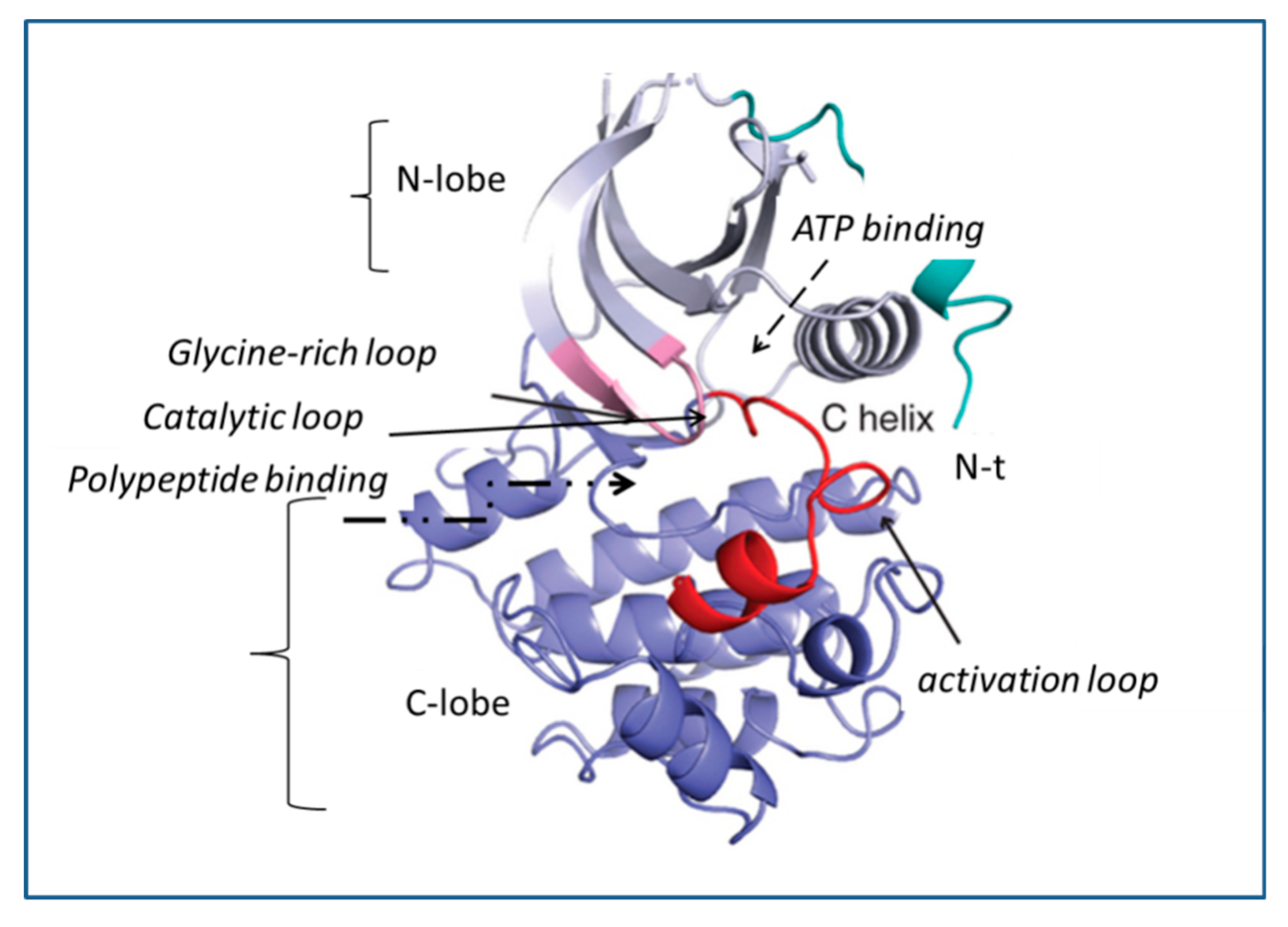
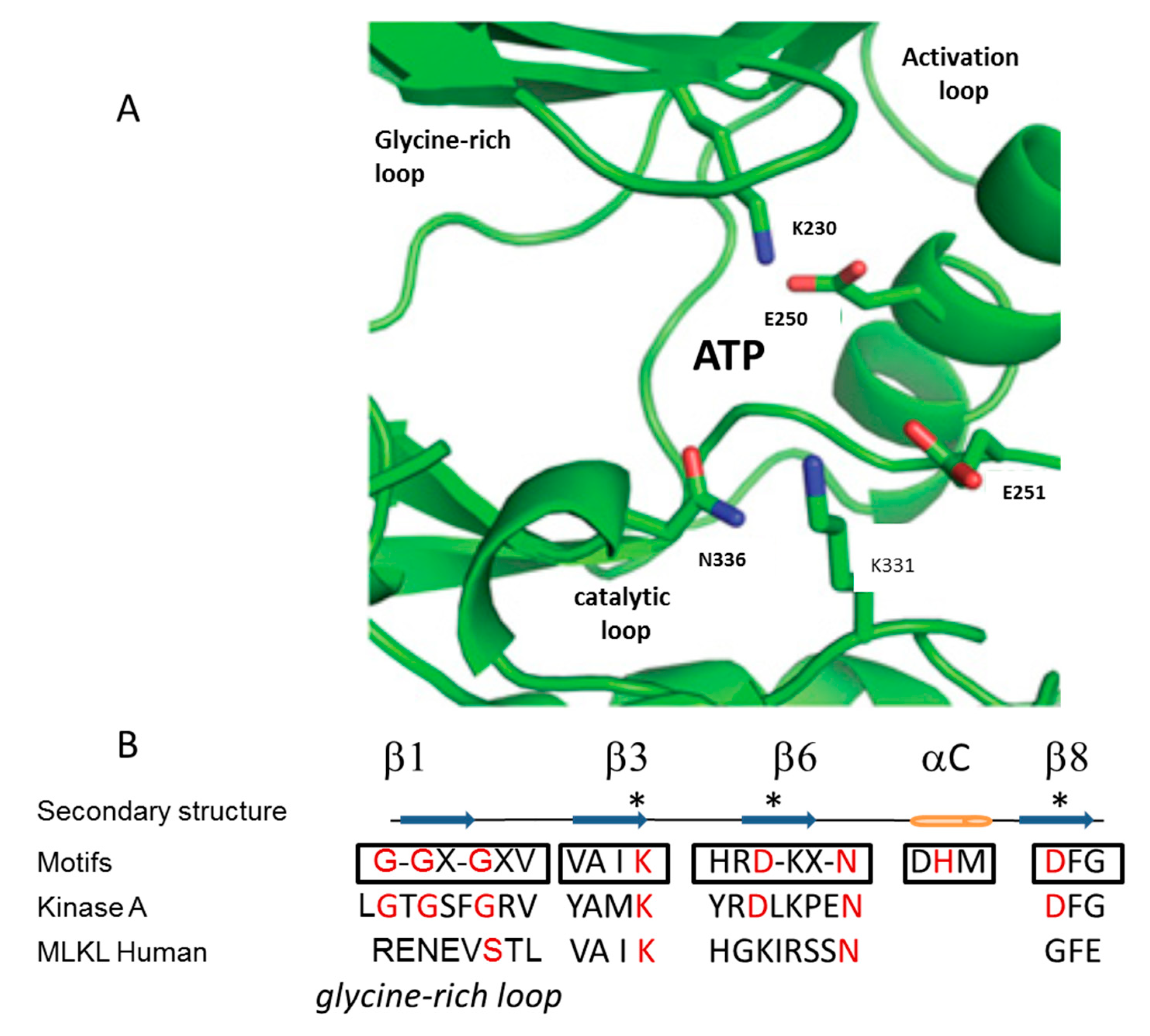
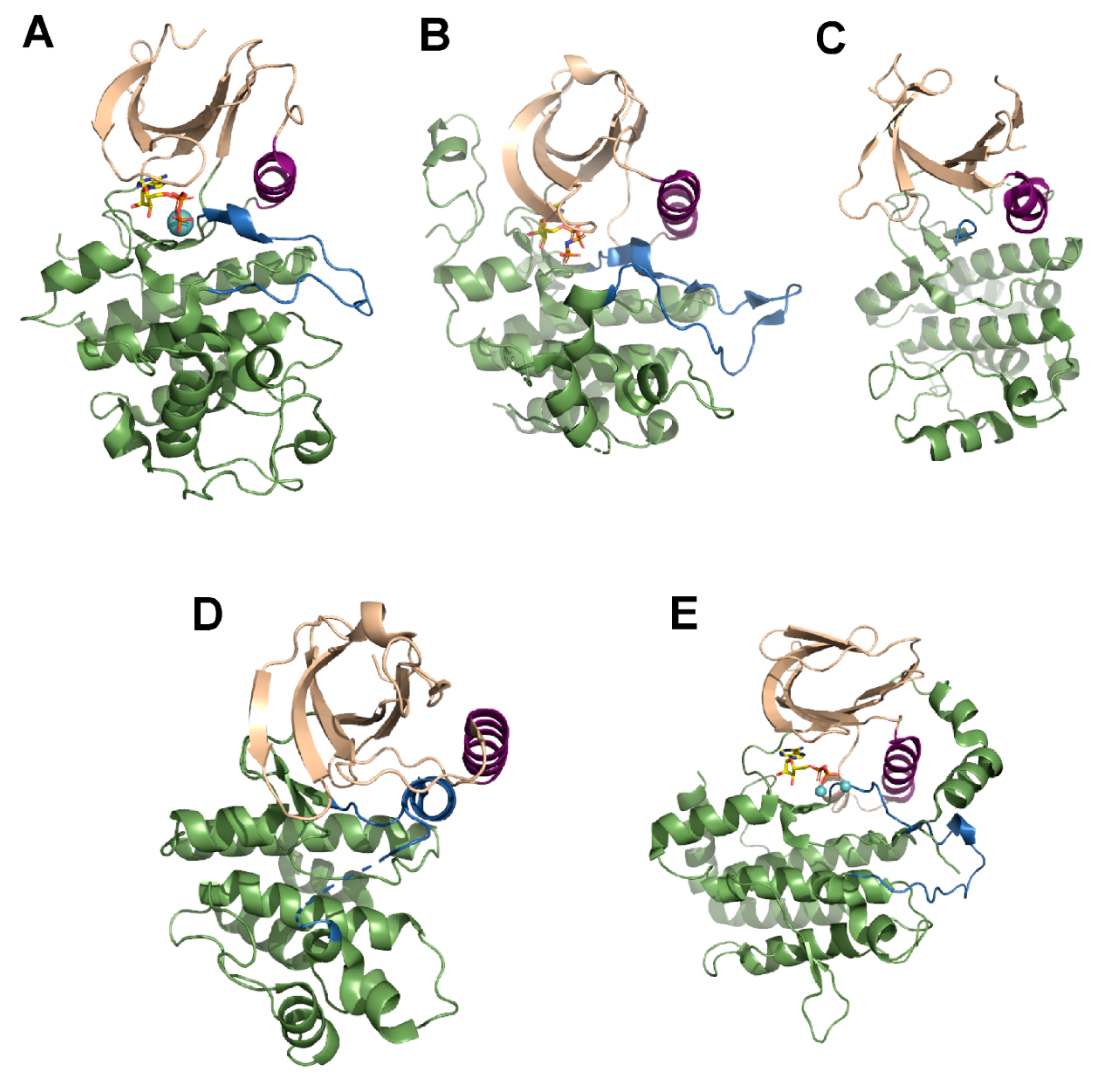
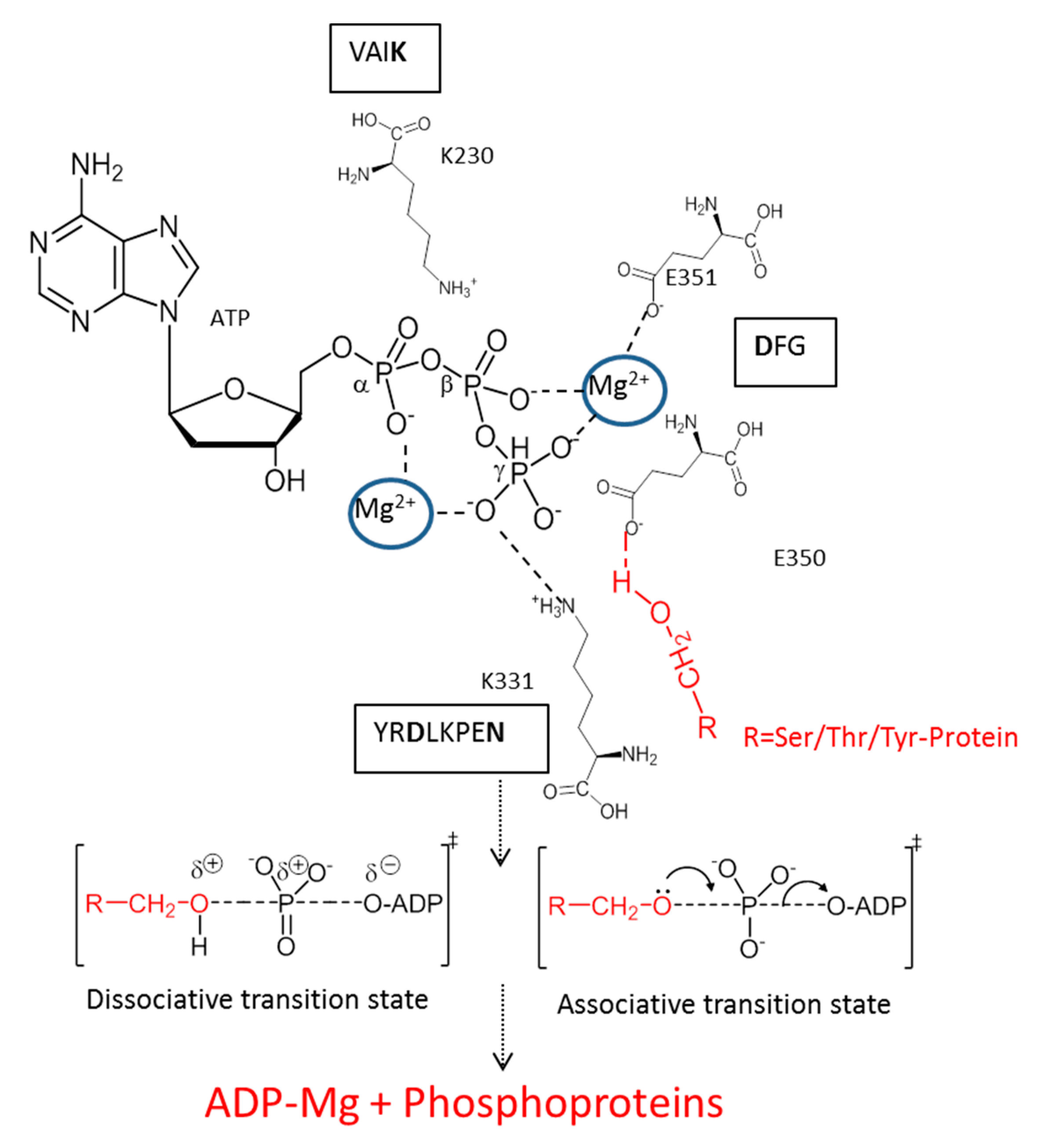
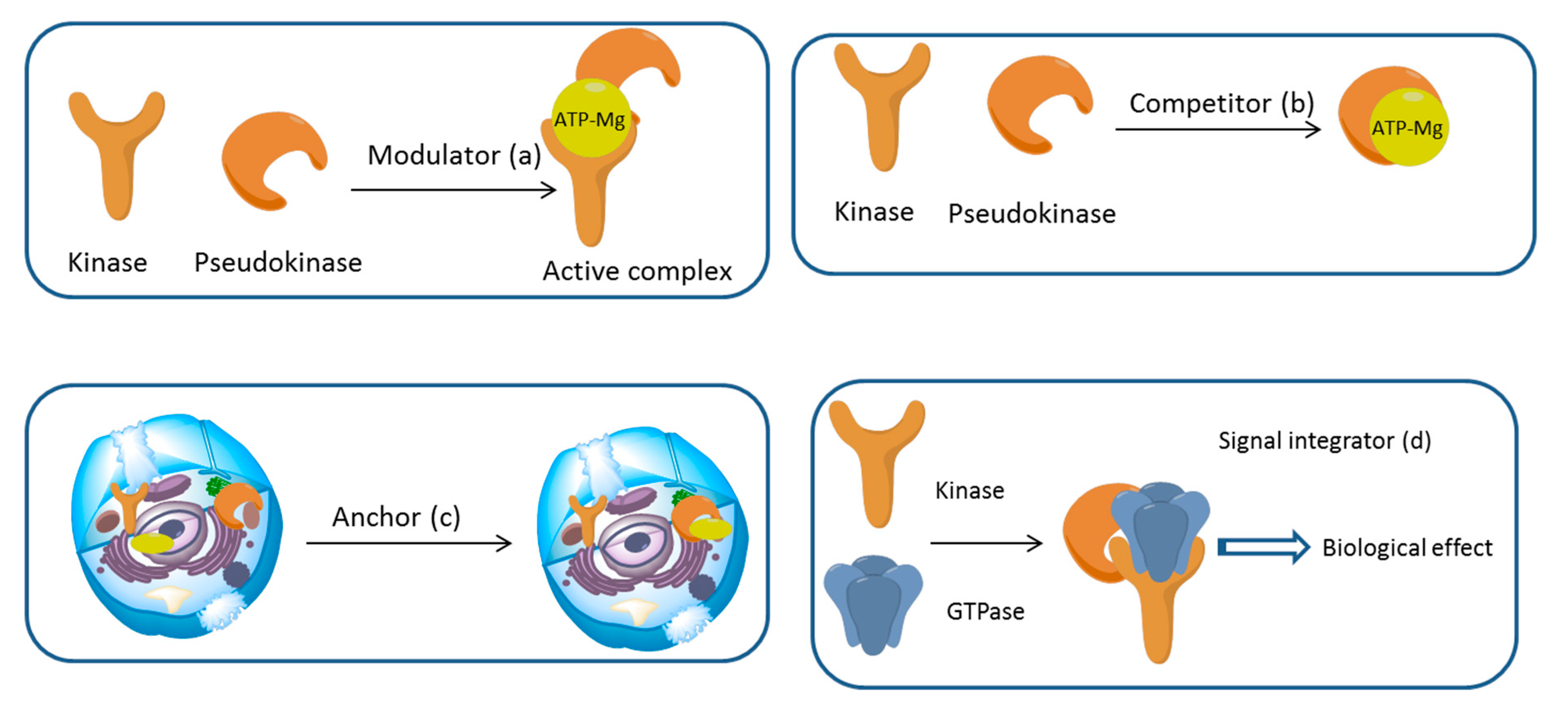
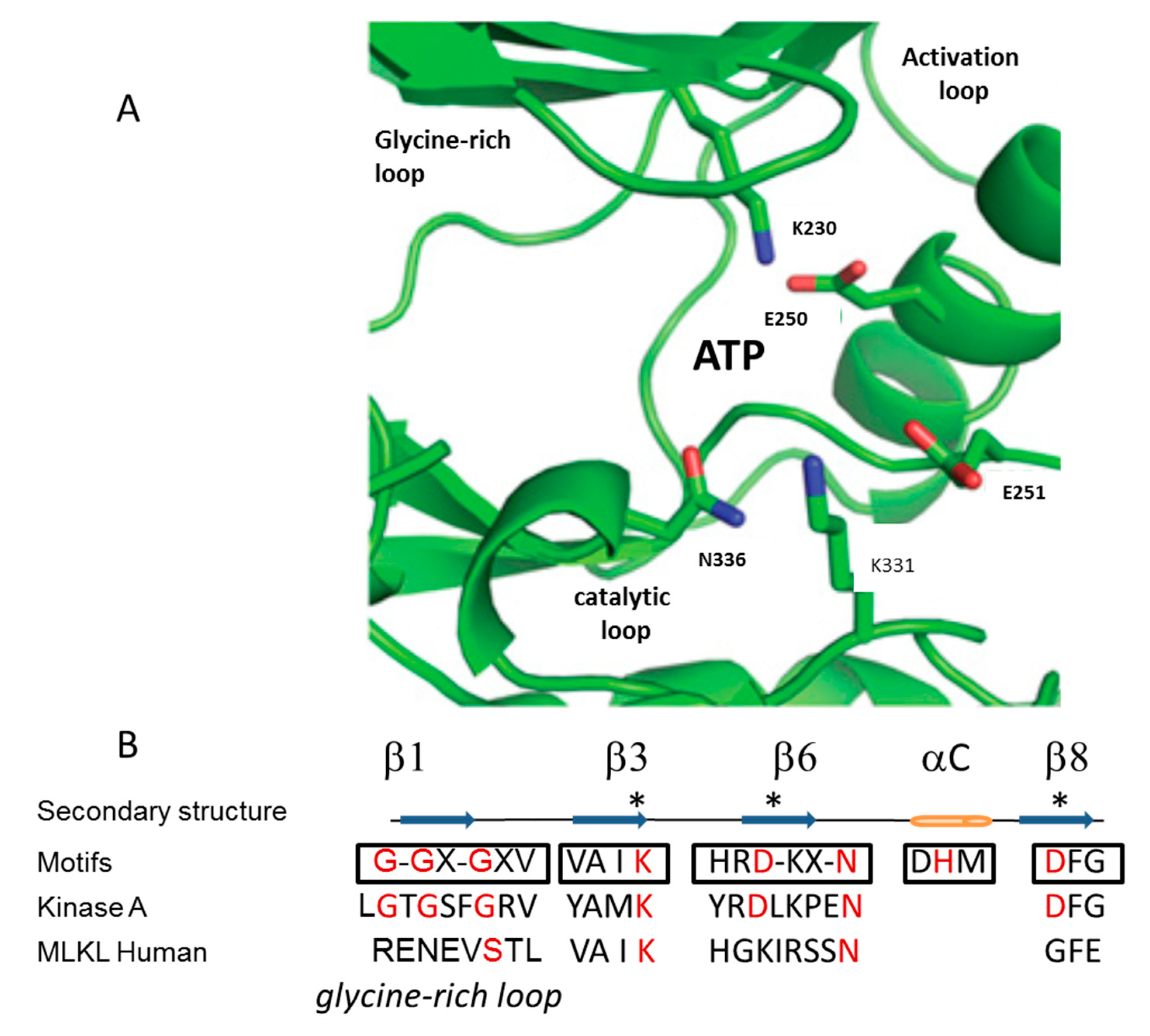
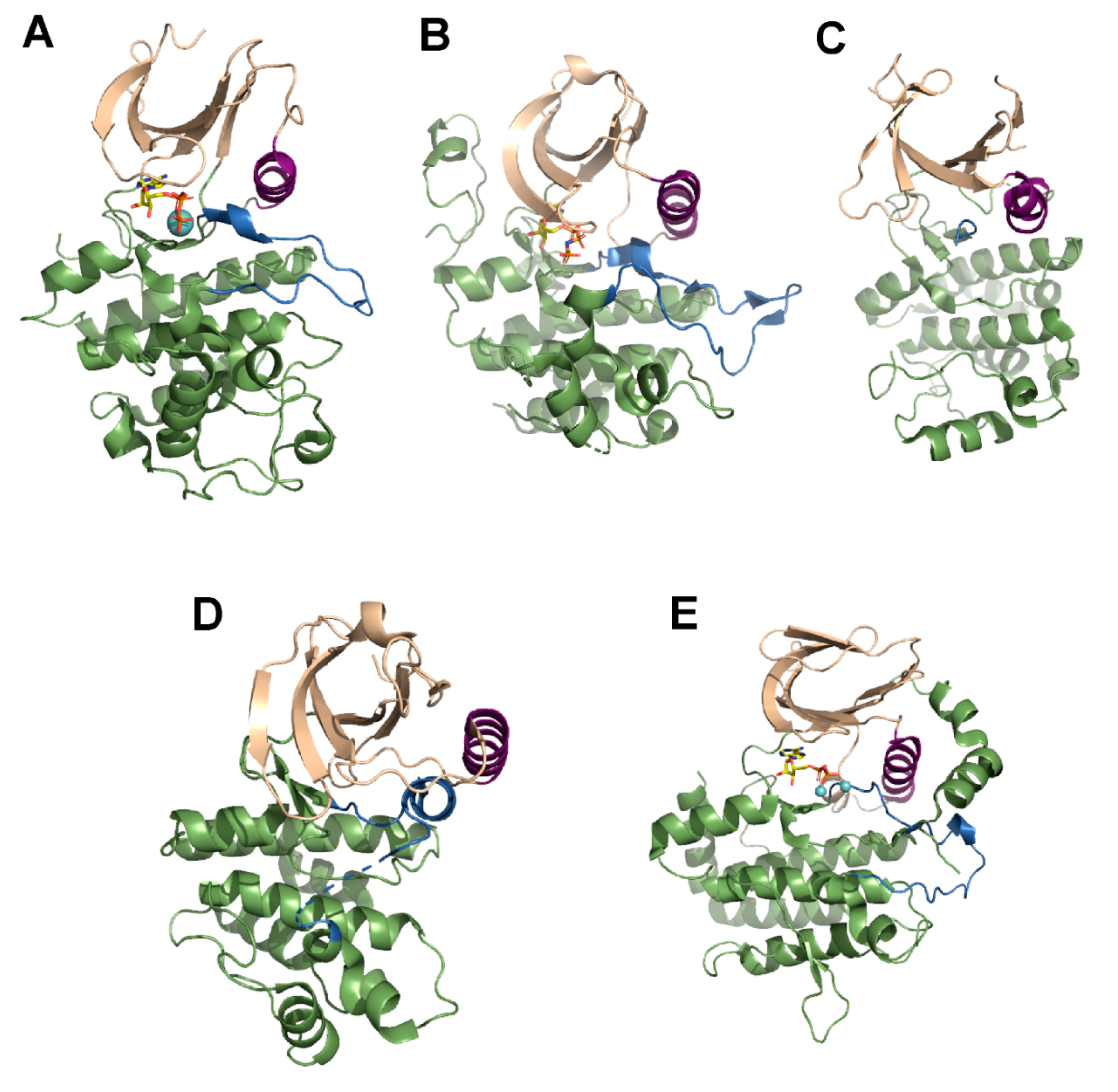
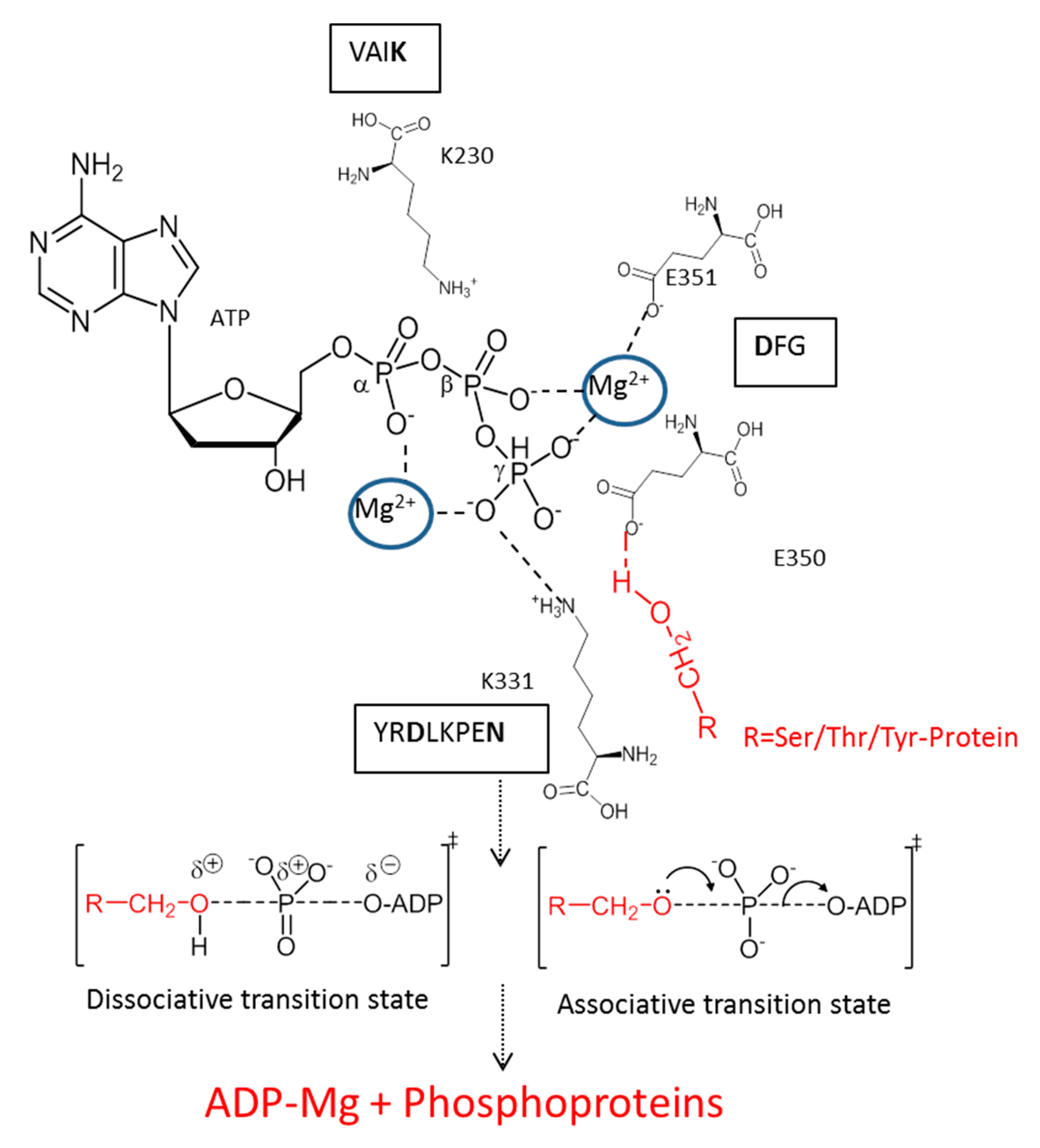
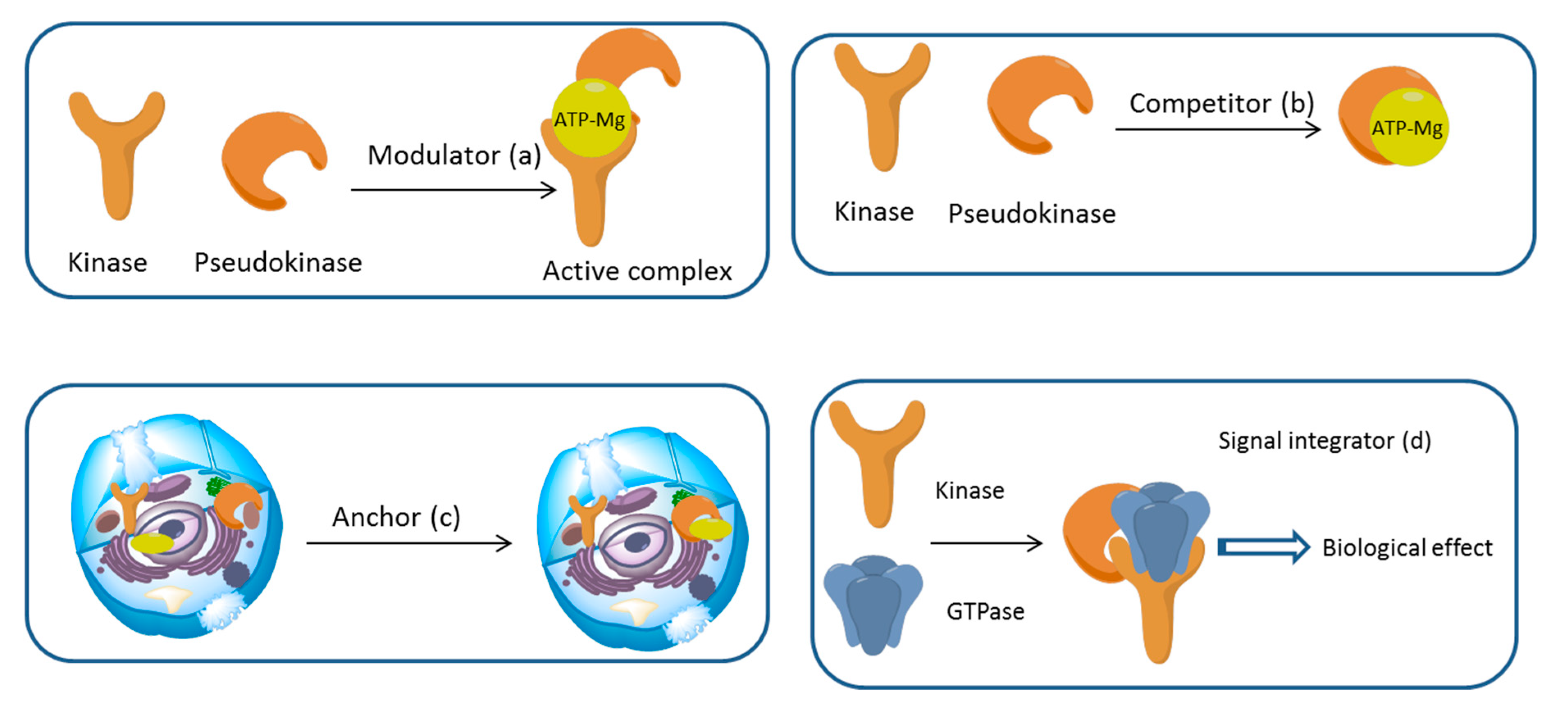
3. Mechanism of Action
4. Pseudokinases and Subcellular Localisation
4.1. Transmembrane Pseudokinases
4.2. Nuclear Pseudokinases
4.3. Cytoplasmic Pseudokinases
5. Pseudokinases and Disease
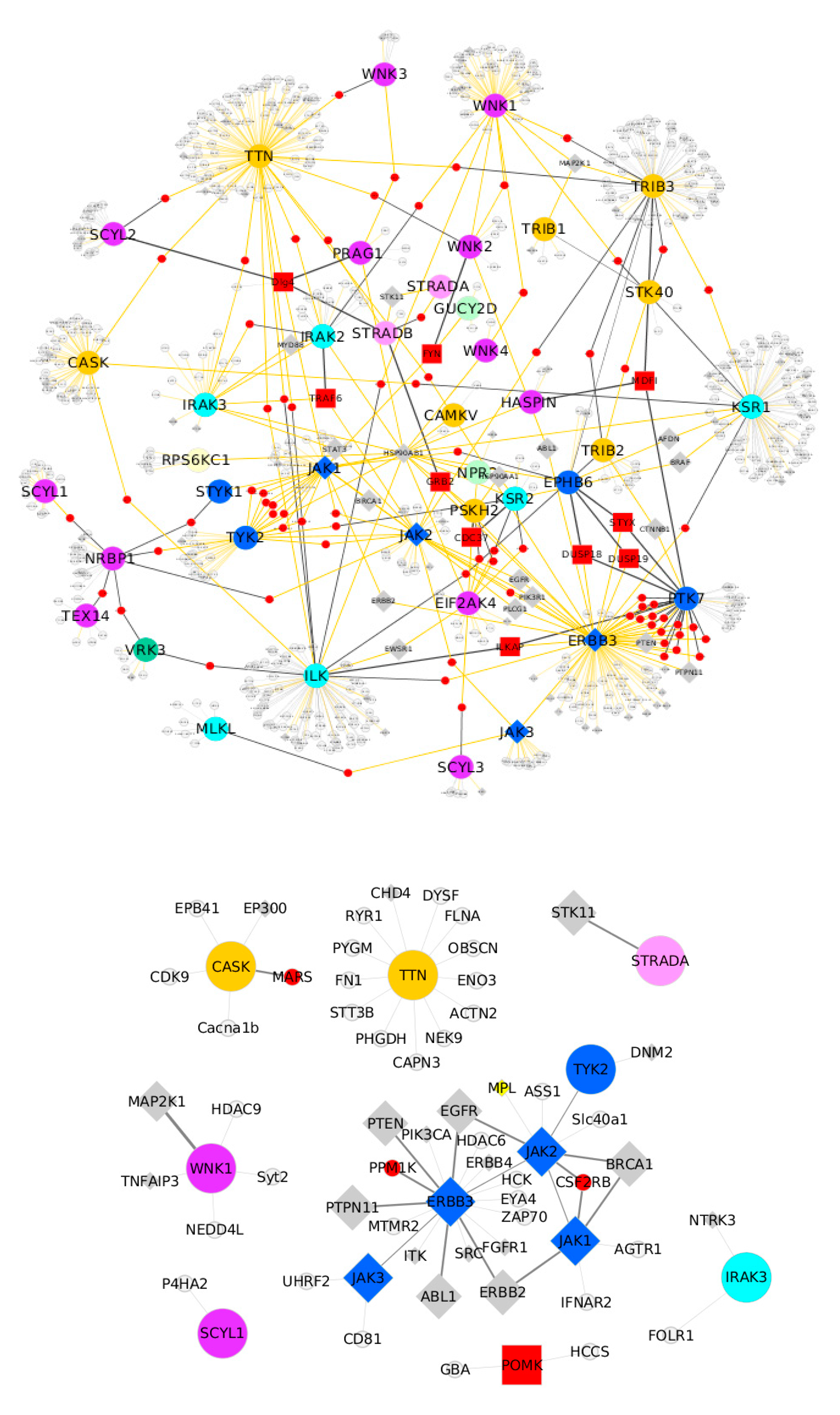
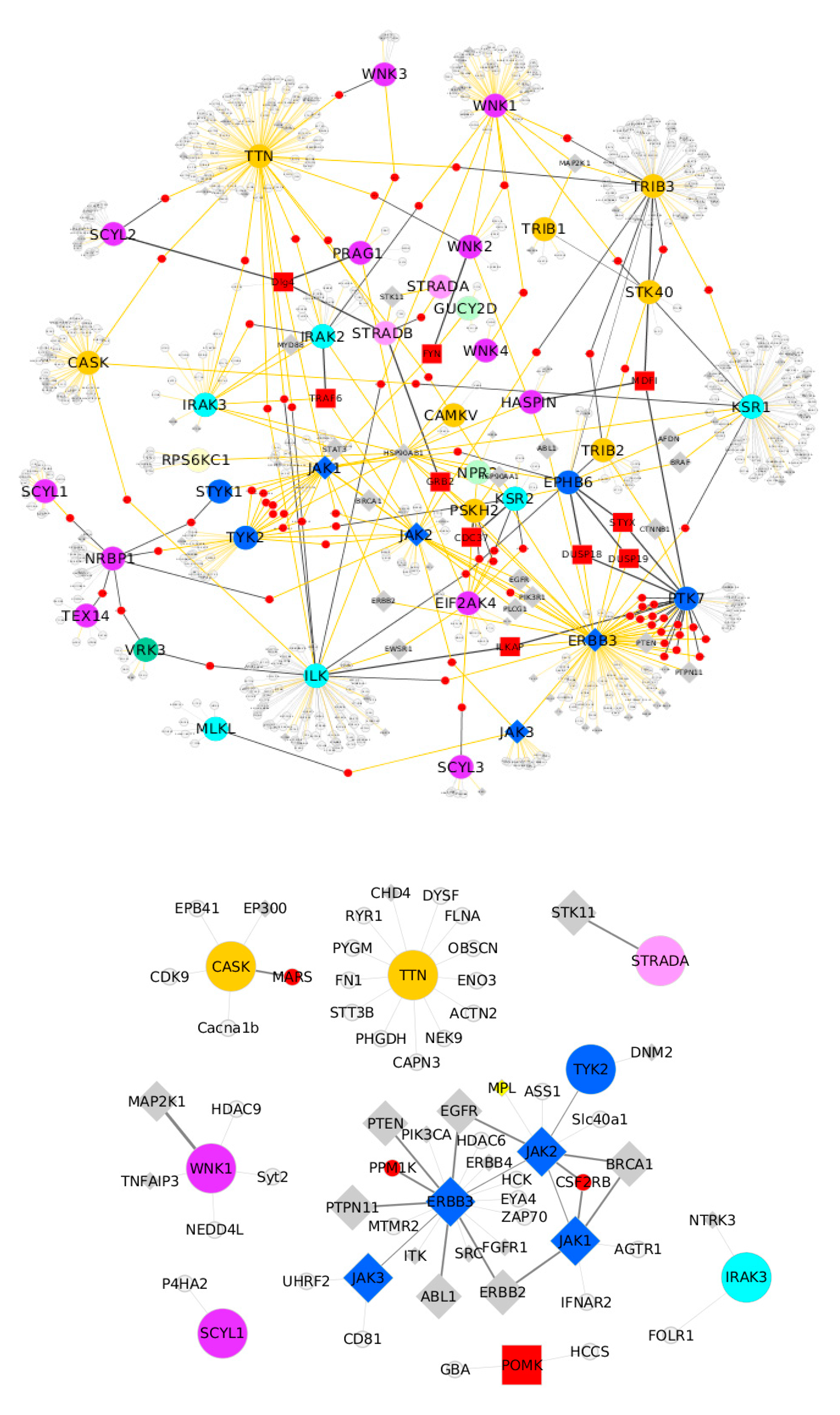
6. Pseudokinases Protein–Protein Interaction Analysis
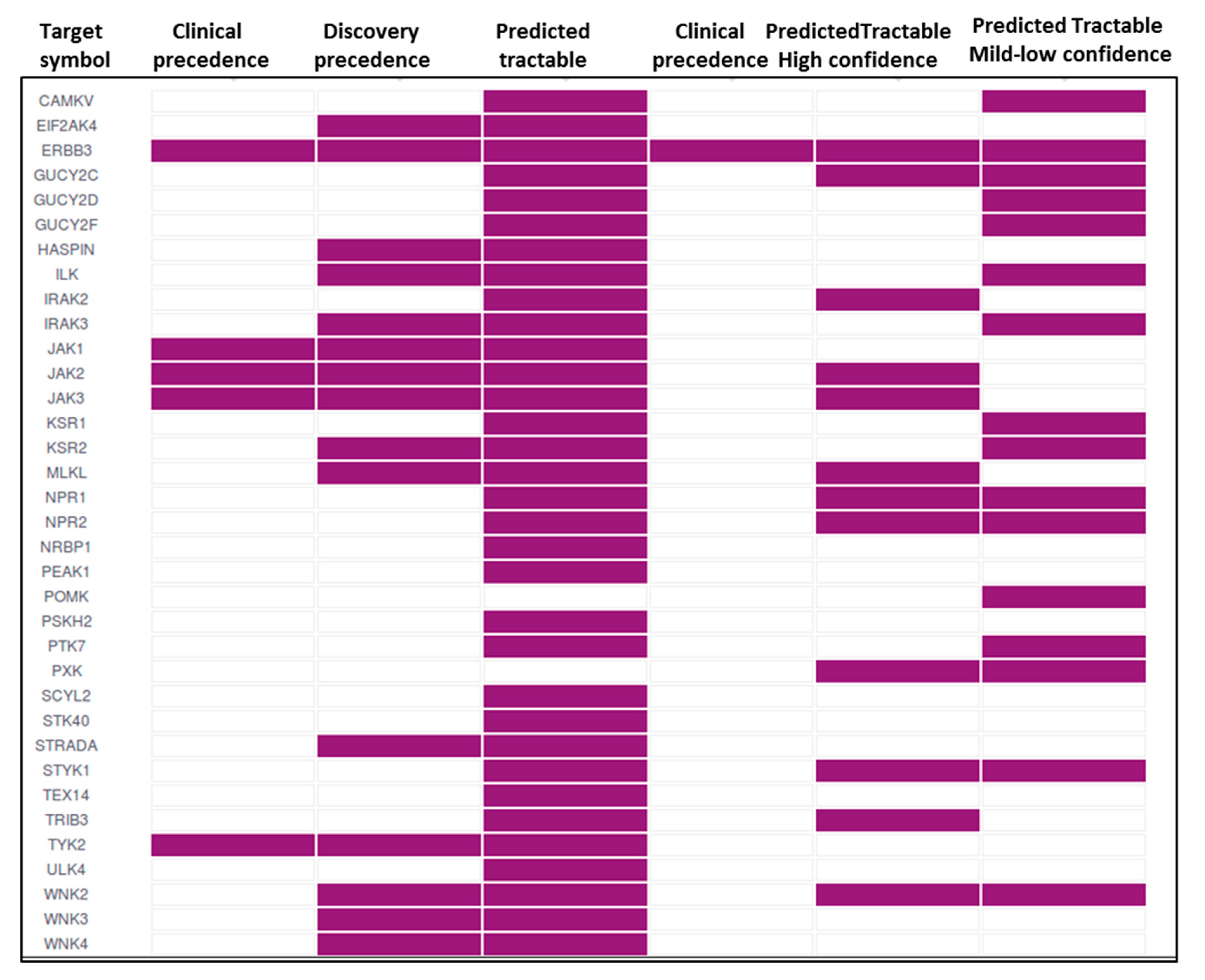
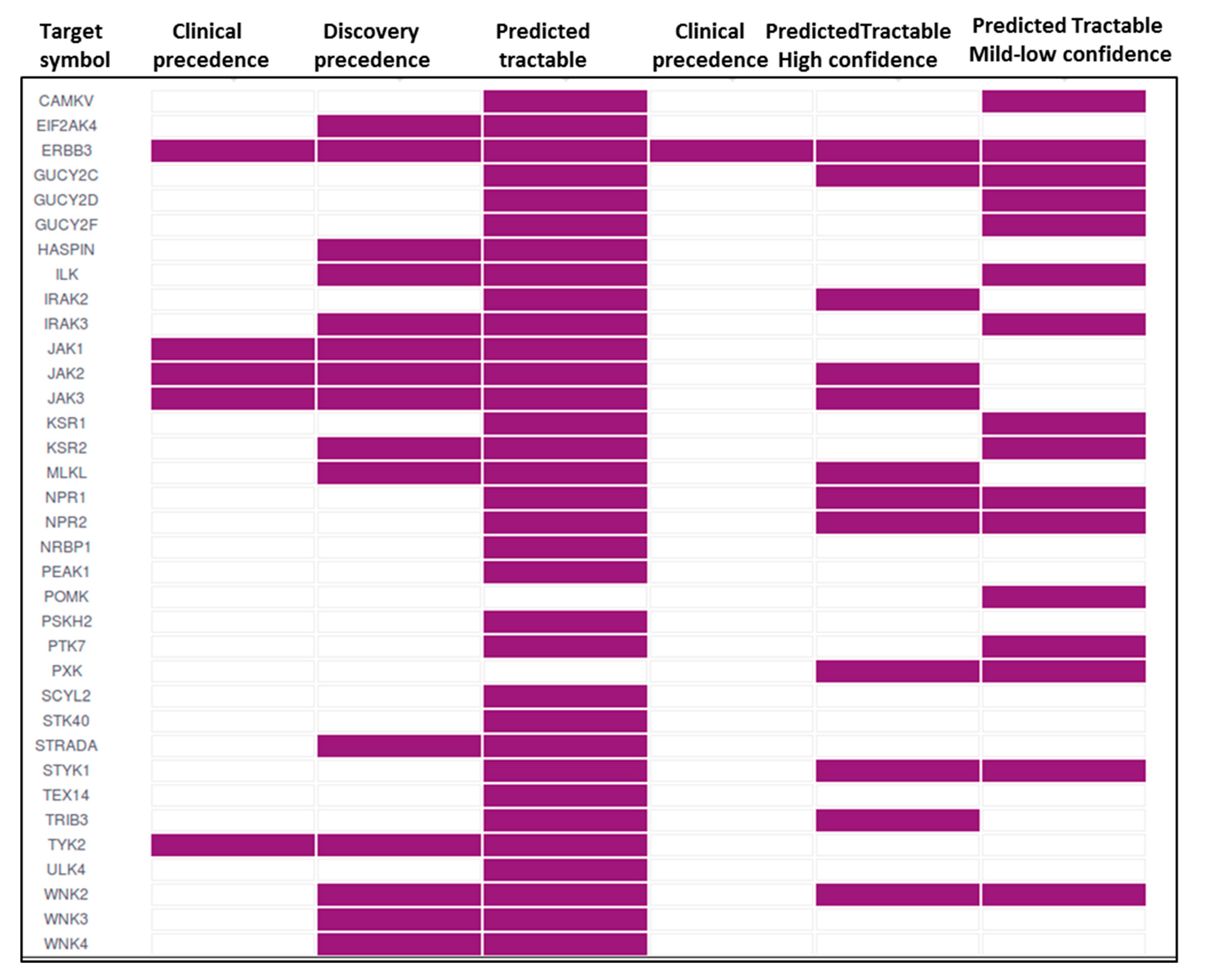
7. Pseudokinases as Drug Targets
8. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Manning, G.; Whyte, D.B.; Martinez, R.; Hunter, T.; Sudarsanam, S. The protein kinase complement of the human genome. Science 2002, 298, 1912–1934. [Google Scholar] [CrossRef] [PubMed]
- Caenepeel, S.; Charydczak, G.; Sudarsanam, S.; Hunter, T.; Manning, G. The mouse kinome: Discovery and comparative genomics of all mouse protein kinases. Proc. Natl. Acad. Sci. USA 2004, 101, 11707–11712. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Plowman, G.D.; Sudarsanam, S.; Bingham, J.; Whyte, D.; Hunter, T. The protein kinases of Caenorhabditis elegans: A model for signal transduction in multicellular organisms. Proc. Natl. Acad. Sci. USA 1999, 96, 13603–13610. [Google Scholar] [CrossRef] [PubMed]
- Giamas, G.; Man, Y.L.; Hirner, H.; Bischof, J.; Kramer, K.; Khan, K.; Ahmed, S.S.; Stebbing, J.; Knippschild, U. Kinases as targets in the treatment of solid tumors. Cell. Signal. 2010, 22, 984–1002. [Google Scholar] [CrossRef] [PubMed]
- Kwon, A.; Scott, S.; Taujale, R.; Yeung, W.; Kochut, K.J.; Eyers, P.A.; Kannan, N. Tracing the origin and evolution of pseudokinases across the tree of life. Sci. Signal. 2019, 12, eaav3810. [Google Scholar] [CrossRef] [PubMed]
- Manning, G.; Reiner, D.S.; Lauwaet, T.; Dacre, M.; Smith, A.; Zhai, Y.; Svard, S.; Gillin, F.D. The minimal kinome of Giardia lamblia illuminates early kinase evolution and unique parasite biology. Genome Biol. 2011, 12, R66. [Google Scholar] [CrossRef] [PubMed]
- Ribeiro, A.J.M.; Das, S.; Dawson, N.; Zaru, R.; Orchard, S.; Thornton, J.M.; Orengo, C.; Zeqiraj, E.; Murphy, J.M.; Eyers, P.A. Emerging concepts in pseudoenzyme classification, evolution, and signaling. Sci. Signal. 2019, 12, eaat9797. [Google Scholar] [CrossRef] [PubMed]
- Sharir-Ivry, A.; Xia, Y. Using Pseudoenzymes to Probe Evolutionary Design Principles of Enzymes. Evol. Bioinform. 2019, 15, 1176934319855937. [Google Scholar] [CrossRef]
- Jeffery, C.J. The demise of catalysis, but new functions arise: Pseudoenzymes as the phoenixes of the protein world. Biochem. Soc. Trans. 2019, 47, 371–379. [Google Scholar] [CrossRef]
- Bailey, F.P.; Byrne, D.P.; McSkimming, D.; Kannan, N.; Eyers, P.A. Going for broke: Targeting the human cancer pseudokinome. Biochem. J. 2015, 465, 195–211. [Google Scholar] [CrossRef]
- Reiterer, V.; Eyers, P.A.; Farhan, H. Day of the dead: Pseudokinases and pseudophosphatases in physiology and disease. Trends Cell Biol. 2014, 24, 489–505. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Photiou, A.; Grothey, A.; Stebbing, J.; Giamas, G. The role of pseudokinases in cancer. Cell. Signal. 2012, 24, 1173–1184. [Google Scholar] [CrossRef] [PubMed]
- Boudeau, J.; Miranda-Saavedra, D.; Barton, G.J.; Alessi, D.R. Emerging roles of pseudokinases. Trends Cell Biol. 2006, 16, 443–452. [Google Scholar] [CrossRef] [PubMed]
- Eyers, P.; Murphy, J.M. Dawn of the dead: Protein pseudokinases signal new adventures in cell biology. Biochem. Soc. Trans. 2013, 41, 969–974. [Google Scholar] [CrossRef] [PubMed]
- Kung, J.E.; Jura, N. Prospects for pharmacological targeting of pseudokinases. Drug Dev. Nat. Rev. Drug Discov. 2019, 18, 501–526. [Google Scholar] [CrossRef] [PubMed]
- Fabbro, D.; Cowan-Jacob, S.W.; Moebitz, H. Ten things you should know about protein kinases: IUPHAR Review 14. Br. J. Pharmacol. 2015, 172, 2675–2700. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Salazar, M.; Lorente, M.; Orea-Soufi, A.; Dávila, D.; Erazo, T.; Lizcano, J.; Carracedo, A.; Kiss-Toth, E.; Velasco, G. Oncosuppressive functions of tribbles pseudokinase 3. Biochem. Soc. Trans. 2015, 43, 1122–1126. [Google Scholar] [CrossRef]
- Wu, P.; Nielsen, T.E.; Clausen, M.H. FDA-approved small-molecule kinase inhibitors. Trends Pharmacol. Sci. 2015, 36, 422–439. [Google Scholar] [CrossRef] [Green Version]
- Knight, Z.A.; Shokat, K.M. Features of Selective Kinase Inhibitors. Chem. Biol. 2005, 12, 621–637. [Google Scholar] [CrossRef] [Green Version]
- Davis, M.I.; Hunt, J.P.; Herrgard, S.; Ciceri, P.; Wodicka, L.M.; Pallares, G.; Hocker, M.; Treiber, D.K.; Zarrinkar, P.P. Comprehensive analysis of kinase inhibitor selectivity. Nat. Biotechnol. 2011, 29, 1046–1051. [Google Scholar] [CrossRef]
- Zhang, X.; Gureasko, J.; Shen, K.; Cole, P.A.; Kuriyan, J. An allosteric mechanism for activation of the kinase domain of epidermal growth factor receptor. Cell 2006, 125, 1137–1149. [Google Scholar] [CrossRef] [PubMed]
- Littlefield, P.; Liu, L.; Mysore, V.; Shan, Y.; Shaw, D.E.; Jura, N. Structural analysis of the EGFR/HER3 heterodimer reveals the molecular basis for activating HER3 mutations. Sci. Signal. 2014, 7, ra114. [Google Scholar] [CrossRef] [PubMed]
- Rajakulendran, T.; Sicheri, F. Allosteric protein kinase regulation by pseudokinases: Insights from STRAD. Sci. Signal. 2010, 3, pe8. [Google Scholar] [CrossRef] [PubMed]
- Vulpetti, A.; Bosotti, R. Sequence and structural analysis of kinase ATP pocket residues. Farmaco 2004, 59, 759–765. [Google Scholar] [CrossRef] [PubMed]
- Huse, M.; Kuriyan, J. The conformational plasticity of protein kinases. Cell 2002, 109, 275–282. [Google Scholar] [CrossRef]
- Taylor, S.S.; Kornev, A.P. Protein kinases: Evolution of dynamic regulatory proteins. Trends Biochem. Sci. 2011, 36, 65–77. [Google Scholar] [CrossRef] [PubMed]
- Bose, R.; Holbert, M.A.; Pickin, K.A.; Cole, P.A. Protein tyrosine kinase-substrate interactions. Curr. Opin. Struct. Biol. 2006, 16, 668–675. [Google Scholar] [CrossRef]
- Murphy, J.M.; Zhang, Q.; Young, S.N.; Reese, M.L.; Bailey, F.P.; Eyers, P.A.; Ungureanu, D.; Hammaren, H.; Silvennoinen, O.; Varghese, L.N.; et al. A robust methodology to subclassify pseudokinases based on their nucleotide-binding properties. Biochem. J. 2013, 457, 323–334. [Google Scholar] [CrossRef]
- Jacobsen, A.V.; Murphy, J.M. The secret life of kinases: Insights into non-catalytic signalling functions from pseudokinases. Biochem. Soc. Trans. 2017, 15, 665–681. [Google Scholar] [CrossRef]
- Scheeff, E.D.; Eswaran, J.; Bunkoczi, G.; Knapp, S.; Manning, G. Structure of the pseudokinase VRK3 reveals a degraded catalytic site, a highly conserved kinase fold, and a putative regulatory binding site. Structure 2009, 17, 128–138. [Google Scholar] [CrossRef]
- Bailey, F.P.; Byrne, D.P.; Oruganty, K.; Eyers, C.E.; Novotny, C.J.; Shokat, K.M.; Kannan, N.; Eyers, P.A. The Tribbles 2 (TRB2) pseudokinase binds to ATP and autophosphorylates in a metal independent manner. Biochem. J. 2015, 467, 47–62. [Google Scholar] [CrossRef] [PubMed]
- Foulkes, D.M.; Byrne, D.P.; Bailey, F.P.; Eyers, P.A. Tribbles pseudokinases: Novel targets for chemical biology and drug discovery? Biochem. Soc. Trans. 2015, 43, 1095–1103. [Google Scholar] [CrossRef] [PubMed]
- Eyers, P.A.; Keeshan, K.; Kannan, N. Tribbles in the 21st Century: The evolving roles of tribbles pseudokinases in biology and disease. Trends Cell Biol. 2017, 27, 284–298. [Google Scholar] [CrossRef] [PubMed]
- Zeqiraj, E.; Filippi, B.M.; Goldie, S.; Navratilova, I.; Boudeau, J.; Deak, M.; Alessi, D.R.; van Aalten, D.M.F. ATP and MO25α regulate the conformational state of the STRADα pseudokinase and activation of the LKB1 tumour suppressor. PLoS Biol. 2009, 7, 1000126. [Google Scholar] [CrossRef] [PubMed]
- Cui, J.; Zhu, Q.; Zhang, H.; Cianfrocco, M.A.; Leschziner, A.E.; Dixon, J.E.; Xiao, J. Structure of Fam20A reveals a pseudokinase featuring unique disulfide pattern and inverted ATP-binding. eLife 2017, 6, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Murphy, J.M.; Lucet, I.S.; Hildebrand, J.M.; Tanzer, M.C.; Young, S.N.; Sharma, P.; Lessene, G.; Warren, S.A.; Babon, J.J.; Silke, J.; et al. Insights into the evolution of divergent nucleotide-binding mechanisms among pseudokinases revealed by crystal structures of human and mouse MLKL. Biochem. J. 2014, 457, 369–377. [Google Scholar] [CrossRef]
- Petrie, E.J.; Sandow, J.J.; Jacobsen, A.V.; Smith, B.J.; Griffin, M.D.W.; Lucet, I.S.; Dai, W.; Young, S.N.; Tanzer, M.C.; Wardak, A.; et al. Conformational switching of the pseudokinase domain promotes human MLKL tetramerization and cell death by necroptosis. Nat. Commun. 2018, 9, 2422. [Google Scholar] [CrossRef]
- Jacobsen, A.V.; Lowes, K.N.; Tanzer, M.Z.; Lucet, I.S.; Hildebrand, J.M.; Petrie, E.J.; van Delft, M.F.; Liu, Z.; Conos, S.A.; Zhang, J.-G.; et al. HSP90 activity is required for MLKL oligomerisation and membrane translocation and the induction of necroptotic cell death. Cell Death Dis. 2016, 7, e2051. [Google Scholar] [CrossRef]
- Ha, B.H.; Boggon, T.J. The crystal structure of pseudokinase PEAK1 (Sugen Kinase 269) reveals an unusual catalytic cleft and a novel mode of kinase fold dimerization. J. Biol. Chem. 2017, 293, 1642–1650. [Google Scholar] [CrossRef]
- Labesse, G.; Gelin, M.; Bessin, Y.; Lebrun, M.; Papoin, J.; Cerdan, R.; Arold, S.T.; Dubremetz, J.F. ROP2 from Toxoplasma gondii: A virulence factor with a protein- kinase foldand no enzymatic activity. Structure 2009, 17, 139–146. [Google Scholar] [CrossRef]
- Yarden, Y.; Sliwkowski, M.X. Untangling the ErbB signaling network. Nat. Rev. Mol. Cell. Biol. 2001, 2, 127–137. [Google Scholar] [CrossRef] [PubMed]
- Zhang, N.; Chang, Y.; Rios, A.; An, Z. HER3/ErbB3, an emerging cancer therapeutic target. Acta Biochim. Biophys. Sin. 2016, 48, 39–48. [Google Scholar] [CrossRef] [PubMed]
- Toms, A.V.; Deshpande, A.; McNally, R.; Jeong, Y.; Rogers, J.M.; Kim, C.U.; Gruner, S.M.; Ficarro, S.B.; Marto, J.A.; Sattler, M.; et al. Structure of a pseudokinase domain switch that controls oncogenic activation of Jak kinases. Nat. Struct. Mol. Biol. 2014, 20, 1221–1223. [Google Scholar] [CrossRef] [PubMed]
- Ungureanu, D.; Wu, J.; Pekkala, T.; Niranjan, Y.; Young, C.; Jensen, O.N.; Xu, C.F.; Neubert, T.A.; Skoda, R.C.; Hubbard, S.R.; et al. The pseudokinase domain of JAK2 is a dual-specificity protein kinase that negatively regulates cytokine signaling. Nat. Struct. Mol. Biol. 2011, 14, 971–976. [Google Scholar] [CrossRef] [PubMed]
- Laurence, A.; Pesu, M.; Silvennoinen, O.; O’shea, J. JAK kinases in health and disease: An update. Open Rheumatol. J. 2012, 6, 232–244. [Google Scholar] [CrossRef]
- Haan, C.; Behrmann, I.; Haan, S. Perspectives for the use of structural information and chemical genetics to develop inhibitors of Janus kinases. J. Cell. Mol. Med. 2010, 14, 504–527. [Google Scholar] [CrossRef] [PubMed]
- Hammarén, H.M.; Ungureanu, D.; Grisouard, J.; Skoda, R.C.; Hubbard, S.R.; Silvennoinena, O. ATP binding to the pseudokinase domain of JAK2 is critical for pathogenic activation. Proc. Natl. Acad. Sci. USA 2015, 112, 4642–4647. [Google Scholar] [CrossRef] [Green Version]
- Notarangelo, L.D.; Mella, P.; Jones, A.; de Saint Basile, G.; Savoldi, G.; Cranston, T.; Vihinen, M.; Schumacher, R.F. Mutations in severe combined immune deficiency (SCID) due to JAK3 deficiency. Hum. Mutat. 2001, 18, 255–263. [Google Scholar] [CrossRef]
- Amand, M.; Erpicum, C.; Bajou, K.; Cerignoli, F.; Blacher, S.; Martin, M.; Dequiedt, F.; Drion, P.; Singh, P.; Zurashvili, T.; et al. DUSP3/VHR is a pro-angiogenic atypical dual-specificity phosphatase. Mol. Cancer 2014, 13, 108–126. [Google Scholar] [CrossRef]
- Yoon, S.; Seger, R. The extracellular signal-regulated kinase: Multiple substrates regulate diverse cellular functions. Growth Factors 2006, 24, 21–44. [Google Scholar] [CrossRef]
- Rauch, J.; Volinsky, N.; Romano, D.; Kolch, W. The secret life of kinases: Functions beyond catalysis. Cell Commun. Signal. 2011, 9, 23. [Google Scholar] [CrossRef] [PubMed]
- Kolch, W. Coordinating ERK/MAPK signalling through scaffolds and inhibitors. Nat. Rev. Mol. Cell. Biol. 2005, 6, 827–837. [Google Scholar] [CrossRef] [PubMed]
- Costanzo-Garvey, D.L.; Pfluger, P.T.; Dougherty, M.K.; Stock, J.L.; Boehm, M.; Chaika, O.; Fernandez, M.R.; Fisher, K.; Kortum, R.L.; Hong, E.G.; et al. KSR2 is an essential regulator of AMP kinase, energy expenditure, and insulin sensitivity. Cell Metab. 2009, 10, 366–378. [Google Scholar] [CrossRef] [PubMed]
- Revelli, J.P.; Smith, D.; Allen, J.; Jeter-Jones, S.; Shadoan, M.K.; Desai, U.; Schneider, M.; van Sligtenhorst, I.; Kirkpatrick, L.; Platt, K.A.; et al. Profound obesity secondary to hyperphagia in mice lacking kinase suppressor of ras 2. Obesity 2011, 19, 1010–1018. [Google Scholar] [CrossRef]
- Pearce, L.R.; Atanassova, N.; Banton, M.C.; Bottomley, B.; van der Klaauw, A.A.; Revelli, J.P.; Hendricks, A.; Keogh, J.M.; Henning, E.; Doree, D.; et al. KSR2 mutations are associated with obesity, insulin resistance, and impaired cellular fuel oxidation. Cell 2013, 7, 765–777. [Google Scholar] [CrossRef] [PubMed]
- Rhyasen, G.W.; Starczynowski, D.T. IRAK signalling in cancer. Br. J. Cancer 2015, 112, 232–237. [Google Scholar] [CrossRef] [PubMed]
- Lin, S.C.; Lo, Y.C.; Wu, H. Helical assembly in the Myd88-IRAK4-IRAK2 complex in TLR/IL-1R signalling. Nature 2010, 465, 885–890. [Google Scholar] [CrossRef] [PubMed]
- Ge, Z.W.; Wang, B.C.; Hu, J.L.; Sun, J.J.; Wang, S.; Chen, X.J.; Meng, S.P.; Liu, L.; Cheng, Z.Y. IRAK3 gene silencing prevents cardiac rupture and ventricular remodeling through negative regulation of the NF-κB signaling pathway in a mouse model of acute myocardial infarction. J. Cell Physiol. 2019, 234, 11722–11733. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, K.; Hernandez, L.D.; Galan, J.E.; Janeway, C.A., Jr.; Medzhitov, R.; Flavell, R.A. IRAK-M is a negative regulator of toll-like receptor signaling. Cell 2002, 110, 191–202. [Google Scholar] [CrossRef]
- Kuhn, M. Molecular physiology of membrane guanylyl cyclase receptors. Physiol. Rev. 2016, 96, 751–804. [Google Scholar] [CrossRef]
- Biswas, K.H.; Shenoy, A.R.; Dutta, A.; Visweswariah, S.S. The evolution of guanylyl cyclases as multidomain proteins: Conserved features of kinase-cyclase domain fusions. J. Mol. Evol. 2009, 68, 587–602. [Google Scholar] [CrossRef] [PubMed]
- Kerr, J.S.; Wilson, C.H. Nuclear receptor-binding protein 1: A novel tumour suppressor and pseudokinase. Biochem. Soc. Trans. 2013, 41, 1055–1060. [Google Scholar] [CrossRef] [PubMed]
- Bolanos-Garcia, V.M.; Blundell, T.L. BUB1 and BUBR1: Multifaceted kinases of the cell cycle. Trends Biochem. Sci. 2001, 36, 141–150. [Google Scholar] [CrossRef] [PubMed]
- Hein, J.B.; Nilsson, J. Stable MCC binding to the APC/C is required for a functional spindle assembly checkpoint. EMBO Rep. 2014, 15, 264–272. [Google Scholar] [CrossRef] [PubMed]
- Alfieri, C.; Chang, L.; Zhang, Z.; Yang, J.; Maslen, S.; Skehel, M.; Barford, D. Molecular basis of APC/C regulation by the spindle assembly checkpoint. Nature 2016, 536, 431–436. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, H.; Hittle, J.; Zappacosta, F.; Annan, R.S.; Hershko, A.; Yen, T.J. Phosphorylation sites in BubR1 that regulate kinetochore attachment, tension, and mitotic exit. J. Cell Biol. 2008, 183, 667–680. [Google Scholar] [CrossRef] [Green Version]
- Chan, G.K.T.; Jablonski, S.A.; Sudakin, V.; Hittle, J.C.; Yen, T.J. Human BUBR1 is a mitotic checkpoint kinase that monitors CENP-E functions at kinetochores and binds the cyclosome/APC. J. Cell Biol. 1999, 146, 941–954. [Google Scholar] [CrossRef]
- Suijkerbuijk, S.J.; van Dam, T.J.; Karagöz, G.E.; von Castelmur, E.; Hubner, N.C.; Duarte, A.M.; Vleugel, M.; Perrakis, A.; Rüdiger, S.G.; Snel, B.; et al. The vertebrate mitotic checkpoint protein BUBR1 is an unusual pseudokinase. Dev. Cell. 2012, 22, 1321–1329. [Google Scholar] [CrossRef]
- Huang, Y.; Lin, L.; Liu, X.; Ye, S.; Yao, P.Y.; Wang, W.; Yang, F.; Gao, X.; Li, J.; Zhang, Y.; et al. BubR1 phosphorylates CENP-E as a switch enabling the transition from lateral association to end-on capture of spindle microtubules. Cell Res. 2019, 29, 562–578. [Google Scholar] [CrossRef]
- Li, H.; Cuenin, C.; Murr, R.; Wang, Z.-Q.; Herceg, Z. HAT cofactor Trrap regulates the mitotic checkpoint by modulation of Mad1 and Mad2 expression. EMBO J. 2004, 23, 4824–4834. [Google Scholar] [CrossRef] [Green Version]
- McMahon, S.B.; Wood, M.A.; Cole, M.D. The essential cofactor TRRAP recruits the histone acetyltransferase hGCN5 to c-Myc. Mol. Cell. Biol. 2000, 20, 556–562. [Google Scholar] [CrossRef] [PubMed]
- Murr, R.; Loizou, J.I.; Yang, Y.-G.; Cuenin, C.; Li, H.; Wang, Z.-Q.; Herceq, Z. Histone acetylation by Trrap-Tip60 modulates loading of repair proteins and repair of DNA double-strand breaks. Nat. Cell Biol. 2006, 8, 91–99. [Google Scholar] [CrossRef] [PubMed]
- Ichim, G.; Mola, M.; Finkbeiner, M.; Cros, M.-P.; Herceg, Z.; Hernandez-Vargas, H. The histone acetyltransferase component TRRAP is targeted for destruction during the cell cycle. Oncogene 2014, 33, 181–192. [Google Scholar] [CrossRef] [PubMed]
- Tapias, A.; Zhou, Z.-W.; Shi, Y.; Chong, Z.; Wang, P.; Groth, M.; Platzer, M.; Huttner, W.; Herceg, Z.; Yang, Y.G.; et al. Trrap-dependent histone acetylation specifically regulates cell-cycle gene transcription to control neural progenitor fate decisions. Cell Stem Cell 2014, 14, 632–643. [Google Scholar] [CrossRef] [PubMed]
- Kung, J.E.; Jura, N. The pseudokinase TRIB1 toggles an intramolecular switch to regulate COP1 nuclear export. EMBO J. 2019, 38, e99708. [Google Scholar] [CrossRef] [PubMed]
- Lange, S.; Xiang, F.; Yakovenko, A.; Vihola, A.; Hackman, P.; Rostkova, E. The kinase domain of Titin controls muscle gene expression and protein turnover. Science 2005, 308, 1599–1603. [Google Scholar] [CrossRef]
- Bogomolovas, J.; Gasch, A.; Simkovic, F.; Rigden, D.J.; Labeit, S.; Mayans, O. Titin kinase is an inactive pseudokinase scaffold that supports MuRF1 recruitment to the sarcomeric M-line. Open Biol. 2014, 4, 140041. [Google Scholar] [CrossRef] [Green Version]
- Puchner, E.M.; Alexandrovich, A.; Kho, A.L.; Hensen, U.; Schafer, L.V.; Brandmeier, B. Mechanoenzymatics of titin kinase. Proc. Natl. Acad. Sci. USA 2008, 105, 13385–13390. [Google Scholar] [CrossRef] [Green Version]
- Pelletier, S. SCYL pseudokinases in neuronal function and survival. Neural Regen. Res. 2016, 11, 42–44. [Google Scholar] [CrossRef]
- A Partnership to Transform Drug Discovery through the Systematic Identification and Prioritisation of Targets. Available online: www.opentargets.org/ (accessed on 22 August 2019).
- Tang, B.L. (WNK)ing at death: With-no-lysine (Wnk) kinases in neuropathies and neuronal survival. Brain Res. Bull. 2016, 125, 92–98. [Google Scholar] [CrossRef]
- Xu, B.-E.; English, J.M.; Wilsbacher, J.L.; Stippec, S.; Goldsmith, E.J.; Cobb, M.H. WNK1, a novel mammalian serine/threonine protein kinase lacking the catalytic lysine in subdomain II. J. Biol. Chem. 2000, 275, 16795–16801. [Google Scholar] [CrossRef] [PubMed]
- Wu, A.; Wolley, M.; Stowasser, M. The interplay of renal potassium and sodium handling in blood pressure regulation: Critical role of the WNK-SPAK-NCC pathway. J. Hum. Hypertens. 2019, 33, 508–523. [Google Scholar] [CrossRef] [PubMed]
- Shekarabi, M.; Zhang, J.; Khanna, A.R.; Ellison, D.H.; Delpire, E.; Kahle, K.T. WNK Kinase Signaling in Ion Homeostasis and Human Disease. Cell Metab. 2017, 25, 285–299. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pommier, Y.; Kiselev, E.; Marchand, C. Interfacial inhibitors. Bioorg. Med. Chem. Lett. 2015, 25, 3961–3965. [Google Scholar] [CrossRef] [PubMed]
- Jin, L.; Wang, W.; Fang, G. Targeting protein-protein interaction by small molecules. Annu. Rev. Pharmacol. Toxicol. 2014, 54, 435–456. [Google Scholar] [CrossRef] [PubMed]
- Jubb, H.; Blundell, T.L.; Ascher, D.B. Flexibility and small pockets at protein-protein interfaces: New insights into druggability. Prog. Biophys. Mol. Biol. 2015, 119, 2–9. [Google Scholar] [CrossRef] [PubMed]
- Karvonen, H.; Perttilä, R.; Niininen, W.; Barker, H.; Ungureanu, D. Targeting Wnt signaling pseudokinases in hematological cancers. Eur. J. Haematol. 2018, 101, 457–465. [Google Scholar] [CrossRef] [PubMed]
- Byrne, D.P.; Foulkes, D.M.; Eyers, P.A. Pseudokinases: Update on their functions and evaluation as new drug targets. Future Med. Chem. 2017, 9, 245–265. [Google Scholar] [CrossRef]
- Cowan-Jacob, S.W.; Jahnke, W.; Knapp, S. Novel approaches for targeting kinases: Allosteric inhibition, allosteric activation and pseudokinases. Future Med. Chem. 2014, 6, 541–561. [Google Scholar] [CrossRef]
- Yokoyama, T.; Toki, T.; Aoki, Y.; Kanezaki, R.; Park, M.J.; Kanno, Y.; Takahara, T.; Yamazaki, Y.; Ito, E.; Hayashi, Y.; et al. Identification of TRIB1 R107L gain-of-function mutation in human acute megakaryocytic leukemia. Blood 2012, 119, 2608–2611. [Google Scholar] [CrossRef]
- Yamada, K.; Park, H.M.; Rigel, D.F.; DiPetrillo, K.; Whalen, E.J.; Anisowicz, A.; Beil, M.; Berstler, J.; Brocklehurst, C.E.; Burdick, D.A.; et al. Small-molecule WNK inhibition regulates cardiovascular and renal function. Nat. Chem. Biol. 2016, 12, 896–898. [Google Scholar] [CrossRef] [PubMed]
- Burke, J.R.; Cheng, L.; Gillooly, K.M.; Strnad, J.; Zupa-Fernandez, A.; Catlett, I.M.; Zhang, Y.; Heimrich, E.M.; McIntyre, K.W.; Cunningham, M.D.; et al. Autoimmune pathways in mice and humans are blocked by pharmacological stabilization of the TYK2 pseudokinase domain. Sci. Transl. Med. 2019, 11, eaaw1736. [Google Scholar] [CrossRef] [PubMed]
- Moslin, R.; Zhang, Y.; Wrobleski, S.T.; Lin, S.; Mertzman, M.; Spergel, S.; Tokarski, J.S.; Strnad, J.; Gillooly, K.; McIntyre, K.W.; et al. Identification of N-Methyl Nicotinamide and N-Methyl Pyridazine-3-Carboxamide Pseudokinase Domain Ligands as Highly Selective Allosteric Inhibitors of Tyrosine Kinase 2 (TYK2). J. Med. Chem. 2019. [Google Scholar] [CrossRef] [PubMed]
- Zhavoronkov, A.; Ivanenkov, Y.A.; Aliper, A.; Veselov, M.S.; Aladinskiy, V.A.; Aladinskaya, A.V.; Terentiev, V.A.; Polykovskiy, D.A.; Kuznetsov, M.D.; Asadulaev, A.; et al. Deep learning enables rapid identification of potent DDR1 kinase inhibitors. Nat. Biotechnol. 2019, 37, 1038–1040. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Target | Max Phase | Molecula Type | Drugs |
|---|---|---|---|
| JAK2 | Phase III | Small molecule | 5 |
| ERBB3 | Phase II | Antibody | 4 |
| JAK3 | Phase II | Small molecule | 4 |
| JAK1 | Phase II | Small molecule | 3 |
| JAK2 | Phase I | Small molecule | 3 |
| JAK1 | Phase III | Small molecule | 3 |
| JAK2 | Phase II | Small molecule | 3 |
| JAK2 | Phase IV | Small molecule | 2 |
| ERBB3 | Phase IV | Small molecule | 2 |
| GUCY2C | Phase IV | protein | 2 |
| ERBB3 | Phase I | Antibody | 2 |
| EPHB6 | Phase IV | Small molecule | 1 |
| JAK1 | Phase IV | Small molecule | 1 |
| ERBB3 | Phase III | Small molecule | 1 |
| ERBB3 | Phase III | Antibody | 1 |
| ERBB3 | Phase II | Small molecule | 1 |
| NPR1 | Phase IV | protein | 1 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tomoni, A.; Lees, J.; Santana, A.G.; Bolanos-Garcia, V.M.; Bastida, A. Pseudokinases: From Allosteric Regulation of Catalytic Domains and the Formation of Macromolecular Assemblies to Emerging Drug Targets. Catalysts 2019, 9, 778. https://doi.org/10.3390/catal9090778
Tomoni A, Lees J, Santana AG, Bolanos-Garcia VM, Bastida A. Pseudokinases: From Allosteric Regulation of Catalytic Domains and the Formation of Macromolecular Assemblies to Emerging Drug Targets. Catalysts. 2019; 9(9):778. https://doi.org/10.3390/catal9090778
Chicago/Turabian StyleTomoni, Andrada, Jonathan Lees, Andrés G. Santana, Victor M. Bolanos-Garcia, and Agatha Bastida. 2019. "Pseudokinases: From Allosteric Regulation of Catalytic Domains and the Formation of Macromolecular Assemblies to Emerging Drug Targets" Catalysts 9, no. 9: 778. https://doi.org/10.3390/catal9090778
APA StyleTomoni, A., Lees, J., Santana, A. G., Bolanos-Garcia, V. M., & Bastida, A. (2019). Pseudokinases: From Allosteric Regulation of Catalytic Domains and the Formation of Macromolecular Assemblies to Emerging Drug Targets. Catalysts, 9(9), 778. https://doi.org/10.3390/catal9090778