Recycling of Gas Phase Residual Dichloromethane by Hydrodechlorination: Regeneration of Deactivated Pd/C Catalysts

 ,
,  ,
,  ,
, 
Abstract
1. Introduction
2. Results and Discussion
2.1. Evolution of Catalytic Activity
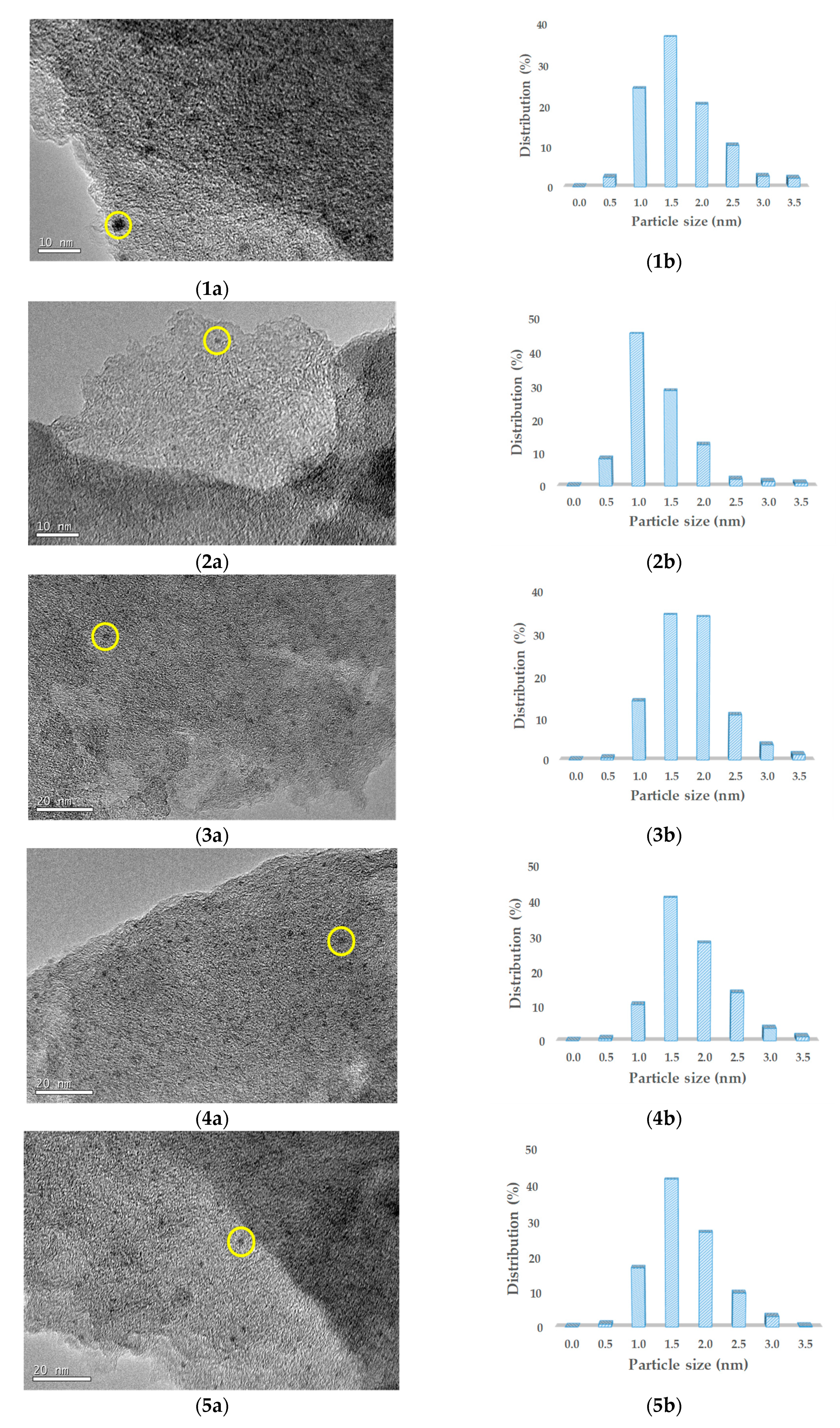
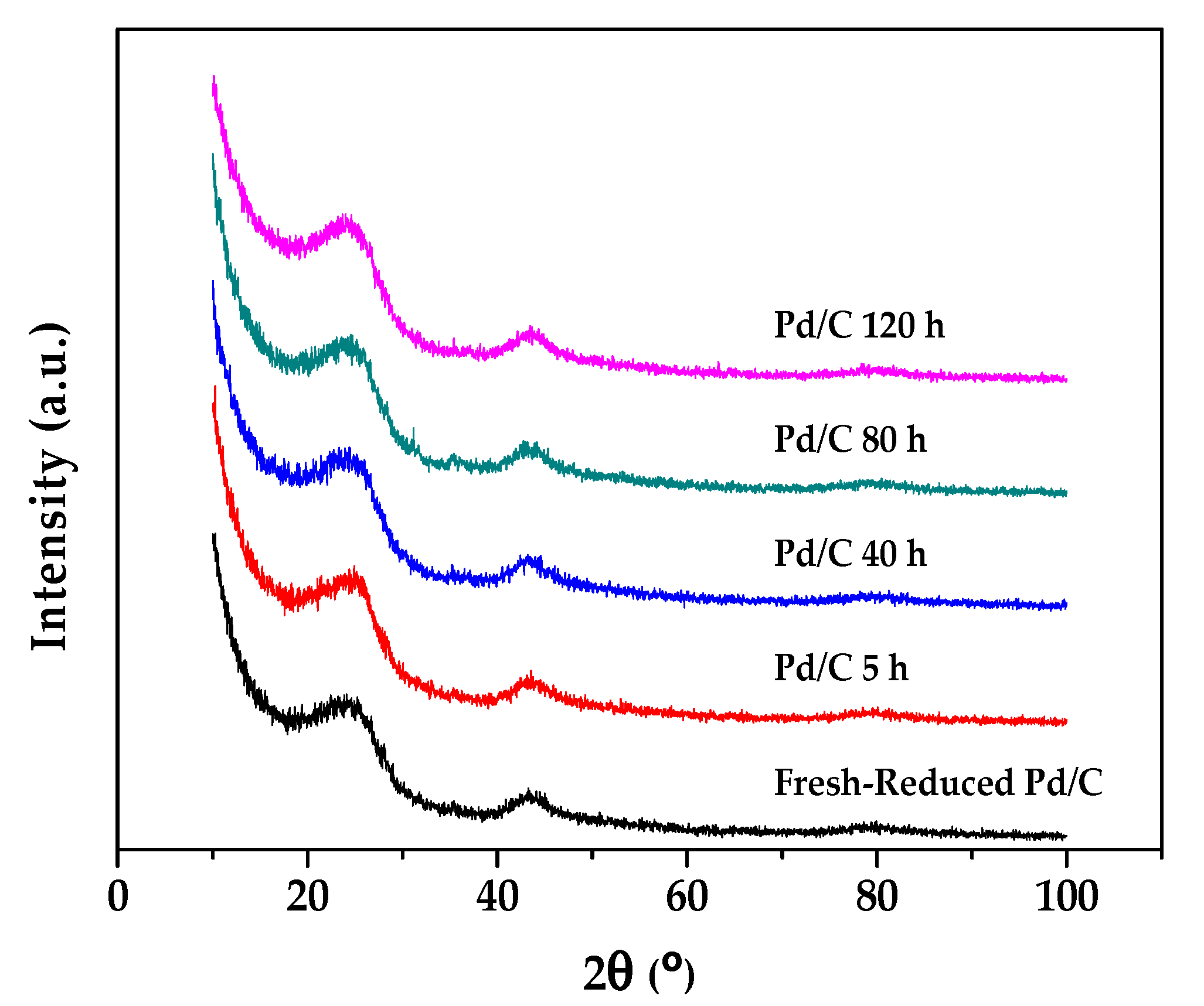
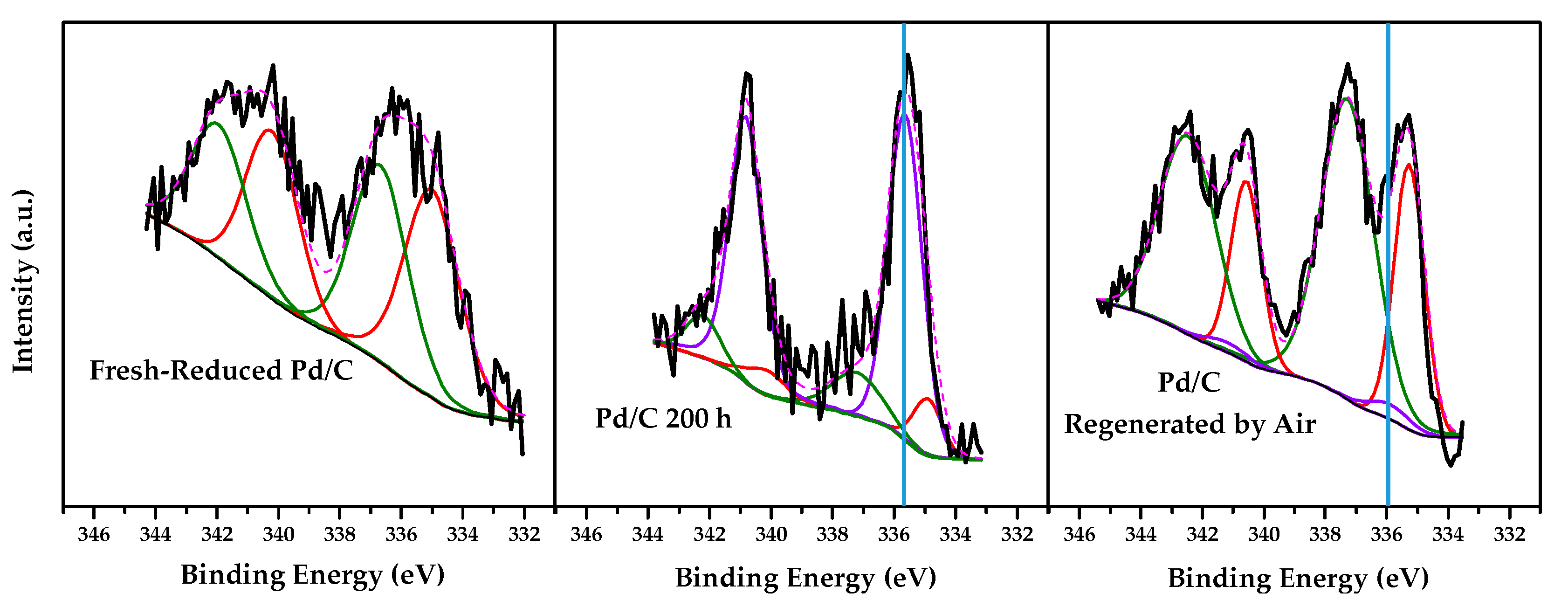
2.2. Characterization Results
3. Materials and Methods
3.1. Catalysts Preparation
3.2. Catalyst Characterization
3.3. Catalytic Activity Experiments
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Marty, M.; Spurlock, F.; Barry, T. Volatile organic compounds from pesticide application and contribution to tropospheric ozone. In Hayes’ Handbook of Pesticide Toxicology, 3rd ed.; Krieger, R., Ed.; Academic Press: London, UK, 2010; Volume 1, pp. 571–585. [Google Scholar] [CrossRef]
- Dobrzynska, E.; Posniak, M.; Szewczynska, M.; Buszewski, B. Chlorinated volatile organic compounds—Old, however, actual analytical and toxicological problem. Crit. Rev. Anal. Chem. 2010, 40, 41–57. [Google Scholar] [CrossRef]
- Ciccioli, P. VOCs and air pollution. In Chemistry and Analysis of Volatile Organic Compounds in the Environment; Bloemen, H.J.T., Burn, J., Eds.; Springer-Science+Business Media, B.V.: Dordrecht, The Netherlands, 1993; pp. 92–174. [Google Scholar] [CrossRef]
- Lewis, N.M.; Gatchett, A.M. U.S. environmental protection agency’s SITE emerging technology program: 1991 update. J. Air Waste Manag. Assoc. 1991, 41, 1645–1653. [Google Scholar] [CrossRef]
- De Pedro, Z.M.; Casas, J.A.; Gomez-Sainero, L.M.; Rodriguez, J.J. Hydrodechlorination of dichloromethane with a Pd/AC catalyst: Reaction pathway and kinetics. Appl. Catal. B Environ. 2010, 98, 79–85. [Google Scholar] [CrossRef]
- Keane, M.A. Supported transition metal catalysts for hydrodechlorination reactions. ChemCatChem 2011, 3, 800–821. [Google Scholar] [CrossRef]
- Álvarez-Montero, M.A.; Gómez-Sainero, L.M.; Juan-Juan, J.; Linares-Solano, A.; Rodriguez, J.J. Gas-Phase hydrodechlorination of dichloromethane with activated carbon-supported metallic catalysts. Chem. Eng. J. 2010, 162, 599–608. [Google Scholar] [CrossRef]
- Ordoñez, S.; Sastre, H.; Diez, F.V. Hydrodechlorination of aliphatic organochlorinated compounds over commercial hydrogenation catalysts. Appl. Catal. B Environ. 2000, 25, 49–58. [Google Scholar] [CrossRef]
- Legawiec-Jarzyna, M.; Srebowata, A.; Juszczyk, W.; Karpinski, Z. Hydrodechlorination of chloroalkanes on supported platinum catalysts. React. Kinet. Catal. Lett. 2006, 87, 291–296. [Google Scholar] [CrossRef]
- Urbano, F.J.; Marinas, J.M. Hydrogenolysis of organohalogen compounds over palladium supported catalysts. J. Mol. Catal. A Chem. 2001, 173, 329–345. [Google Scholar] [CrossRef]
- Kalnes, T.N.; James, R.B. Hydrogenation and recycle of organic waste streams. Environ. Prog. 1988, 7, 185–191. [Google Scholar] [CrossRef]
- Carpenter, B.H.; Wilson, D.L. Technical/Economic assessment of selected PCB decontamination processes. J. Hazard. Mater. 1988, 17, 125–148. [Google Scholar] [CrossRef]
- Karasek, F.W.; Clement, R.E.; Viau, A.C. Distribution of PCDDs and other toxic compounds generated on fly ash particulates in municipal incinerators. J. Chromatogr. 1982, 239, 173–180. [Google Scholar] [CrossRef]
- Martin-Martinez, M.; Álvarez-Montero, A.; Gómez-Sainero, L.M.; Baker, R.T.; Palomar, J.; Omar, S.; Eser, S.; Rodriguez, J.J. Deactivation behavior of Pd/C and Pt/C catalysts in the gas-phase hydrodechlorination of chloromethanes: Structure reactivity relationship. Appl. Catal. B Environ. 2015, 162, 532–543. [Google Scholar] [CrossRef]
- Martin-Martinez, M.; Gómez-Sainero, L.M.; Alvarez-Montero, M.A.; Bedia, J.; Rodriguez, J.J. Comparison of different precious metals in activated carbon-supported catalysts for the gas-phase hydrodechlorination of chloromethanes. Appl. Catal. B Environ. 2013, 132, 256–265. [Google Scholar] [CrossRef]
- Fernandez-Ruiz, C.; Bedia, J.; Bonal, P.; Rodriguez, J.J.; Gómez-Sainero, L.M. Chloroform conversion into ethane and propane by catalytic hydrodechlorination with Pd supported on activated carbons from lignin. Catal. Sci. Technol. 2018, 8, 3926–3935. [Google Scholar] [CrossRef]
- Gómez-Sainero, L.M.; Palomar, J.; Omar, S.; Fernández, C.; Bedia, J.; Álvarez-Montero, A.; Rodriguez, J.J. Valorization of chloromethanes by hydrodechlorination with metallic catalysts. Catal. Today 2018, 310, 75–85. [Google Scholar] [CrossRef]
- Mori, T.; Hirose, K.; Kikuchi, T.; Kubo, J.; Morikawa, Y. Formation of higher hydrocarbons from chloromethanes via hydrodechlorination over Pd/SiO2 catalyst. J. Jpn. Petrol. Inst. 2002, 45, 256–259. [Google Scholar] [CrossRef]
- Lee, S.R.; Cho, J.M.; Son, M.; Park, M.J.; Kim, W.Y.; Kim, S.Y.; Bae, J.W. Selective hydrodechlorination of trichloromethane to dichloromethane over bimetallic Pt-Pd/KIT-6: Catalytic activity and reaction kinetics. Chem. Eng. J. 2018, 331, 556–569. [Google Scholar] [CrossRef]
- Simagina, V.I.; Netskina, O.V.; Tayban, E.S.; Komova, O.V.; Grayfer, E.D.; Ischenko, A.V.; Pazhetnov, E.M. The effect of support properties on the activity of Pd/C catalysts in the liquid-phase hydrodechlorination of chlorobenzene. Appl. Catal. A Gen. 2010, 379, 87–94. [Google Scholar] [CrossRef]
- Concibido, N.C.; Okuda, T.; Nishijima, W.; Okada, M. Deactivation and reactivation of Pd/C catalyst used in repeated batch hydrodechlorination of PCE. Appl. Catal. B Environ. 2007, 71, 64–69. [Google Scholar] [CrossRef]
- Moon, D.J.; Chung, M.J.; Park, K.Y.; Hong, S.I. Deactivation of Pd catalysts in the hydrodechlorination of chloropentafluoroethane. Appl. Catal. A Gen. 1998, 168, 159–170. [Google Scholar] [CrossRef]
- Zheng, X.; Xiao, Q.; Zhang, Y.; Zhang, X.L.; Zhong, Y.J.; Zhu, W.D. Deactivation of Pd/C Catalysts in the Hydrodechlorination of the Chlorofluorocarbons CFC-115 and CFC-12. Catal. Today 2011, 175, 615–618. [Google Scholar] [CrossRef]
- Ziemecki, S.B.; Jones, G.A. Formation of interstitial palladium-carbon phase by interaction of ethylene, acetylene, and carbon monoxide with palladium. J. Catal. 1985, 95, 621–622. [Google Scholar] [CrossRef]
- Ha, J.M.; Kim, D.; Kim, J.; Kim, S.K.; Ahn, B.S.; Kang, J.W. Supercritical-Phase-Assisted highly selective and active catalytic hydrodechlorination of the ozone-depleting refrigerant CHClF2. Chem. Eng. J. 2012, 213, 346–355. [Google Scholar] [CrossRef]
- Van de Sandt, E.J.A.X.; Wiersma, A.; Makkee, M.; van Bekkum, H.; Moulijn, J.A. Palladium black as model catalyst in the hydrogenolysis of CC12F2 (CFC-12) into CH2F2 (HFC-32). Appl. Catal. A Gen. 1997, 155, 59–73. [Google Scholar] [CrossRef]
- Legawiec-Jarzyna, M.; Srebowata, A.; Juszczyk, W.; Karpinski, Z. Hydrodechlorination over Pd–Pt/Al2O3 catalysts a comparative study of chlorine removal from dichlorodifluoromethane, carbon tetrachloride and 1,2-dichloroethane. Appl. Catal. A Gen. 2004, 271, 61–68. [Google Scholar] [CrossRef]
- Ordóñez, S.; Díaz, E.; Díez, F.V.; Sastre, H. Regeneration of Pd/Al2O3 catalysts used for tetrachloroethylene hydrodechlorination. React. Kinet. Catal. Lett. 2007, 90, 101–106. [Google Scholar] [CrossRef]
- González, C.A.; Bartoszek, M.; Martin, A.; Montes de Correa, C. Hydrodechlorination of light organochlorinated compounds and their mixtures over Pd/TiO2-washcoated minimonoliths. Ind. Eng. Chem. Res. 2009, 48, 2826–2835. [Google Scholar] [CrossRef]
- Golubina, E.V.; Lokteva, E.S.; Lunin, V.V.; Turakulova, A.O.; Simagina, V.I.; Stoyanova, I.V. Modification of the supported palladium catalysts surface during hydrodechlorination of carbon tetrachloride. Appl. Catal. A Gen. 2003, 241, 123–132. [Google Scholar] [CrossRef]
- De Pedro, Z.M.; Gómez-Sainero, L.M.; Gonzalez-Serrano, E.; Rodríguez, J.J. Gas-Phase hydrodechlorination of dichloromethane at low concentrations with palladium/carbon catalysts. Ind. Eng. Chem. Res. 2006, 45, 7760–7766. [Google Scholar] [CrossRef]
- Moulder, J.F.; Stickle, W.F.; Sobol, P.E.; Bomben, K.D. Standard XPS spectra of the elements. In Handbook of X-ray Photoelectron Spectroscopy: A Reference Book of Standard Spectra for Identification and Interpretation of XPS Data; Chastain, J., Ed.; Perkin-Elmer Corporation, Physical Electronics Division: Eden Prairie, MN, USA, 1992; pp. 118–119. [Google Scholar]
- Gómez-Sainero, L.M.; Grau, J.M.; Arcoya, A.; Seoane, X.L. Carbon-Supported palladium catalysts for liquid-phase hydrodechlorination of carbon tetrachloride to chloroform. Stud. Surf. Sci. Catal. 2000, 130, 2009–2014. [Google Scholar] [CrossRef]
- Gómez-Sainero, L.M.; Xosé, L.; Seoane, X.L.; Fierro, J.L.G.; Arcoya, A. Liquid-Phase hydrodechlorination of CCl4 to CHCl3 on Pd/carbon catalysts: Nature and role of Pd active species. J. Catal. 2002, 209, 279–288. [Google Scholar] [CrossRef]
- Setiawan, A.; Kennedy, E.M.; Dlugogorski, B.Z.; Adesina, A.A.; Tkachenko, O.; Stockenhuber, M. Evidence of the formation of surface palladium carbide during the catalytic combustion of lean methane/air mixtures. Energy Tech. 2014, 2, 243–249. [Google Scholar] [CrossRef]
- Álvarez-Montero, M.A.; Gómez-Sainero, L.M.; Mayoral, A.; Diaz, I.; Baker, R.T.; Rodriguez, J.J. Hydrodechlorination of chloromethanes with a highly stable Pt on activated carbon catalyst. J. Catal. 2011, 279, 389–396. [Google Scholar] [CrossRef]
- Omar, S.; Palomar, J.; Gómez-Sainero, L.M.; Álvarez-Montero, M.A.; Martin-Martinez, M.; Rodriguez, J.J. Density functional theory analysis of dichloromethane and hydrogen interaction with Pd clusters: First step to simulate catalytic hydrodechlorination. J. Phys. Chem. C 2011, 115, 14180–14192. [Google Scholar] [CrossRef]
- Wagner, C.D.; Davis, L.E.; Zeller, M.V.; Taylor, J.A.; Raymond, R.H.; Gale, L.H. Empirical atomic sensitivity factors for quantitative analysis by electron spectroscopy for chemical analysis. Surf. Interface Anal. 1981, 3, 211–225. [Google Scholar] [CrossRef]
- Álvarez-Montero, M.A.; Martin-Martinez, M.; Gómez-Sainero, L.M.; Arevalo-Bastante, A.; Bedia, J.; Rodriguez, J.J. kinetic study of the hydrodechlorination of chloromethanes with activated-carbon-supported metallic catalysts. Ind. Eng. Chem. Res. 2015, 54, 2023–2029. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
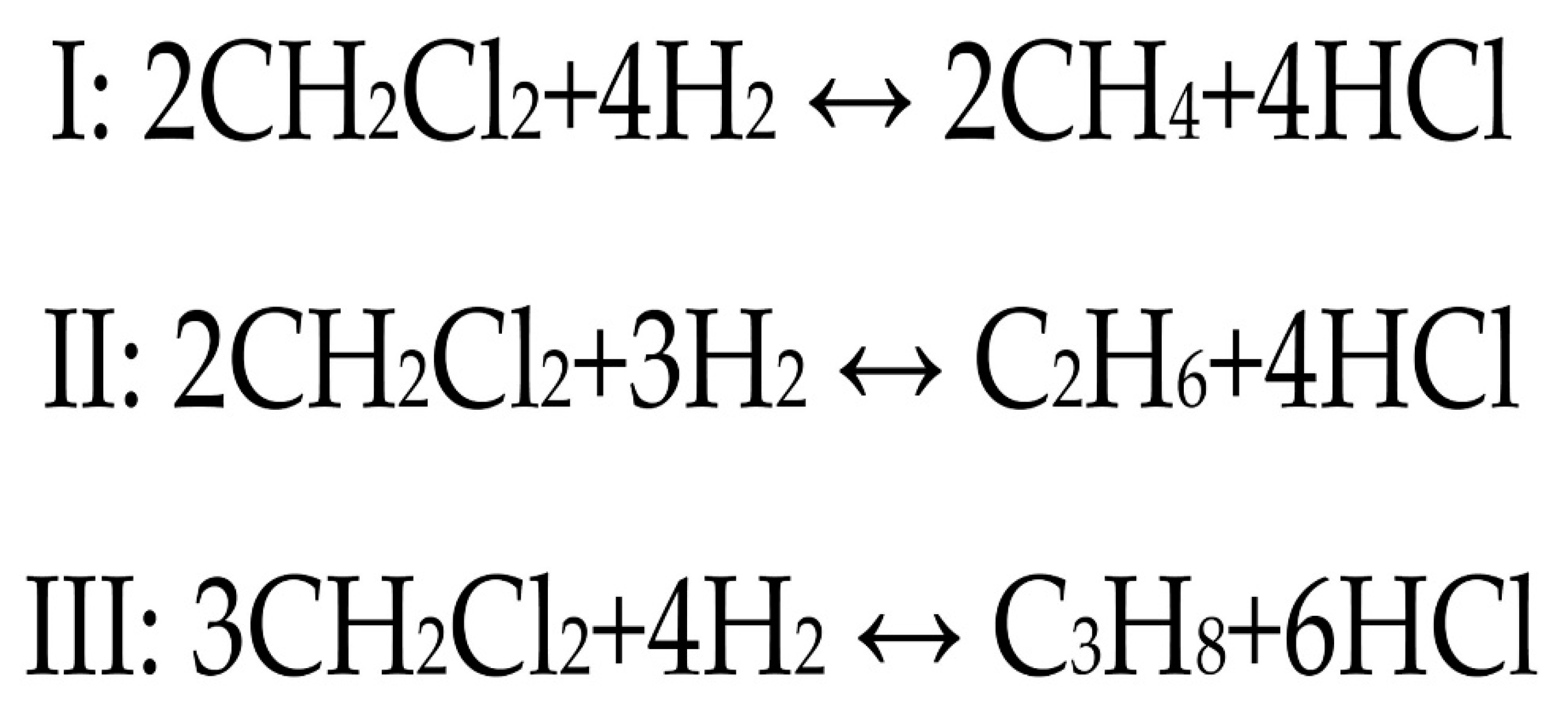{kind=link}
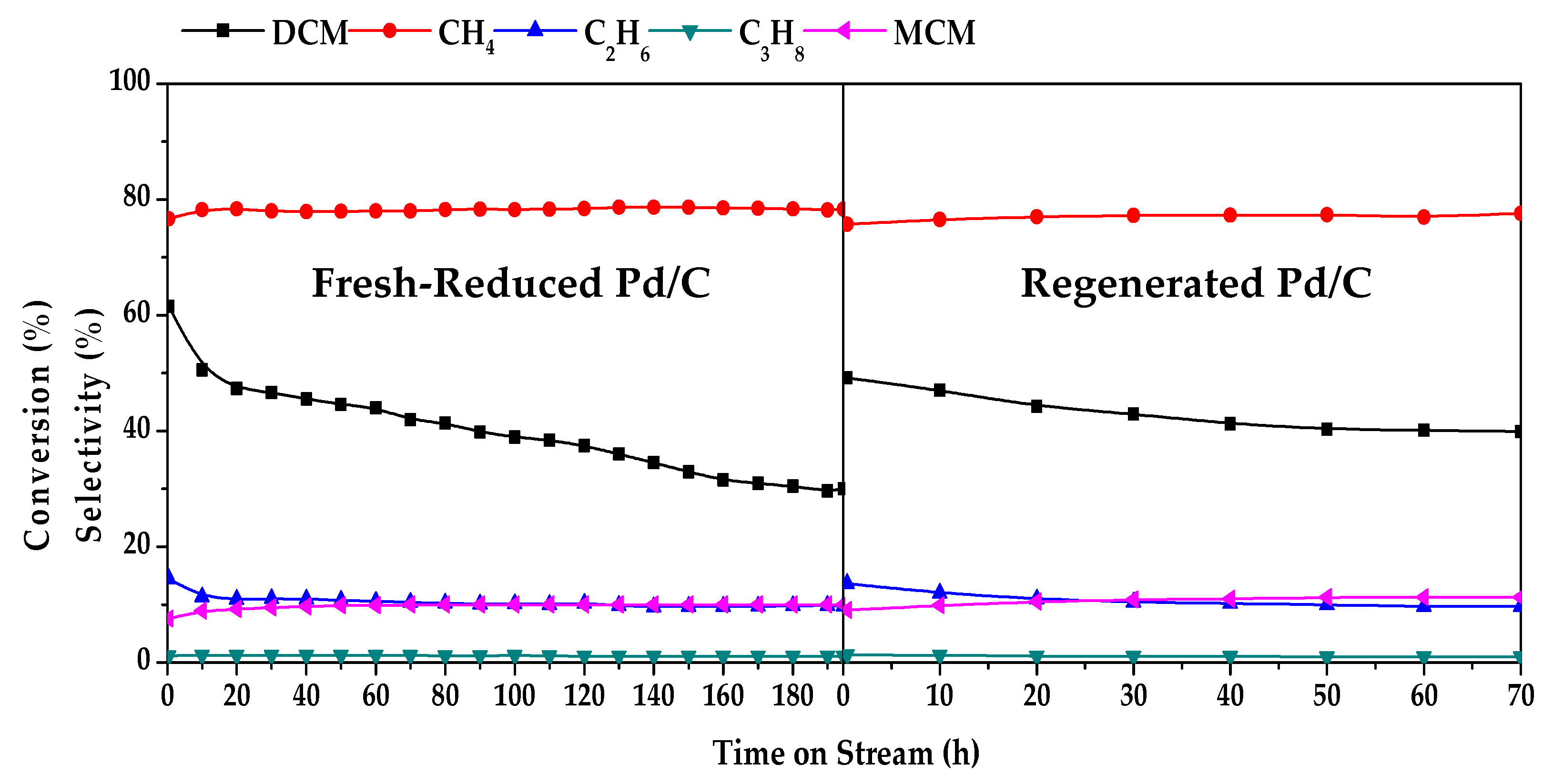
| Duration (h) | Conversion (%) | Selectivity (%) | |||
|---|---|---|---|---|---|
| DCM | CH4 | C2H6 | C3H8 | MCM | |
| 0 | 100.0 | 77.8 | 16.5 | 1.1 | 4.6 |
| 5 | 98.0 | 78.4 | 13.4 | 1.0 | 7.1 |
| 40 | 96.8 | 78.3 | 14.0 | 1.0 | 6.7 |
| 80 | 90.5 | 78.5 | 13.4 | 1.0 | 7.1 |
| 120 | 81.7 | 78.5 | 13.1 | 1.1 | 7.3 |
| Catalyst | BET Surface Area (m2 g−1) | Micropore Volume (cm3 g−1) |
|---|---|---|
| Fresh-reduced Pd/C | 1161 | 0.53 |
| Pd/C 5 h | 1155 | 0.53 |
| Pd/C 40 h | 1129 | 0.52 |
| Pd/C 80 h | 1132 | 0.52 |
| Pd/C 120 h | 1114 | 0.51 |
| Catalyst | Surface Composition | Surface Species | ||||||
|---|---|---|---|---|---|---|---|---|
| PdXPS (%) | ClXPS (%) | Pd0 (%) | Pdn+ (%) | PdCx (%) | Clinorg (%) | Clorg 1 (%) | ||
| Experiments at different time on stream | Fresh-Reduced Pd/C | 0.10 | 0.07 | 52.5 | 47.5 | 0.0 | 63.2 | 36.8 |
| Used Pd/C 5 h | 0.08 | 0.33 | 11.5 | 50.7 | 37.8 | 69.4 | 30.6 | |
| Used Pd/C 40 h | 0.10 | 0.10 | 11.9 | 44.1 | 44.0 | 57.9 | 42.1 | |
| Used Pd/C 80 h | 0.08 | 0.09 | 11.0 | 43.3 | 45.7 | 49.1 | 50.9 | |
| Used Pd/C 120 h | 0.09 | 0.13 | 11.1 | 46.1 | 42.8 | 51.2 | 48.8 | |
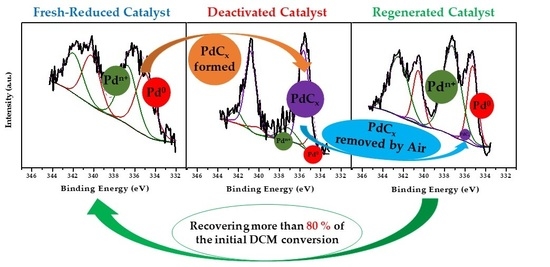
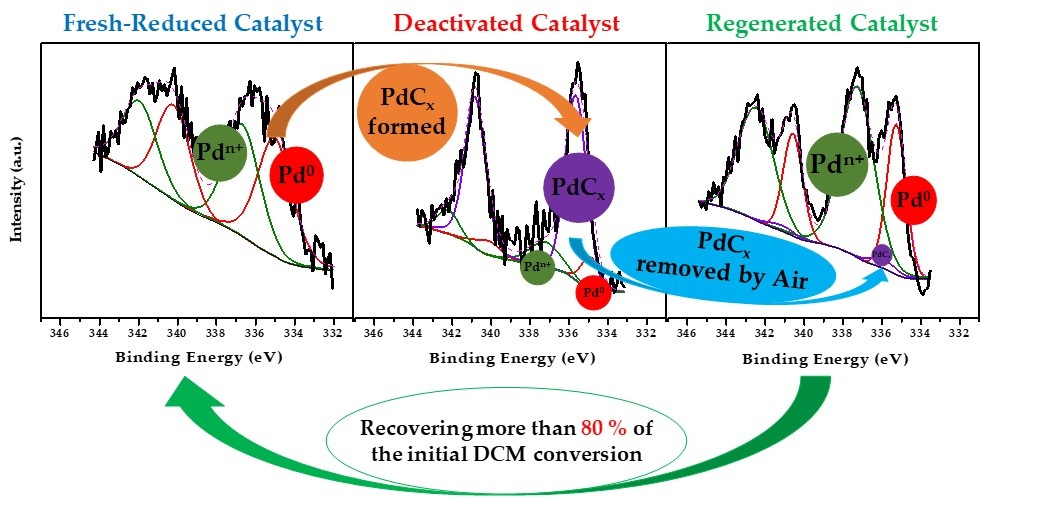
| Deactivation–regeneration study | Deactivated Pd/C 200 h | 0.11 | 0.12 | 10.0 | 14.1 | 75.9 | 51.5 | 48.5 |
| Regenerated Pd/C by Air | 0.37 | 0.20 | 31.8 | 65.7 | 2.5 | 46.7 | 53.3 | |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, S.; Martin-Martinez, M.; Álvarez-Montero, M.A.; Arevalo-Bastante, A.; Rodriguez, J.J.; Gómez-Sainero, L.M. Recycling of Gas Phase Residual Dichloromethane by Hydrodechlorination: Regeneration of Deactivated Pd/C Catalysts. Catalysts 2019, 9, 733. https://doi.org/10.3390/catal9090733
Liu S, Martin-Martinez M, Álvarez-Montero MA, Arevalo-Bastante A, Rodriguez JJ, Gómez-Sainero LM. Recycling of Gas Phase Residual Dichloromethane by Hydrodechlorination: Regeneration of Deactivated Pd/C Catalysts. Catalysts. 2019; 9(9):733. https://doi.org/10.3390/catal9090733
Chicago/Turabian StyleLiu, Sichen, María Martin-Martinez, María Ariadna Álvarez-Montero, Alejandra Arevalo-Bastante, Juan José Rodriguez, and Luisa María Gómez-Sainero. 2019. "Recycling of Gas Phase Residual Dichloromethane by Hydrodechlorination: Regeneration of Deactivated Pd/C Catalysts" Catalysts 9, no. 9: 733. https://doi.org/10.3390/catal9090733
APA StyleLiu, S., Martin-Martinez, M., Álvarez-Montero, M. A., Arevalo-Bastante, A., Rodriguez, J. J., & Gómez-Sainero, L. M. (2019). Recycling of Gas Phase Residual Dichloromethane by Hydrodechlorination: Regeneration of Deactivated Pd/C Catalysts. Catalysts, 9(9), 733. https://doi.org/10.3390/catal9090733