Surface-Doped Graphitic Carbon Nitride Catalyzed Photooxidation of Olefins and Dienes: Chemical Evidence for Electron Transfer and Singlet Oxygen Mechanisms




Abstract
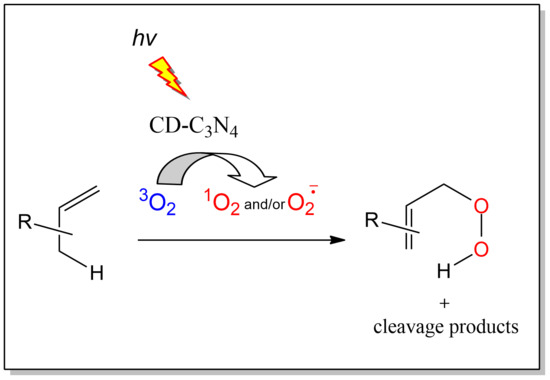
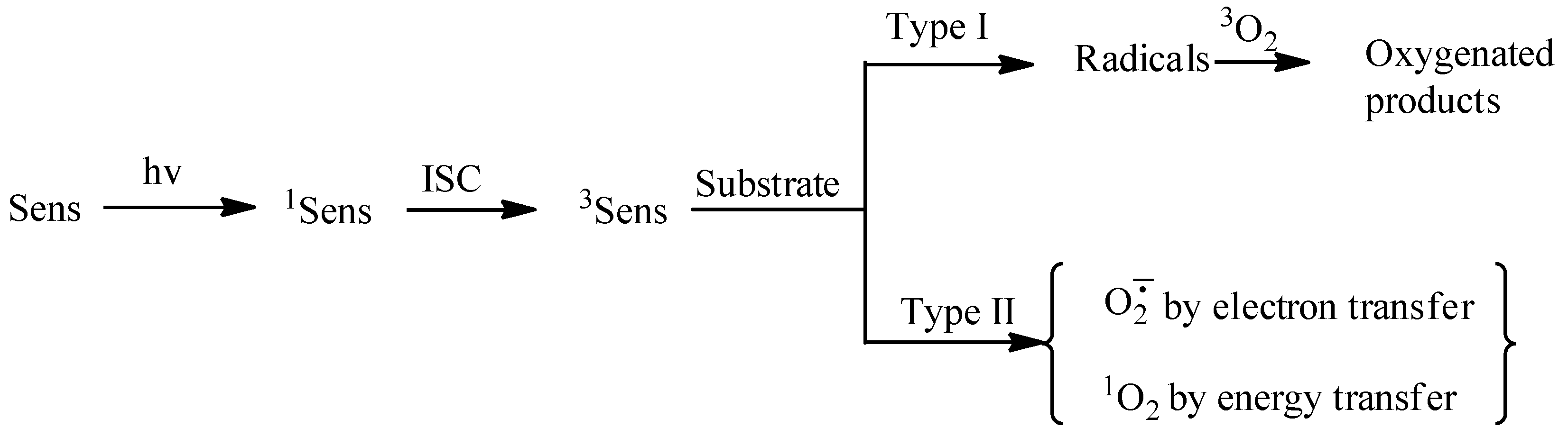
1. Introduction
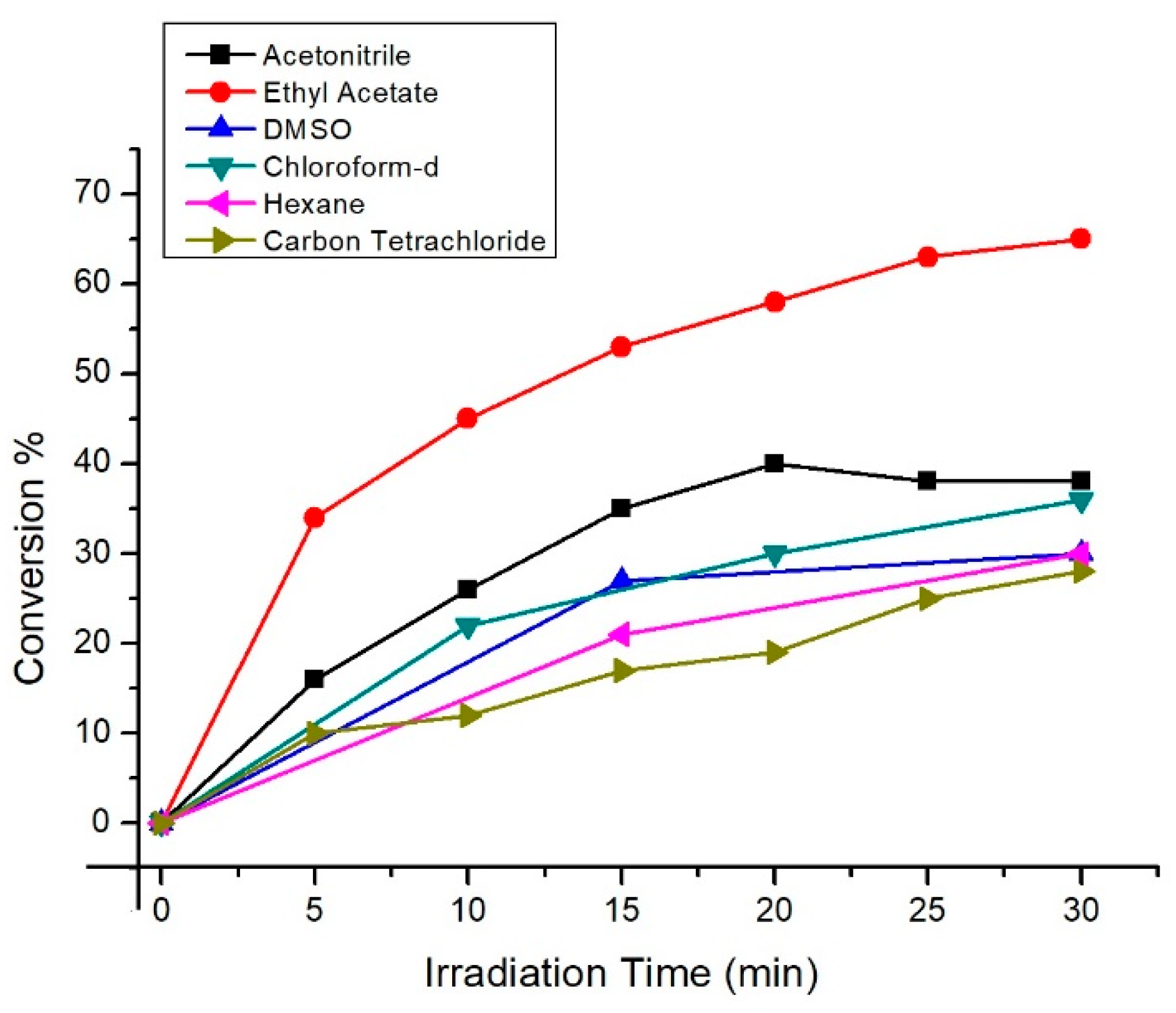
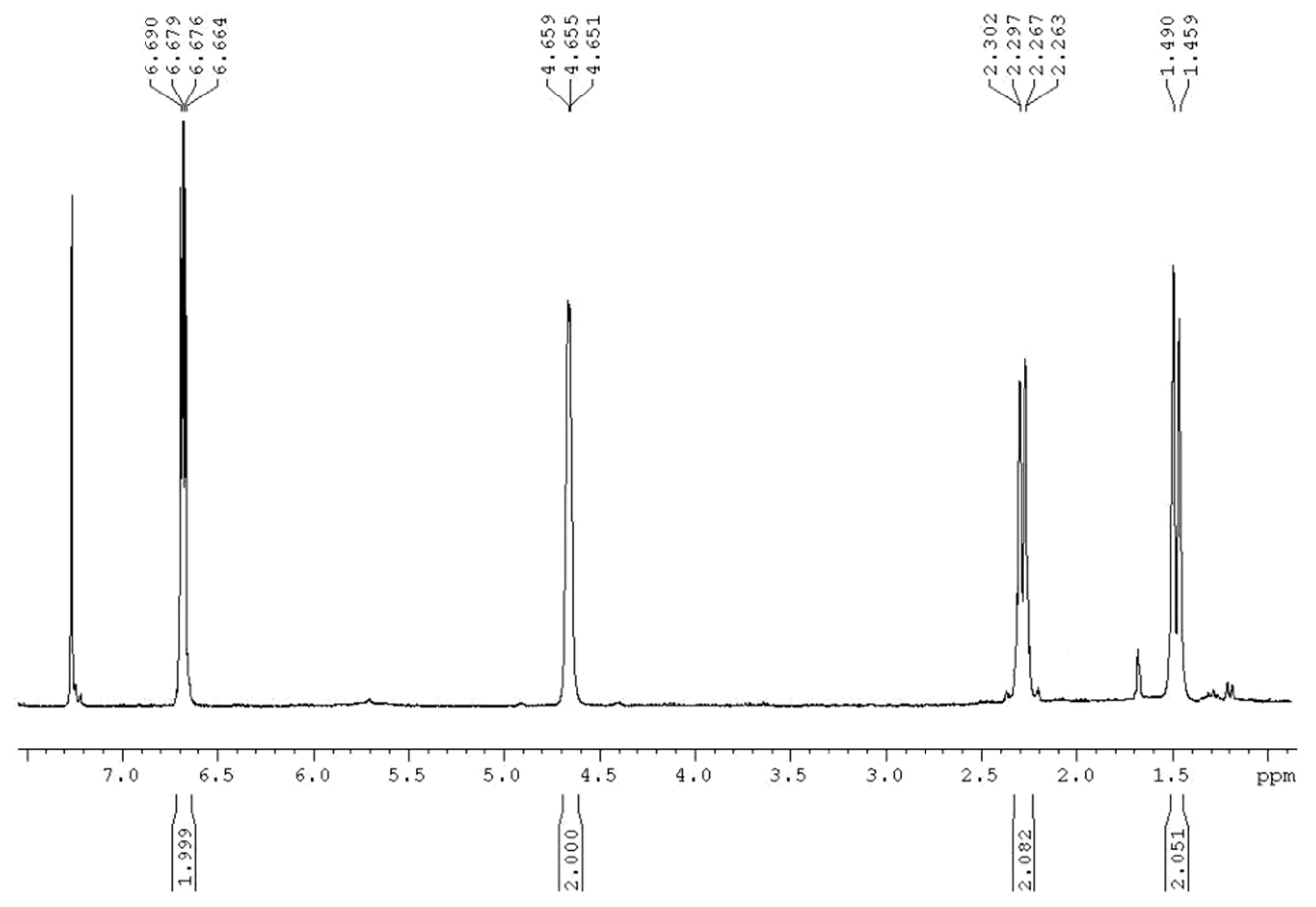
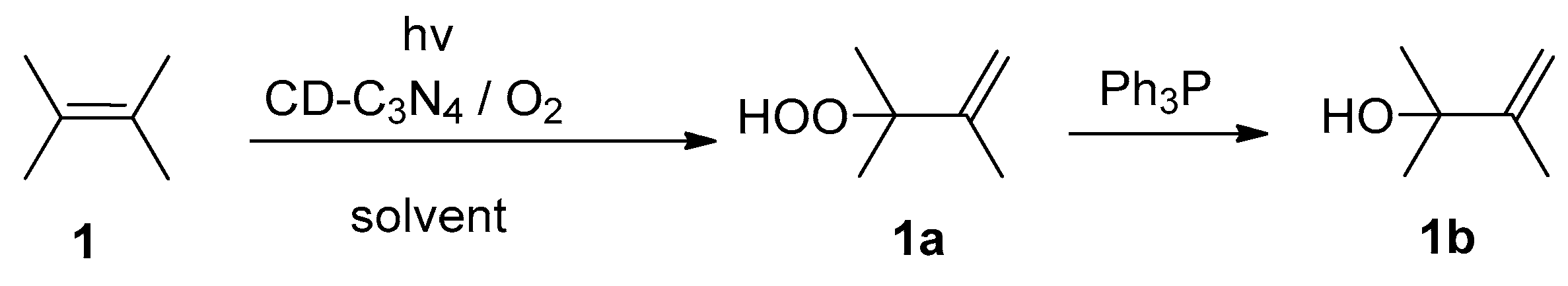
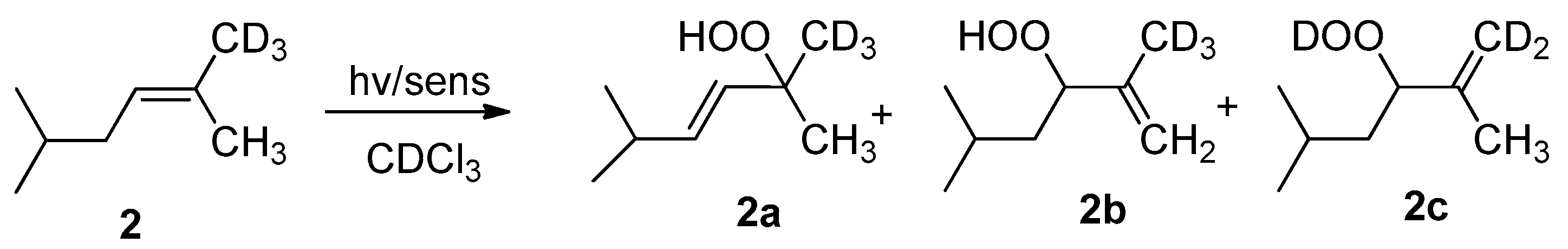
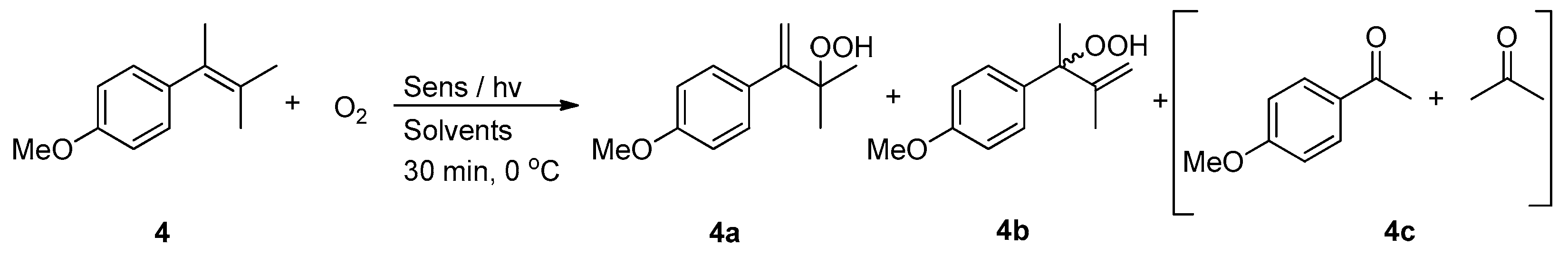
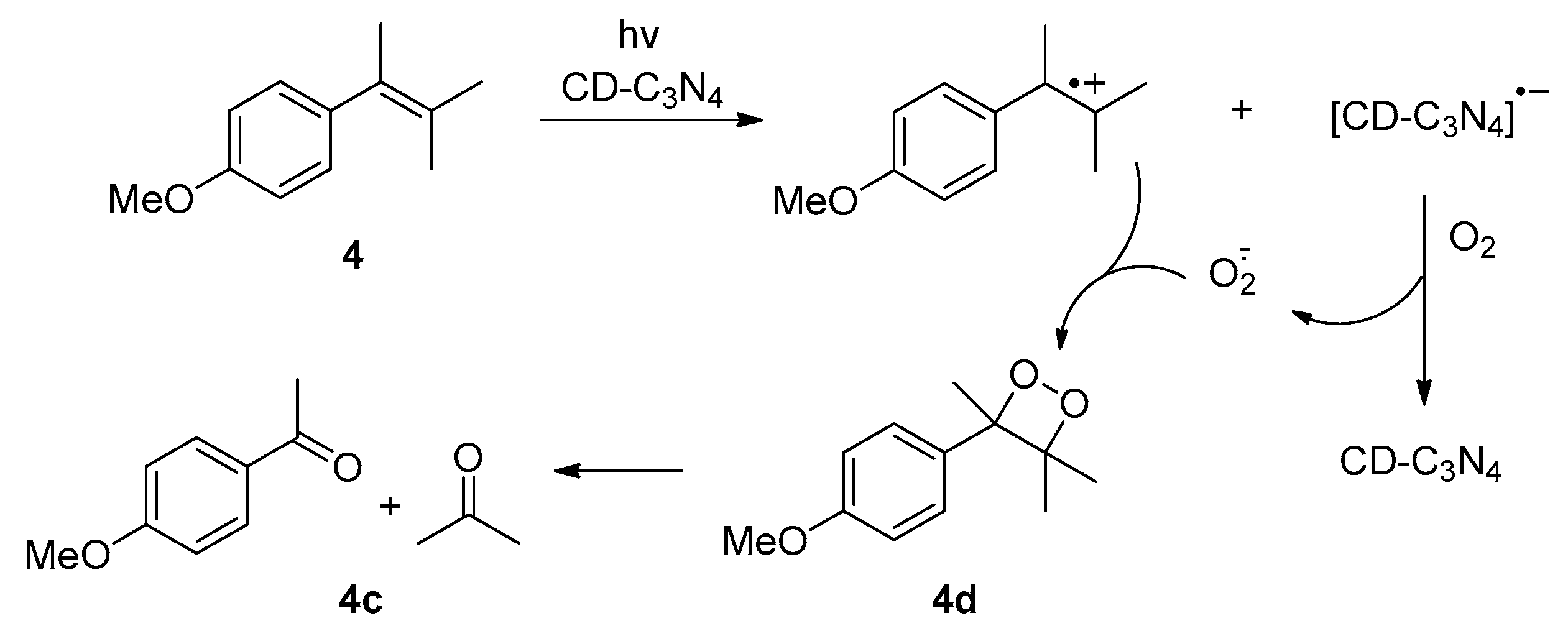
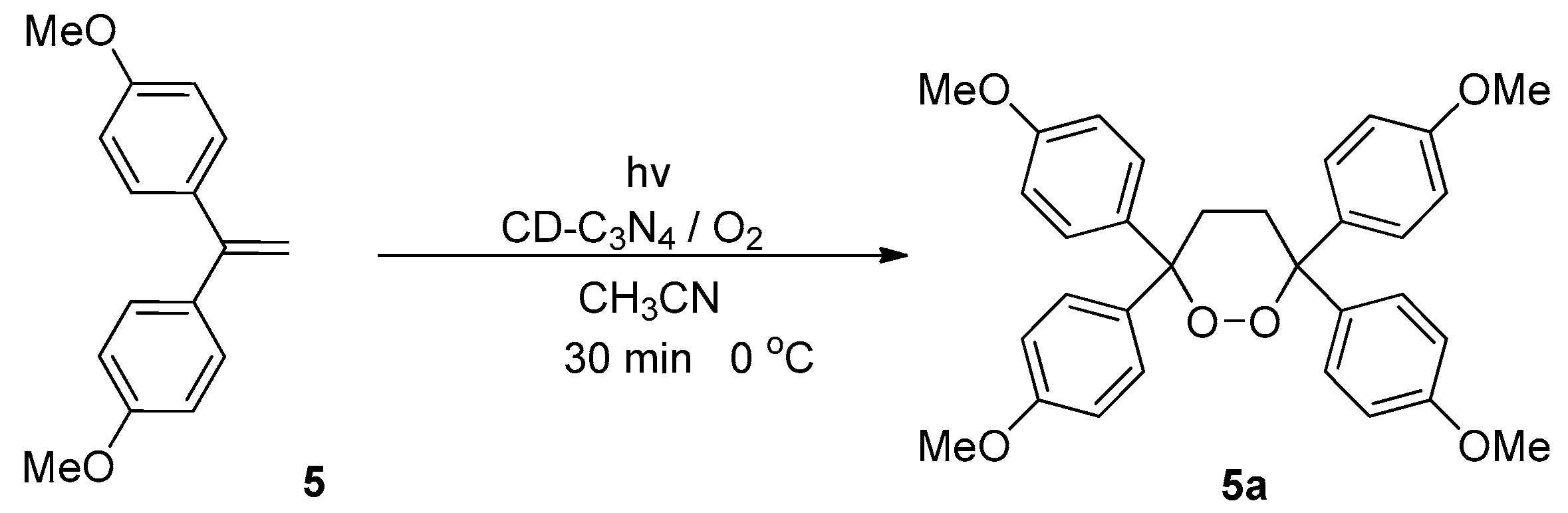
2. Results and Discussion
3. Materials and Methods
3.1. General
3.2. General Photooxidation Method
3.3. Synthesis of 2-(4-methoxyphenyl)-3-methylbut-2-ene (4)
3.4. Synthesis of 2-(1,1,1-trideuteromethyl)-5-methylhex-2-ene (2)
3.5. Synthesis of 1,1-di(p-anisyl)ethylene (5)
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Kroke, E.; Schwarz, M. Novel group 14 nitrides. Coord. Chem. Rev. 2004, 248, 493–532. [Google Scholar] [CrossRef]
- Liu, J.; Wang, H.; Antonietti, M. Graphitic carbon nitride “reloaded”: Emerging applications beyond (photo)catalysis. Chem. Soc. Rev. 2016, 45, 2308–2326. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Zhang, Y.; Lu, L.; Wu, G.; Chen, W. Self-regenerated solar-driven photocatalytic water-splitting by urea derived graphitic carbon nitride with platinum nanoparticles. Chem. Commun. 2012, 48, 8826–8828. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Maeda, K.; Thomas, A.; Takanabe, K.; Xin, G.; Carlsson, J.M.; Domen, K.; Antonietti, M. A metal-free polymeric photocatalyst for hydrogen production from water under visible light. Nat. Mater. 2009, 8, 76–80. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Li, Y.; Bai, X.; Cai, Q.; Liu, C.; Zuo, Y.; Kang, S.; Cui, L. Facile synthesis of Y-doped graphitic carbon nitride with enhanced photocatalytic performance. Catal. Commun. 2016, 84, 179–182. [Google Scholar] [CrossRef]
- Wang, Q.; Chen, A.; Wang, X.; Zhang, J.; Yang, J.; Li, X.A. Fe-species-loaded graphitic carbon nitride with enhanced photocatalytic performance under visible-light irradiation. J. Mol. Cat. A Chem. 2016, 420, 159–166. [Google Scholar] [CrossRef]
- Li, X.; Bi, W.; Zhang, L.; Tao, S.; Chu, W.; Zhang, Q.; Luo, Y.; Wu, C.; Xie, Y. Single-Atom Pt as Co-Catalyst for Enhanced Photocatalytic H2 Evolution. Adv. Mater. 2016, 28, 2427–2431. [Google Scholar] [CrossRef]
- Han, C.; Wu, L.; Ge, L.; Li, Y.; Zhao, Z. AuPd bimetallic nanoparticles decorated graphitic carbon nitride for highly efficient reduction of water to H2 under visible light irradiation. Carbon 2015, 92, 31–40. [Google Scholar] [CrossRef]
- Xiong, T.; Cen, W.; Zhang, Y.; Dong, F. Bridging the g-C3N4 Interlayers for Enhanced Photocatalysis. ACS Catal. 2016, 6, 2462–2472. [Google Scholar] [CrossRef]
- Fu, Y.; Huang, T.; Zhang, L.; Zhu, J.; Wang, X. Ag/g-C3N4 catalyst with superior catalytic performance for the degradation of dyes: A borohydride-generated superoxide radical approach. Nanoscale 2015, 7, 13723–13733. [Google Scholar] [CrossRef]
- Kuriki, R.; Matsunaga, H.; Nakashima, T.; Wada, K.; Yamakata, A.; Ishitani, O.; Maeda, K. Nature-Inspired, Highly Durable CO2 Reduction System Consisting of a Binuclear Ruthenium(II) Complex and an Organic Semiconductor Using Visible Light. J. Am. Chem. Soc. 2016, 138, 5159–5170. [Google Scholar] [CrossRef] [PubMed]
- Chai, B.; Peng, T.; Mao, J.; Li, K.; Zan, L. Graphitic carbon nitride (g-C3N4)–Pt-TiO2 nanocomposite as an efficient photocatalyst for hydrogen production under visible light irradiation. Phys. Chem. Chem. Phys. 2012, 14, 16745–16752. [Google Scholar] [CrossRef] [PubMed]
- Idrees, F.; Dillert, R.; Bahnemann, D.; Butt, F.K.; Tahir, M. In-Situ Synthesis of Nb2O5/g-C3N4 Heterostructures as Highly Efficient Photocatalysts for Molecular H2 Evolution under Solar Illumination. Catalysts 2019, 9, 169. [Google Scholar] [CrossRef]
- Hong, J.; Wang, Y.; Wang, Y.; Zhang, W.; Xu, R. Noble-Metal-Free NiS/C3N4 for Efficient Photocatalytic Hydrogen Evolution from Water. ChemSusChem 2013, 6, 2263–2268. [Google Scholar] [CrossRef] [PubMed]
- Xiang, Q.; Yu, J.; Jaroniec, M. Preparation and Enhanced Visible-Light Photocatalytic H2-Production Activity of Graphene/C3N4 Composites. J. Phys. Chem. C 2011, 115, 7355–7363. [Google Scholar] [CrossRef]
- Suryawanshi, A.; Dhanasekaran, P.; Mhamane, D.; Kelkar, S.; Patil, S.; Gupta, N.; Ogale, S. Doubling of photocatalytic H2 evolution from g-C3N4via its nanocomposite formation with multiwall carbon nanotubes: Electronic and morphological effects. Int. J. Hydrogen Energy 2012, 37, 9584–9589. [Google Scholar] [CrossRef]
- Ong, W.-J.; Tan, L.-L.; Ng, Y.H.; Yong, S.-T.; Chai, S.-P. Graphitic Carbon Nitride (g-C3N4)-Based Photocatalysts for Artificial Photosynthesis and Environmental Remediation: Are We a Step Closer To Achieving Sustainability? Chem. Rev. 2016, 116, 7159–7329. [Google Scholar] [CrossRef]
- Liu, J.; Liu, Y.; Liu, N.; Han, Y.; Zhang, X.; Huang, H.; Lifshitz, Y.; Lee, S.-T.; Zhong, J.; Kang, Z. Metal-free efficient photocatalyst for stable visible water splitting via a two-electron pathway. Science 2015, 347, 970–974. [Google Scholar] [CrossRef]
- Wen, J.; Xie, J.; Chen, X.; Li, X. A review on g-C3N4-based photocatalysts. Appl. Surf. Sci. 2017, 391, 72–123. [Google Scholar] [CrossRef]
- Khan, M.A.; Teixeira, I.F.; Li, M.M.J.; Koito, Y.; Tsang, S.C.E. Graphitic carbon nitride 10atalyzed photoacetalization of aldehydes/ketones under ambient conditions. Chem. Commun. 2016, 52, 2772–2775. [Google Scholar] [CrossRef]
- Goettmann, F.; Fischer, A.; Antonietti, M.; Thomas, A. Chemical Synthesis of Mesoporous Carbon Nitrides Using Hard Templates and Their Use as a Metal-Free Catalyst for Friedel–Crafts Reaction of Benzene. Angew. Chem. Int. ed. 2006, 45, 4467–4471. [Google Scholar] [CrossRef] [PubMed]
- Verma, S.; Nasir Baig, R.B.; Nadagouda, M.N.; Varma, R.S. Selective Oxidation of Alcohols using Photoactive VO@g-C3N4. ACS Sus. Chem. Eng. 2016, 4, 1094–1098. [Google Scholar] [CrossRef]
- Han, H.; Ding, G.; Wu, T.; Yang, D.; Jiang, T.; Han, B. Cu and Boron Doped Carbon Nitride for Highly Selective Oxidation of Toluene to Benzaldehyde. Molecules 2015, 20, 12686–12697. [Google Scholar] [CrossRef] [PubMed]
- Ding, Z.; Chen, X.; Antonietti, M.; Wang, X. Synthesis of Transition Metal-Modified Carbon Nitride Polymers for Selective Hydrocarbon Oxidation. ChemSusChem 2011, 4, 274–281. [Google Scholar] [CrossRef] [PubMed]
- Verma, S.; Nasir Baig, R.B.; Nadagouda, M.N.; Varma, R.S.; Photocatalytic, C.-H. Activation of Hydrocarbons over VO@g-C3N4. ACS Sus. Chem. Eng. 2016, 4, 2333–2336. [Google Scholar] [CrossRef]
- Zhang, P.; Deng, J.; Mao, J.; Li, H.; Wang, Y. Selective aerobic oxidation of alcohols by a mesoporous graphitic carbon nitride/N-hydroxyphthalimide system under visible-light illumination at room temperature. Chinese J. Catal. 2015, 36, 1580–1586. [Google Scholar] [CrossRef]
- Su, F.Z.; Mathew, S.C.; Lipner, G.; Fu, X.Z.; Antonietti, M.; Blechert, S.; Wang, X.C. mpg-C3N4-Catalyzed Selective Oxidation of Alcohols Using O2 and Visible Light. J. Am. Chem. Soc. 2010, 132, 16299–16301. [Google Scholar] [CrossRef] [PubMed]
- Long, B.; Ding, Z.; Wang, X. Carbon Nitride for the Selective Oxidation of Aromatic Alcohols in Water under Visible Light. ChemSusChem 2013, 6, 2074–2078. [Google Scholar] [CrossRef]
- Zhang, P.; Wang, Y.; Yao, J.; Wang, C.; Yan, C.; Antonietti, M.; Li, H. Visible-Light-Induced Metal-Free Allylic Oxidation Utilizing a Coupled Photocatalytic System of g-C3N4 and N-Hydroxy Compounds. Adv. Synth. Catal. 2011, 353, 1447–1451. [Google Scholar] [CrossRef]
- Zhang, W.; Bariotaki, A.; Smonou, I.; Hollmann, F. Visible-light-driven photooxidation of alcohols using surface-doped graphitic carbon nitride. Green Chem. 2017, 19, 2096–2100. [Google Scholar] [CrossRef]
- Wasserman, H.H.; Ives, J.L. Singlet Oxygen in Organic Synthesis. Tetrahedron 1980, 37, 1825–1832. [Google Scholar] [CrossRef]
- Foote, C.S.; Clennan, E.L. Properties and Reactions of Singlet Dioxygen. In Active Oxygen in Chemistry; Foote, C.S., Valentine, J.S., Greenberg, A., Liebman, J.F., Eds.; Chapman and Hall: London, UK, 1995; pp. 105–140. [Google Scholar]
- Chin, K.K.; Trevithick-Sutton, C.C.; McCallum, J.; Jockusch, S.; Turro, N.J.; Scaiano, J.C.; Foote, C.S.; Garcia-Garibay, M.A. Quantitative Determination of Singlet Oxygen Generated by Excited State Aromatic Amino Acids, Proteins, and Immunoglobulins. J. Am. Chem. Soc. 2008, 130, 6912–6913. [Google Scholar] [CrossRef] [PubMed]
- Kuimova, M.K.; Botchway, S.W.; Parker, A.W.; Balaz, M.; Collins, H.A.; Anderson, H.L.; Suhling, K.; Ogilby, P.R. Imaging intracellular viscosity of a single cell during photoinduced cell death. Nat. Chem. 2009, 1, 69–73. [Google Scholar] [CrossRef] [PubMed]
- Dolmans, D.E.; Fukumura, D.; Jain, R.K. Photodynamic therapy for cancer. Nat. Rev. Cancer 2003, 3, 380–387. [Google Scholar] [CrossRef] [PubMed]
- Ranby, B.; Rabek, J.F. Photodegradation, Photooxidation and Photostabilization of Polymers. In Singlet Oxygen Reactions with Organic Compounds & Polymers; Wiley: London, UK; John Wiley & Sons: New York, NY, USA, 1978. [Google Scholar] [CrossRef]
- Abdou, M.S.A.; Holdcroft, S. Mechanisms of photodegradation of poly(3-alkylthiophenes) in solution. Macromolecules 1993, 26, 2954–2962. [Google Scholar] [CrossRef]
- Scurlock, R.D.; Wang, B.; Ogilby, P.R.; Sheats, J.R.; Clough, R.L. Singlet Oxygen as a Reactive Intermediate in the Photodegradation of an Electroluminescent Polymer. J. Am. Chem. Soc. 1995, 117, 10194–10202. [Google Scholar] [CrossRef]
- Herzberg, G. Photography of the Infra-Red Solar spectrum to Wave-length 12,900 A. Nature 1934, 133, 759. [Google Scholar] [CrossRef]
- Kautsky, H. Quenching of luminescence by oxygen. Trans. Faraday Soc. 1939, 35, 216–219. [Google Scholar] [CrossRef]
- Foote, C.S.; Wexler, S. Olefin Oxidations with Excited Singlet Molecular Oxygen. J. Am. Chem. Soc. 1964, 86, 3879–3880. [Google Scholar] [CrossRef]
- Foote, C.S.; Wexler, S. Singlet Oxygen. A Probable Intermediate in Photosensitized Autoxidations. J. Am. Chem. Soc. 1964, 86, 3880–3881. [Google Scholar] [CrossRef]
- Montagnon, T.; Tofi, M.; Vassilikogiannakis, G. Using Singlet Oxygen to Synthesize Polyoxygenated Natural Products from Furans. Acc. Chem. Res. 2008, 41, 1001–1011. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, N. Photochemical Reactions as Key Steps in Organic Synthesis. Chem. Rev. 2008, 108, 1052–1103. [Google Scholar] [CrossRef]
- Martin, D.J.; Qiu, K.; Shevlin, S.A.; Handoko, A.D.; Chen, X.; Guo, Z.; Tang, J. Highly Efficient Photocatalytic H2 Evolution from Water using Visible Light and Structure-Controlled Graphitic Carbon Nitride. Angew. Chem. Int. Ed. 2014, 53, 9240–9245. [Google Scholar] [CrossRef]
- Pan, J.; Sheng, Y.; Zhang, J.; Wei, J.; Huang, P.; Zhang, X.; Feng, B. Preparation of carbon quantum dots/ TiO2 nanotubes composites and their visible light catalytic applications. J. Mater. Chem. A 2014, 2, 18082–18086. [Google Scholar] [CrossRef]
- Alberti, M.N.; Orfanopoulos, M. Recent Mechanistic Insights in the Singlet Oxygen Ene Reaction. Synlett 2010, 999–1026. [Google Scholar] [CrossRef]
- Stratakis, M.; Orfanopoulos, M. Regioselectivity in the Ene Reaction of Singlet Oxygen with Alkenes. Tetrahedron 2000, 56, 1595–1615. [Google Scholar] [CrossRef]
- Singleton, D.A.; Hang, C.; Szymanski, M.J.; Meyer, M.P.; Leach, A.G.; Kuwata, K.T.; Chen, J.S.; Greer, A.; Foote, C.S.; Houk, K.N. Mechanism of Ene Reactions of Singlet Oxygen. A Two-Step No-Intermediate Mechanism. J. Am. Chem. Soc. 2003, 125, 1319–1328. [Google Scholar] [CrossRef] [PubMed]
- Sheppard, A.N.; Acevedo, O. Multidimensional Exploration of Valley−Ridge Inflection Points on Potential-Energy Surfaces. J. Am. Chem. Soc. 2009, 131, 2530–2540. [Google Scholar] [CrossRef] [PubMed]
- An, Y.-Z.; Chen, C.H.B.; Anderson, J.L.; Sigman, D.S.; Foote, C.S.; Rubin, Y. Sequence-specific modification of guanosine in DNA by a C60-linked deoxyoligonucleotide: Evidence for a non-singlet oxygen mechanism. Tetrahedron 1996, 52, 5179–5189. [Google Scholar] [CrossRef]
- Foote, C.S. Quenching of Singlet Oxygen. In Singlet Oxygen; Wasserman, H.H., Murray, R.W., Eds.; Academic Press: New York, NY, USA, 1979; pp. 139–171. [Google Scholar]
- Orfanopoulos, M.; Grdina, M.B.; Stephenson, L.M. Site specificity in the singlet oxygen-trisubstituted olefin reaction. J. Am. Chem. Soc. 1979, 101, 275–276. [Google Scholar] [CrossRef]
- Foote, C.S. Definition of Type I and Type II Photosensitized Oxidation. Photochem. Photobiol. 1991, 54, 659. [Google Scholar] [CrossRef] [PubMed]
- Eriksen, J.; Foote, C.S. Electron-transfer photooxygenation. 5. Oxidation of phenyl-substituted alkenes sensitized by cyanoanthracenes. J. Am. Chem. Soc. 1980, 102, 6083–6088. [Google Scholar] [CrossRef]
- Stephenson, L.M.; Grdina, M.J.; Orfanopoulos, M. Mechanism of the ene reaction between singlet oxygen and olefins. Acc. Chem. Res. 1980, 13, 419–425. [Google Scholar] [CrossRef]
- Gollnick, K.; Schnatterer, A. Formation of a 1,2-dioxane by electron transfer photooxygenation of 1,1-di(p-anisyl)ethylene. Tetrahedron Lett. 1984, 25, 185–188. [Google Scholar] [CrossRef]
- Gollnick, K.; Schnatterer, A. Formation of 1,2-dioxanes by electron-transfer photooxygenation of 1,1-disubstituted ethylenes. Tetrahedron Lett. 1984, 25, 2735–2738. [Google Scholar] [CrossRef]
- Orfanopoulos, M.; Stratakis, M.; Elemes, Y.; Jensen, F. Do rotational barriers dictate the regioselectivity in the ene reactions of singlet oxygen and triazolinedione with alkenes? J. Am. Chem. Soc. 1991, 113, 3180–3181. [Google Scholar] [CrossRef]











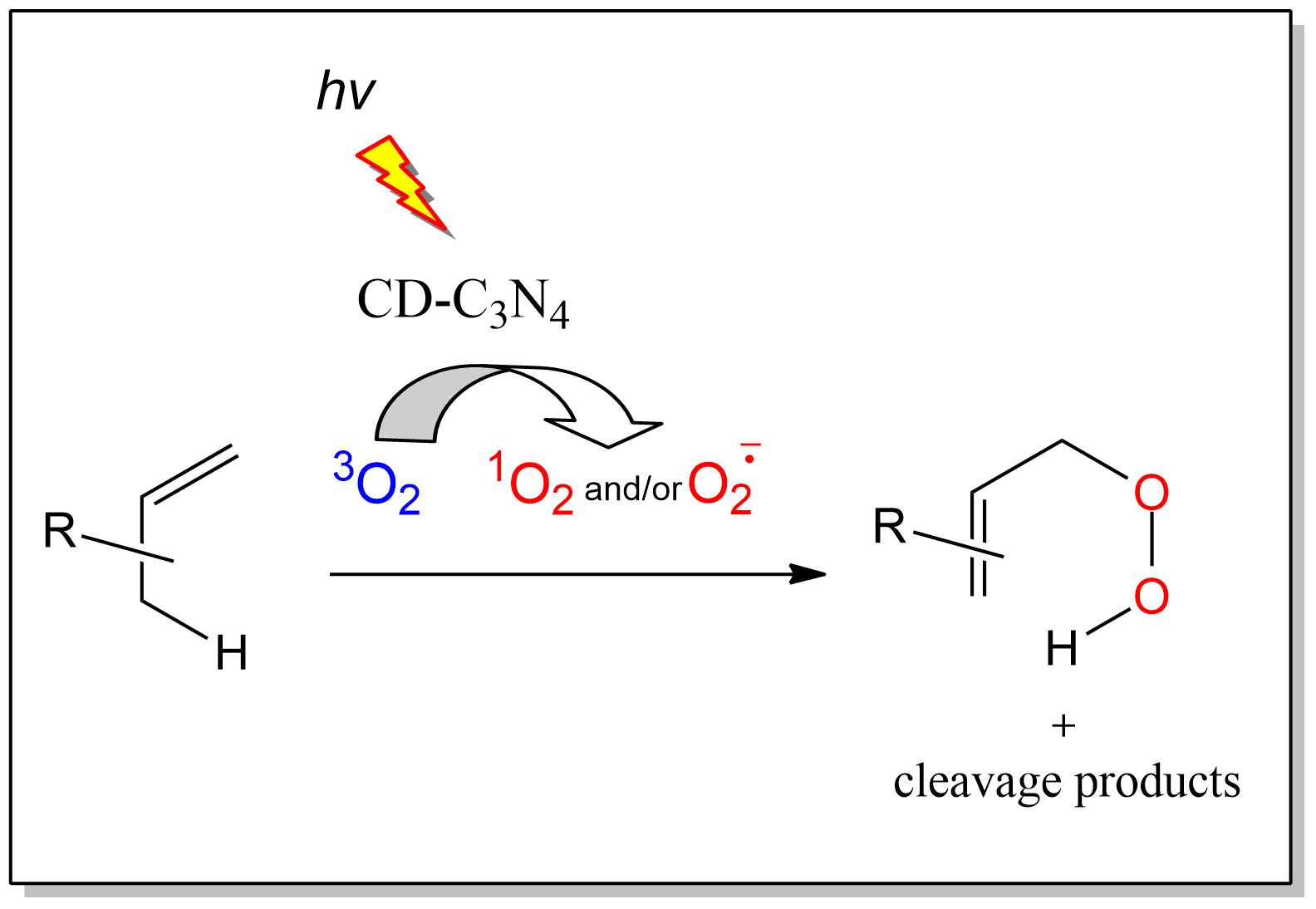{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
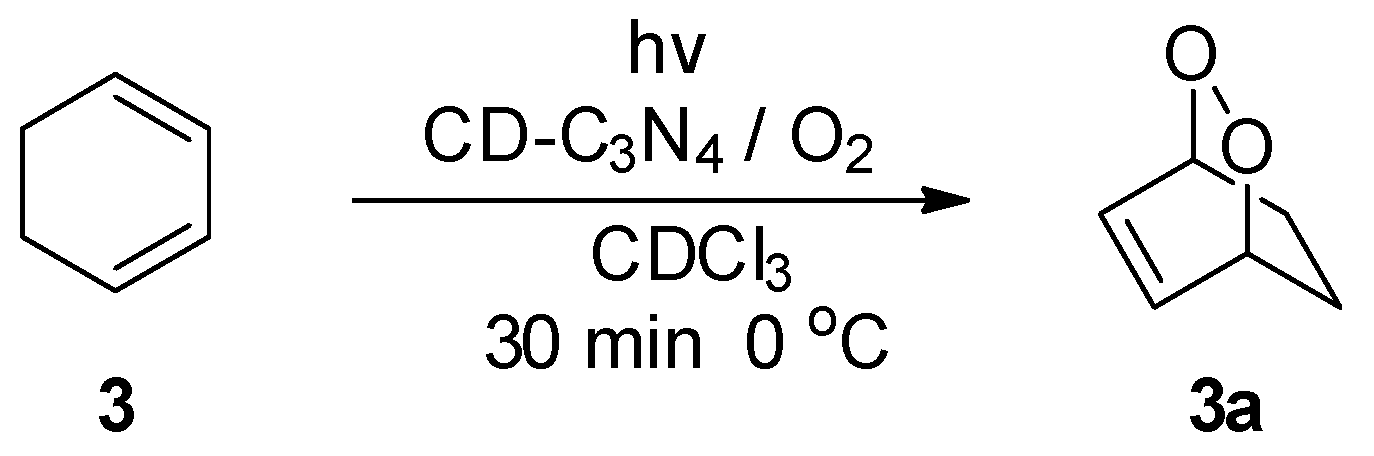{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Entry | Photocatalyst | Conversion (%) a | Irradiation Time (min) | Regioselectivities (%) b | ||
|---|---|---|---|---|---|---|
| 2a | 2b | 2c | ||||
| 1 | TPP | 100 | 5 | 56 | 31 | 13 |
| 2 | CD-C3N4 | 34 | 30 | 58 | 29 | 13 |
| Entry | Photocatalyst | Solvents | 4a % a | 4b % b | 4c % c | Conversion % |
|---|---|---|---|---|---|---|
| 1 | TPP | CCl4 | 76 | 24 | - | 100d |
| 2 | RB | CDCl3 | 70 | 30 | - | 100d |
| 3 | MB | CH3CN | 66 | 34 | - | 100d |
| 4 | DCA | CD3CN | 61 | 27 | 12 | 87d |
| 5 | CD-C3N4 | CDCl3 | 53 | 31 | 16 | 80 |
| 6 | CD-C3N4 | CD3CN | 58 | 27 | 15 | 70 |
| 7 | CD-C3N4 | Benzene | 53 | 31 | 16 | 67 |
| 8 | CD-C3N4 | EtOAc | 53 | 29 | 18 | 65 |
| 9 | CD-C3N4 | EtOH | - | - | 100 | 100 |
| 10 | CD-C3N4 | CDCl3 | - | - | - | - e |
| 11 | no catalyst | CDCl3 | traces | traces | - | <1 f |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chatzoudis, A.; Giannopoulos, V.; Hollmann, F.; Smonou, I. Surface-Doped Graphitic Carbon Nitride Catalyzed Photooxidation of Olefins and Dienes: Chemical Evidence for Electron Transfer and Singlet Oxygen Mechanisms. Catalysts 2019, 9, 639. https://doi.org/10.3390/catal9080639
Chatzoudis A, Giannopoulos V, Hollmann F, Smonou I. Surface-Doped Graphitic Carbon Nitride Catalyzed Photooxidation of Olefins and Dienes: Chemical Evidence for Electron Transfer and Singlet Oxygen Mechanisms. Catalysts. 2019; 9(8):639. https://doi.org/10.3390/catal9080639
Chicago/Turabian StyleChatzoudis, Apostolos, Vasileios Giannopoulos, Frank Hollmann, and Ioulia Smonou. 2019. "Surface-Doped Graphitic Carbon Nitride Catalyzed Photooxidation of Olefins and Dienes: Chemical Evidence for Electron Transfer and Singlet Oxygen Mechanisms" Catalysts 9, no. 8: 639. https://doi.org/10.3390/catal9080639
APA StyleChatzoudis, A., Giannopoulos, V., Hollmann, F., & Smonou, I. (2019). Surface-Doped Graphitic Carbon Nitride Catalyzed Photooxidation of Olefins and Dienes: Chemical Evidence for Electron Transfer and Singlet Oxygen Mechanisms. Catalysts, 9(8), 639. https://doi.org/10.3390/catal9080639