Kinetic Resolution of Racemic Amines to Enantiopure (S)-amines by a Biocatalytic Cascade Employing Amine Dehydrogenase and Alanine Dehydrogenase

 ,
,  ,
, 
Abstract
1. Introduction
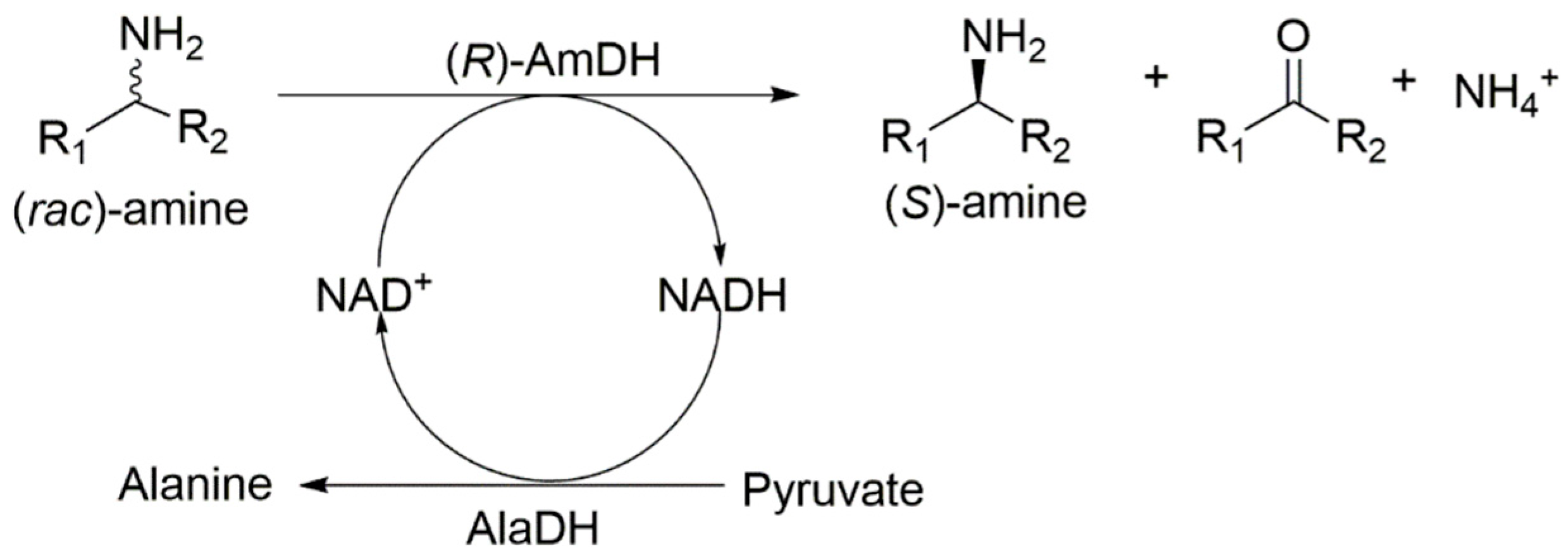
2. Results and Discussion
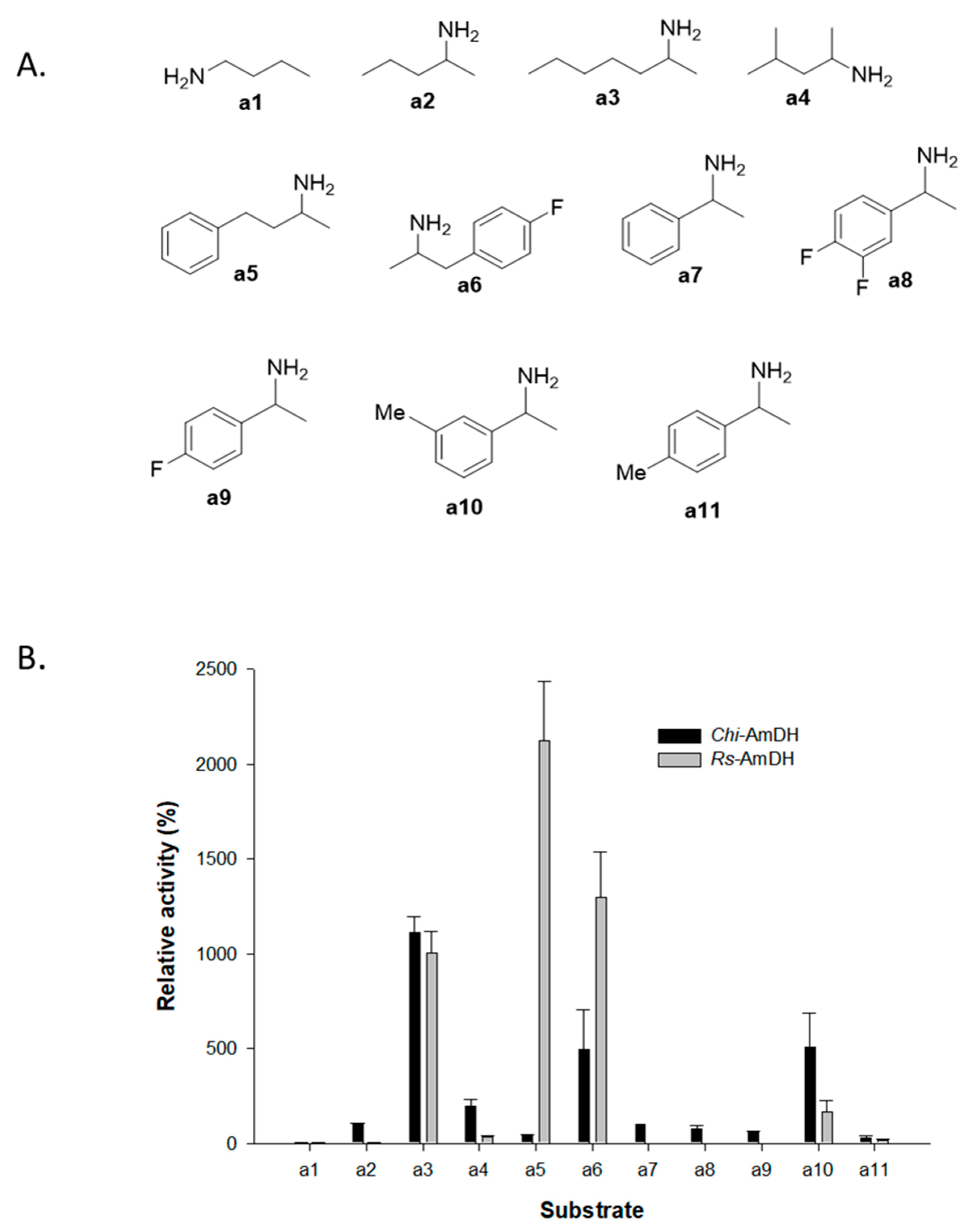
2.1. Reactivity of Amine Dehydrogenases toward Racemic Amines
2.2. Kinetic Resolution of Racemic Amines Using a Purified AmDH/AlaDH Coupling System
2.3. Kinetic Resolution of Racemic Amines Using E. coli Whole Cells Co-Expressing AmDH and AlaDH
3. Materials and Methods
3.1. Chemicals and Media
3.2. Enzyme Expression and Purification
3.3. Co-Expression of Chi-AmDH and AlaDH
3.4. Representative Procedure for Whole-Cell Biotransformations for the Kinetic Resolution of Racemic Amines
3.5. Analysis of Amines
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Bommarius, A.S. Amine dehydrogenases occur in nature. Nature Catal. 2019, 2, 288. [Google Scholar] [CrossRef]
- Ghislieri, D.; Turner, N.J. Biocatalytic Approaches to the Synthesis of Enantiomerically Pure Chiral Amines. Top. Catal. 2014, 57, 284–300. [Google Scholar] [CrossRef]
- Mathew, S.; Yun, H. ω-Transaminases for the Production of Optically Pure Amines and Unnatural Amino Acids. ACS Catal. 2012, 2, 993–1001. [Google Scholar] [CrossRef]
- Constable, D.J.; Dunn, P.J.; Hayler, J.D.; Humphrey, G.R.; Leazer, J.L., Jr.; Linderman, R.J.; Lorenz, K.; Manley, J.; Pearlman, B.A.; Wells, A.; et al. Key green chemistry research areas—A perspective from pharmaceutical manufacturers. Green Chem. 2007, 9, 411–420. [Google Scholar] [CrossRef]
- Patil, M.D.; Grogan, G.; Yun, H. Biocatalyzed C−C Bond Formation for the Production of Alkaloids. ChemCatChem 2018, 10, 4783–4804. [Google Scholar] [CrossRef]
- Lalonde, J. Highly engineered biocatalysts for efficient small molecule pharmaceutical synthesis. Curr. Opin. Biotechnol. 2016, 42, 152–158. [Google Scholar] [CrossRef] [PubMed]
- Roper, L.; Grogan, G. Biocatalysis for Organic Chemists: Hydroxylations. In Organic Synthesis Using Biocatalysis; Goswami, A., Stewart, J., Eds.; Elsevier: Amsterdam, The Netherlands, 2015; pp. 213–241. [Google Scholar]
- Sun, H.; Zhang, H.; Ang, E.; Zhao, H. Biocatalysis for the Synthesis of Pharmaceuticals and Pharmaceutical Intermediates. Bioorg. Med. Chem. 2018, 26, 1275–1284. [Google Scholar] [CrossRef] [PubMed]
- Truppo, M.D. Biocatalysis in the Pharmaceutical Industry—The Need for Speed. ACS Med. Chem. Lett. 2017, 8, 476–480. [Google Scholar] [CrossRef] [PubMed]
- Hwang, E.T.; Lee, S. Multi-Enzymatic Cascade Reactions via Enzyme Complex by Immobilization. ACS Catal. 2019, 9, 4402–4425. [Google Scholar] [CrossRef]
- Kurti, L. Streamlining Amine Synthesis. Science 2015, 348, 863–864. [Google Scholar] [CrossRef]
- Busto, E.; Simon, R.C.; Richter, N.; Kroutil, W. Enzymatic Synthesis of Chiral Amines using ω -Transaminases, Amine Oxidases, and the Berberine Bridge Enzyme. In Green Biocatalysis, 1st ed.; Patel, R.N., Ed.; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2016; pp. 17–57. [Google Scholar]
- Kohls, H.; Steffen-Munsberg, F.; Hohne, M. Recent Achievements in Developing the Biocatalytic Toolbox for Chiral Amine Synthesis. Curr. Opin. Chem. Biol. 2014, 19, 180–192. [Google Scholar] [CrossRef] [PubMed]
- Brundiek, H.; Hohne, M. Transaminases—A Biosynthetic Route for Chiral Amines. In Applied Biocatalysis: From Fundamental Science to Industrial Applications; Hilterhaus, L., Liese, A., Kettling, U., Antranikian, G., Eds.; Wiley—VCH Verlag GmbH & Co.: Weinheim, Germany, 2016; pp. 199–218. [Google Scholar]
- Patil, M.D.; Grogan, G.; Bommarius, A.; Yun, H. Recent Advances in ω-Transaminase-Mediated Biocatalysis for the Enantioselective Synthesis of Chiral Amines. Catalysts 2018, 8, 254. [Google Scholar] [CrossRef]
- Sharma, M.; Mangas-Sanchez, J.; Turner, N.J.; Grogan, G. NAD(P)H-Dependent Dehydrogenases for the Asymmetric Reductive Amination of Ketones: Structure, Mechanism, Evolution and Application. Adv. Synth. Catal. 2017, 359, 2011–2025. [Google Scholar] [CrossRef] [PubMed]
- Batista, V.F.; Galman, J.L.; GA Pinto, D.C.; Silva, A.M.; Turner, N.J. Monoamine oxidase: Tunable activity for amine resolution and functionalization. ACS Catal. 2018, 8, 11889–11907. [Google Scholar] [CrossRef]
- Durairaj, P.; Hur, J.S.; Yun, H. Versatile Biocatalysis of Fungal Cytochrome P450 Monooxygenases. Microb. Cell Fact. 2016, 15, 125. [Google Scholar] [CrossRef] [PubMed]
- Sanfilippo, C.; Paternò, A.A.; Patti, A. Resolution of racemic amines via lipase-catalyzed benzoylation: Chemoenzymatic synthesis of the pharmacologically active isomers of labetalol. Mol. Catal. 2018, 449, 79–84. [Google Scholar] [CrossRef]
- Schmidt, N.G.; Eger, E.; Kroutil, W. Building bridges: Biocatalytic C–C-bond formation toward multifunctional products. ACS Catal. 2016, 6, 4286–4311. [Google Scholar] [CrossRef] [PubMed]
- Abrahamson, M.J.; Vázquez-Figueroa, E.; Woodall, N.B.; Moore, J.C.; Bommarius, A.S. Development of an Amine Dehydrogenase for Synthesis of Chiral Amines. Angew. Chem. Int. Ed. 2012, 51, 3969–3972. [Google Scholar] [CrossRef]
- Itoh, N.; Yachi, C.; Kudome, T. Determining a Novel NAD+-Dependent Amine Dehydrogenase with a Broad Substrate Range from Streptomyces virginiae IFO 12827: Purification and Characterization. J. Mol. Catal. B: Enzym. 2000, 10, 281–290. [Google Scholar] [CrossRef]
- Mayol, O.; David, S.; Darii, E.; Debard, A.; Mariage, A.; Pellouin, V.; Petit, J.L.; Salanoubat, M.; de Berardinis, V.; Zaparucha, A.; et al. Asymmetric Reductive Amination by a Wild-Type Amine Dehydrogenase from the Thermophilic Bacteria Petrotoga mobilis. Catal. Sci. Technol. 2016, 6, 7421–7428. [Google Scholar] [CrossRef]
- Abrahamson, M.J.; Wong, J.W.; Bommarius, A.S. The Evolution of an Amine Dehydrogenase Biocatalyst for the Asymmetric Production of Chiral Amines. Adv. Synth. Catal. 2013, 355, 1780–1786. [Google Scholar] [CrossRef]
- Bommarius, B.R.; Schurmann, M.; Bommarius, A.S. A Novel Chimeric Amine Dehydrogenase Shows Altered Substrate Specificity Compared to its Parent Enzymes. Chem. Commun. 2014, 50, 14953–14955. [Google Scholar] [CrossRef] [PubMed]
- Ye, L.J.; Toh, H.H.; Yang, Y.; Adams, J.P.; Snajdrova, R.; Li, Z. Engineering of Amine Dehydrogenase for Asymmetric Reductive Amination of Ketone by Evolving Rhodococcus Phenylalanine Dehydrogenase. ACS Catal. 2015, 5, 1119–1122. [Google Scholar] [CrossRef]
- Chen, F.F.; Liu, Y.Y.; Zheng, G.W.; Xu, J.H. Asymmetric Amination of Secondary Alcohols by using a Redox-Neutral Two- Enzyme Cascade. ChemCatChem 2015, 7, 3838–3841. [Google Scholar] [CrossRef]
- Pushpanath, A.; Siirola, E.; Bornadel, A.; Woodlock, D.; Schell, U. Understanding and Overcoming the Limitations of Bacillus badius and Caldalkalibacillus thermarum Amine Dehydrogenases for Bio-catalytic Reductive Amination. ACS Catal. 2017, 7, 3204–3209. [Google Scholar] [CrossRef]
- Chen, F.F.; Zheng, G.W.; Liu, L.; Li, H.; Chen, Q.; Li, F.L.; Li, C.X.; Xu, J.H. Reshaping the active pocket of amine dehydrogenases for asymmetric synthesis of bulky aliphatic amines. ACS Catal. 2018, 8, 2622–2628. [Google Scholar] [CrossRef]
- Mutti, F.G.; Knaus, T.; Scrutton, N.S.; Breuer, M.; Turner, N.J. Conversion of Alcohols to Enantiopure Amines Through Dual-Enzyme Hydrogen-Borrowing Cascades. Science 2015, 349, 1525–1529. [Google Scholar] [CrossRef]
- Mayol, O.; Bastard, K.; Beloti, L.; Frese, A.; Turkenburg, J.P.; Petit, J.L.; Mariage, A.; Debard, A.; Pellouin, V.; Perret, A.; et al. A family of native amine dehydrogenases for the asymmetric reductive amination of ketones. Nature Catal. 2019, 2, 324–333. [Google Scholar] [CrossRef]
- Jeon, H.; Yoon, S.; Ahsan, M.; Sung, S.; Kim, G.H.; Sundaramoorthy, U.; Rhee, S.K.; Yun, H. The Kinetic resolution of racemic amines using a whole-cell biocatalyst co-expressing amine dehydrogenase and NADH oxidase. Catalysts 2017, 7, 251. [Google Scholar] [CrossRef]
- Yoon, S.; Patil, M.D.; Sarak, S.; Jeon, H.; Kim, G.H.; Khobragade, T.P.; Sung, S.; Yun, H. Deracemization of Racemic Amines to Enantiopure (R)-and (S)-amines by Biocatalytic Cascade Employing ω-Transaminase and Amine Dehydrogenase. ChemCatChem 2019, 11, 1898–1902. [Google Scholar] [CrossRef]
- Li, J.; Pan, J.; Zhang, J.; Xu, J.H. Stereoselective synthesis of L-tert-leucine by a newly cloned leucine dehydrogenase from Exiguobacterium sibiricum. J. Mol. Catal. B: Enzym. 2014, 105, 11–17. [Google Scholar] [CrossRef]
- Brunhuber, N.M.; Thoden, J.B.; Blanchard, J.S.; Vanhooke, J.L. Rhodococcus L-phenylalanine dehydrogenase: Kinetics, mechanism, and structural basis for catalytic specifity. Biochemistry 2000, 39, 9174–9187. [Google Scholar] [CrossRef] [PubMed]
- Au, S.K.; Bommarius, B.R.; Bommarius, A.S. Biphasic reaction system allows for conversion of hydrophobic substrates by amine dehydrogenases. ACS Catal. 2014, 4, 4021–4026. [Google Scholar] [CrossRef]
- Patil, M.D.; Dev, M.J.; Shinde, A.S.; Bhilare, K.D.; Patel, G.; Chisti, Y.; Banerjee, U.C. Surfactant-mediated permeabilization of Pseudomonas putida KT2440 and use of the immobilized permeabilized cells in biotransformation. Process Biochem. 2017, 63, 113–121. [Google Scholar] [CrossRef]
- Patil, M.D.; Shinde, A.S.; Dev, M.J.; Patel, G.; Bhilare, K.D.; Banerjee, U.C. Combined effect of attrition and ultrasound on the disruption of Pseudomonas putida for the efficient release of arginine deiminase. Biotechnol. Progr. 2018, 34, 1185–1194. [Google Scholar] [CrossRef] [PubMed]
- Patil, M.D.; Dev, M.J.; Tangadpalliwar, S.; Patel, G.; Garg, P.; Chisti, Y.; Banerjee, U.C. Ultrasonic disruption of Pseudomonas putida for the release of arginine deiminase: Kinetics and predictive models. Bioresour. Technol. 2017, 233, 74–83. [Google Scholar] [CrossRef] [PubMed]
- Patil, M.D.; Rathod, V.P.; Bihade, U.R.; Banerjee, U.C. Purification and characterization of arginine deiminase from Pseudomonas putida: Structural insights of the differential affinities of L-arginine analogues. J. Biosci. Bioeng. 2019, 127, 129–137. [Google Scholar] [CrossRef]
- Bhilare, K.D.; Patil, M.D.; Tangadpalliwar, S.; Dev, M.J.; Garg, P.; Banerjee, U.C. Machine learning modelling for the high-pressure homogenization-mediated disruption of recombinant E. coli. Process Biochem. 2018, 71, 182–190. [Google Scholar] [CrossRef]
- Patil, M.D.; Patel, G.; Surywanshi, B.; Shaikh, N.; Garg, P.; Chisti, Y.; Banerjee, U.C. Disruption of Pseudomonas putida by high pressure homogenization: A comparison of the predictive capacity of three process models for the efficient release of arginine deiminase. AMB Express 2016, 6, 84. [Google Scholar] [CrossRef]
- Sheldon, R.A.; Brady, D. The limits to biocatalysis: Pushing the envelope. Chem. Commun. 2018, 54, 6088–6104. [Google Scholar] [CrossRef]
- Wachtmeister, J.; Rother, D. Recent advances in whole cell biocatalysis techniques bridging from investigative to industrial scale. Curr. Opin. Biotechnol. 2016, 42, 169–177. [Google Scholar] [CrossRef] [PubMed]
- Kim, G.H.; Jeon, H.; Khobragade, T.P.; Patil, M.D.; Sung, S.; Yoon, S.; Won, Y.; Sarak, S.; Yun, H. Glutamate as an Efficient Amine Donor for the Synthesis of Chiral β-and γ-Amino Acids Using Transaminase. ChemCatChem 2019, 11, 1437–1440. [Google Scholar] [CrossRef]
- Bhilare, K.D.; Patil, M.D.; Tangadpalliwar, S.; Shinde, A.; Garg, P.; Banerjee, U.C. Machine learning modelling for the ultrasonication-mediated disruption of recombinant E. coli for the efficient release of nitrilase. Ultrasonics 2019, 98, 72–81. [Google Scholar] [CrossRef] [PubMed]
- Molnár, Z.; Farkas, E.; Lakó, Á.; Erdélyi, B.; Kroutil, W.; Vértessy, B.G.; Paizs, C.; Poppe, L. Immobilized Whole-Cell Transaminase Biocatalysts for Continuous-Flow Kinetic Resolution of Amines. Catalysts 2019, 9, 438. [Google Scholar] [CrossRef]
- Ahsan, M.M.; Patil, M.D.; Jeon, H.; Sung, S.; Chung, T.; Yun, H. Biosynthesis of Nylon 12 Monomer, ω-Aminododecanoic Acid Using Artificial Self-Sufficient P450, AlkJ and ω-TA. Catalysts 2018, 8, 400. [Google Scholar] [CrossRef]
- Ahsan, M.M.; Jeon, H.P.; Nadarajan, S.P.; Chung, T.; Yoo, H.W.; Kim, B.G.; Patil, M.D.; Yun, H. Biosynthesis of the nylon 12 monomer, ω-Aminododecanoic acid with novel CYP153A, AlkJ, and ω-TA enzymes. Biotechnol. J. 2018, 13, 1700562. [Google Scholar] [CrossRef] [PubMed]
- Patil, M.D.; Grogan, G.; Bommarius, A.; Yun, H. Oxidoreductase-catalyzed synthesis of chiral amines. ACS Catal. 2018, 8, 10985–11015. [Google Scholar] [CrossRef]
- Kim, G.H.; Jeon, H.; Khobragade, T.P.; Patil, M.D.; Sung, S.; Yoon, S.; Won, Y.; Choi, I.S.; Yun, H. Enzymatic synthesis of sitagliptin intermediate using a novel ω-transaminase. Enzyme Microb. Technol. 2019, 120, 52–60. [Google Scholar] [CrossRef]
- Schmid-Dannert, C.; López-Gallego, F. Advances and opportunities for the design of self-sufficient and spatially organized cell-free biocatalytic systems. Curr. Opin. Chem. Biol. 2019, 49, 97–104. [Google Scholar] [CrossRef]
- Velasco-Lozano, S.; López-Gallego, F. Wiring step-wise reactions with immobilized multi-enzyme systems. Biocatal. Biotransform. 2018, 36, 184–194. [Google Scholar] [CrossRef]
- Beniítez-Mateos, A.I.; Contente, M.L.; Velasco-Lozano, S.; Paradisi, F.; López-Gallego, F. Self-sufficient flow-biocatalysis by coimmobilization of pyridoxal 5′-phosphate and ω-transaminases onto porous carriers. ACS Sustain. Chem. Eng. 2018, 6, 13151–13159. [Google Scholar] [CrossRef]
- Velasco-Lozano, S.; Benítez-Mateos, A.I.; López-Gallego, F. Co-immobilized Phosphorylated Cofactors and Enzymes as Self-Sufficient Heterogeneous Biocatalysts for Chemical Processes. Angew. Chem. Int. Ed. 2017, 56, 771–775. [Google Scholar] [CrossRef] [PubMed]
- Sung, S.; Jeon, H.; Sarak, S.; Ahsan, M.M.; Patil, M.D.; Kroutil, W.; Kim, B.G.; Yun, H. Parallel anti-sense two-step cascade for alcohol amination leading to ω-amino fatty acids and α, ω-diamines. Green Chem. 2018, 20, 4591–4595. [Google Scholar] [CrossRef]
- Ahsan, M.; Sung, S.; Jeon, H.; Patil, M.; Chung, T.; Yun, H. Biosynthesis of medium-to long-chain α, ω-diols from free fatty acids using CYP153A monooxygenase, carboxylic acid reductase, and E. coli endogenous aldehyde reductases. Catalysts 2017, 8, 4. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Substrate | Conv. (%) [a] | eeS (%) |
|---|---|---|
| a1[b] | 17 | 18.5 |
| a2[b] | 37 | 59 |
| a4[b] | 51 | >99 |
| a5[b] | 51 | >99 |
| a6[b] | 51 | >99 |
| a8[c] | 51 | >99 |
| a9[c] | 51 | >99 |
| a10[c] | 52 | >99 |
| a11[c] | 51 | >99 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Patil, M.D.; Yoon, S.; Jeon, H.; Khobragade, T.P.; Sarak, S.; Pagar, A.D.; Won, Y.; Yun, H. Kinetic Resolution of Racemic Amines to Enantiopure (S)-amines by a Biocatalytic Cascade Employing Amine Dehydrogenase and Alanine Dehydrogenase. Catalysts 2019, 9, 600. https://doi.org/10.3390/catal9070600
Patil MD, Yoon S, Jeon H, Khobragade TP, Sarak S, Pagar AD, Won Y, Yun H. Kinetic Resolution of Racemic Amines to Enantiopure (S)-amines by a Biocatalytic Cascade Employing Amine Dehydrogenase and Alanine Dehydrogenase. Catalysts. 2019; 9(7):600. https://doi.org/10.3390/catal9070600
Chicago/Turabian StylePatil, Mahesh D., Sanghan Yoon, Hyunwoo Jeon, Taresh P. Khobragade, Sharad Sarak, Amol D. Pagar, Yumi Won, and Hyungdon Yun. 2019. "Kinetic Resolution of Racemic Amines to Enantiopure (S)-amines by a Biocatalytic Cascade Employing Amine Dehydrogenase and Alanine Dehydrogenase" Catalysts 9, no. 7: 600. https://doi.org/10.3390/catal9070600
APA StylePatil, M. D., Yoon, S., Jeon, H., Khobragade, T. P., Sarak, S., Pagar, A. D., Won, Y., & Yun, H. (2019). Kinetic Resolution of Racemic Amines to Enantiopure (S)-amines by a Biocatalytic Cascade Employing Amine Dehydrogenase and Alanine Dehydrogenase. Catalysts, 9(7), 600. https://doi.org/10.3390/catal9070600