Synthesis of the First Resorcin[4]arene-Functionalized Triazolium Salts and Their Use in Suzuki–Miyaura Cross-Coupling Reactions

Abstract

1. Introduction
2. Results
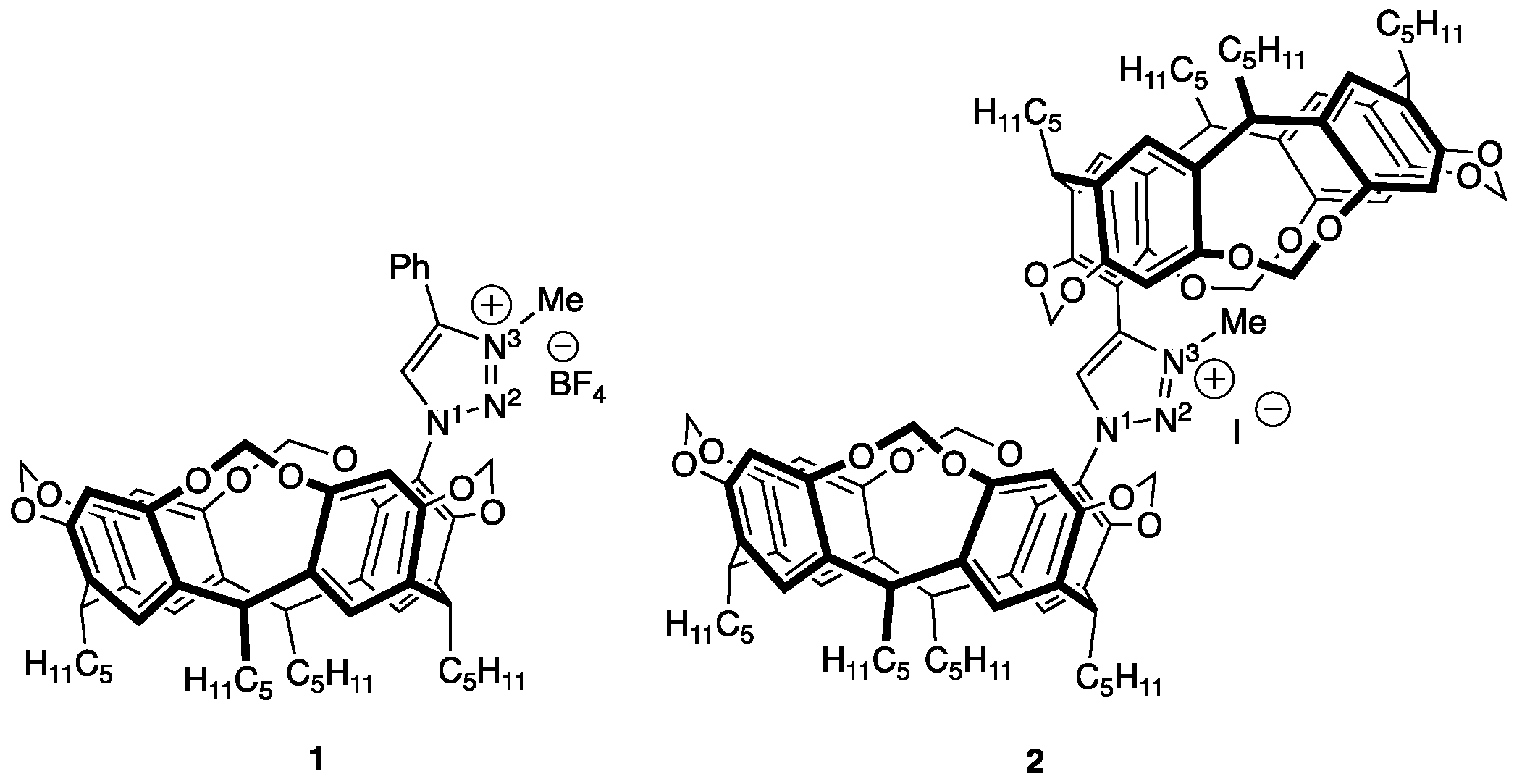
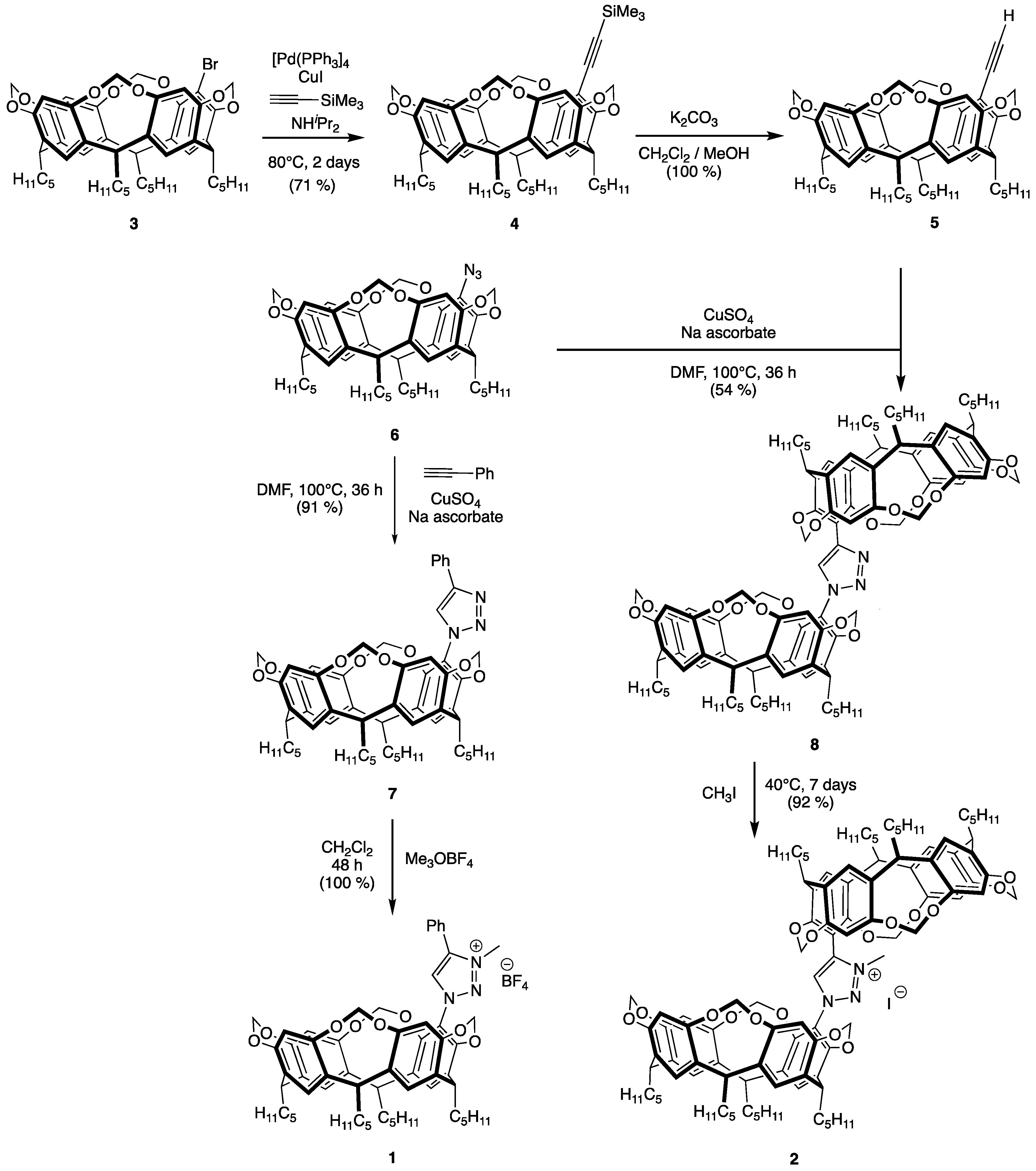
2.1. Synthesis of Triazolium Salts 1 and 2
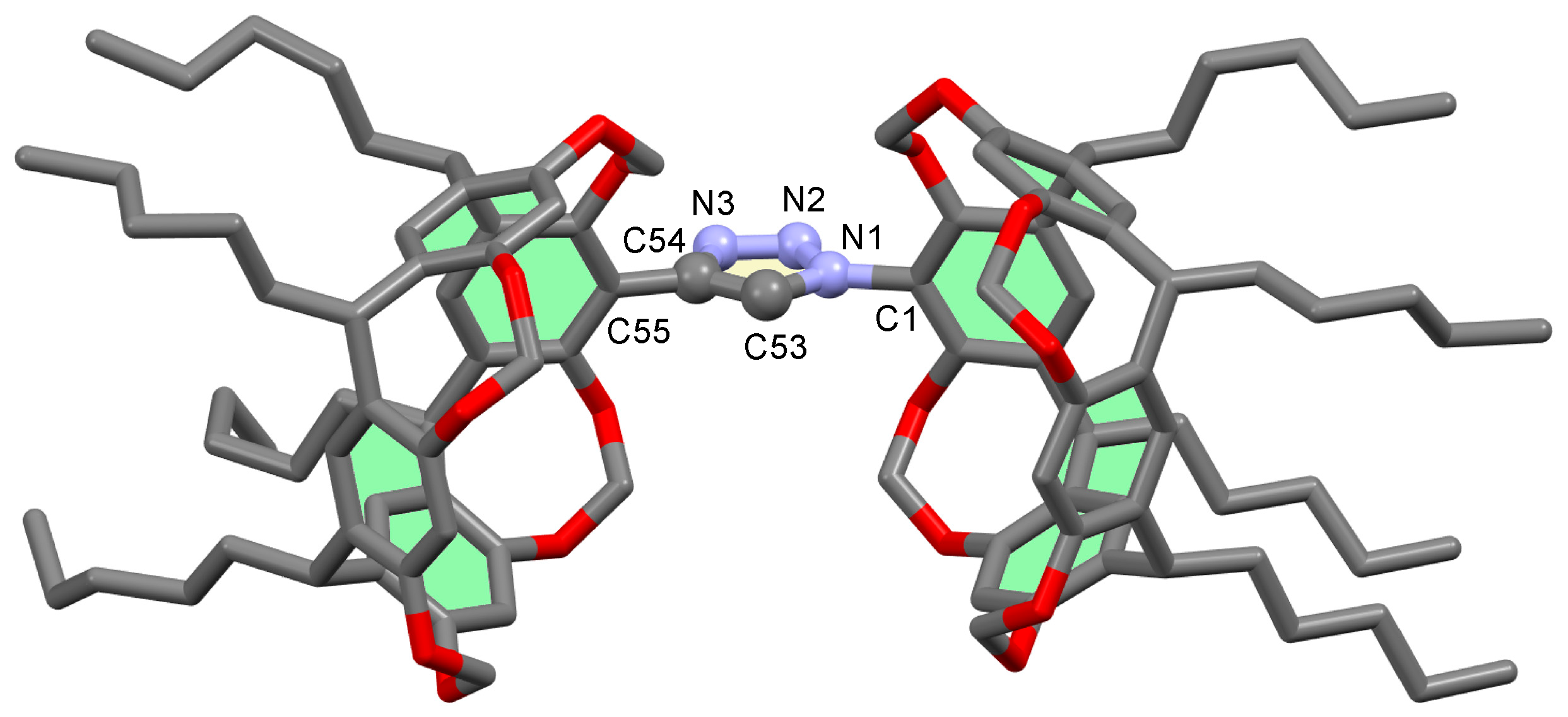
2.2. Crystal Structures of Triazole 8
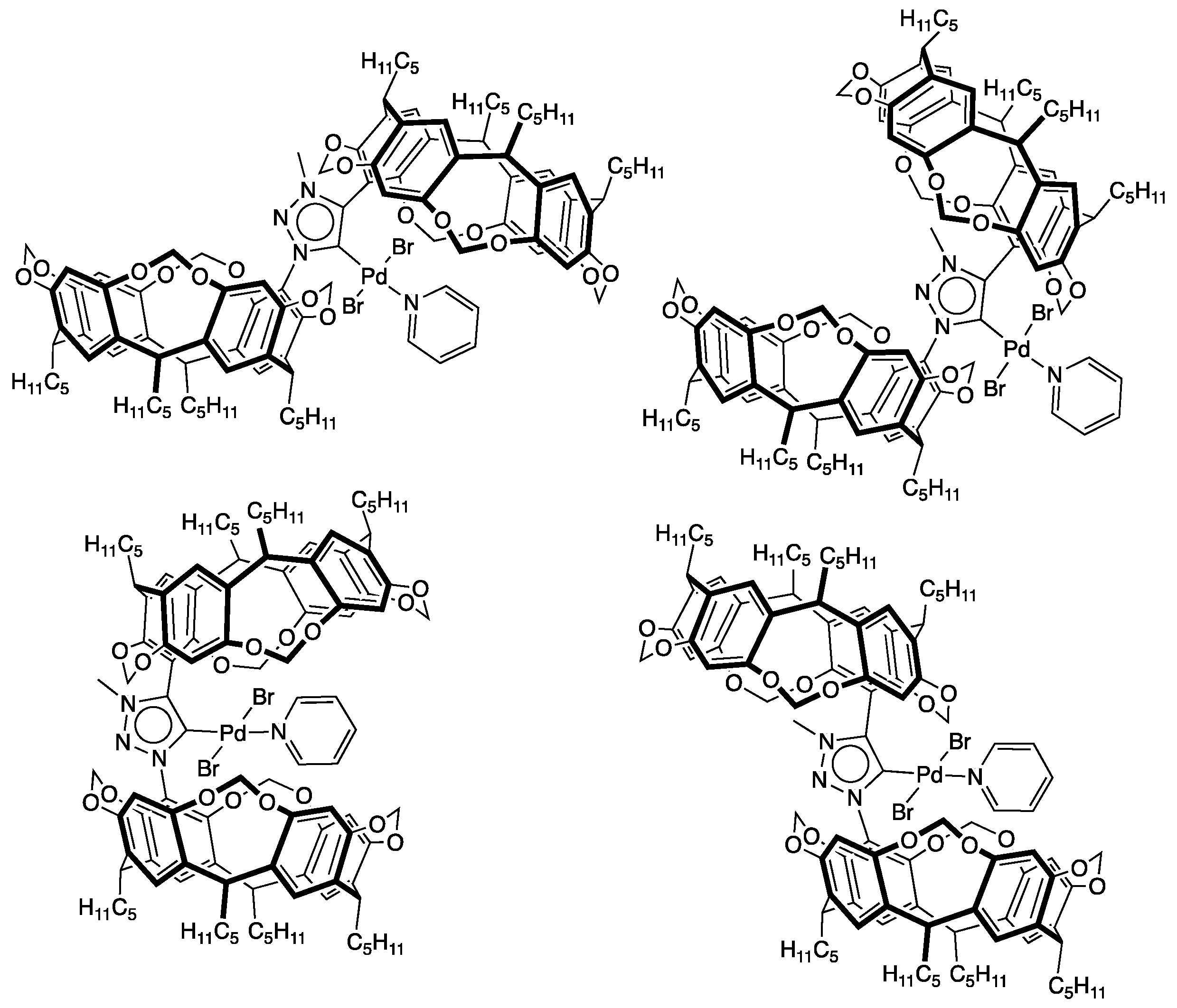
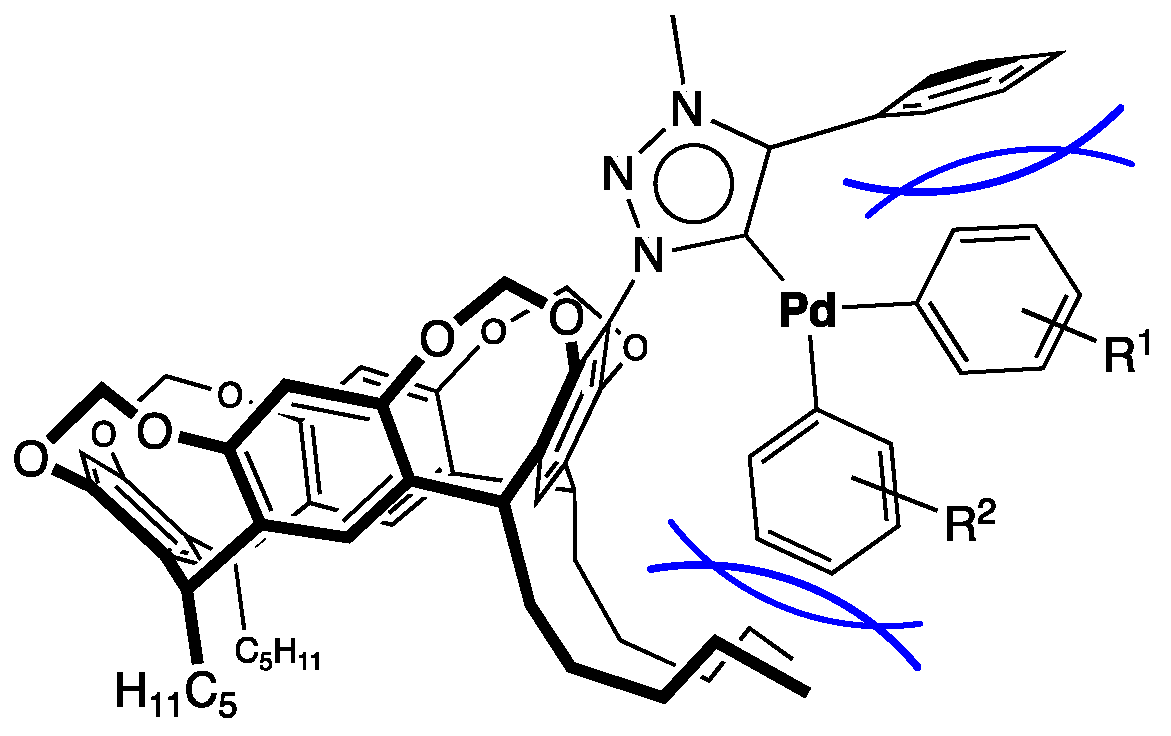
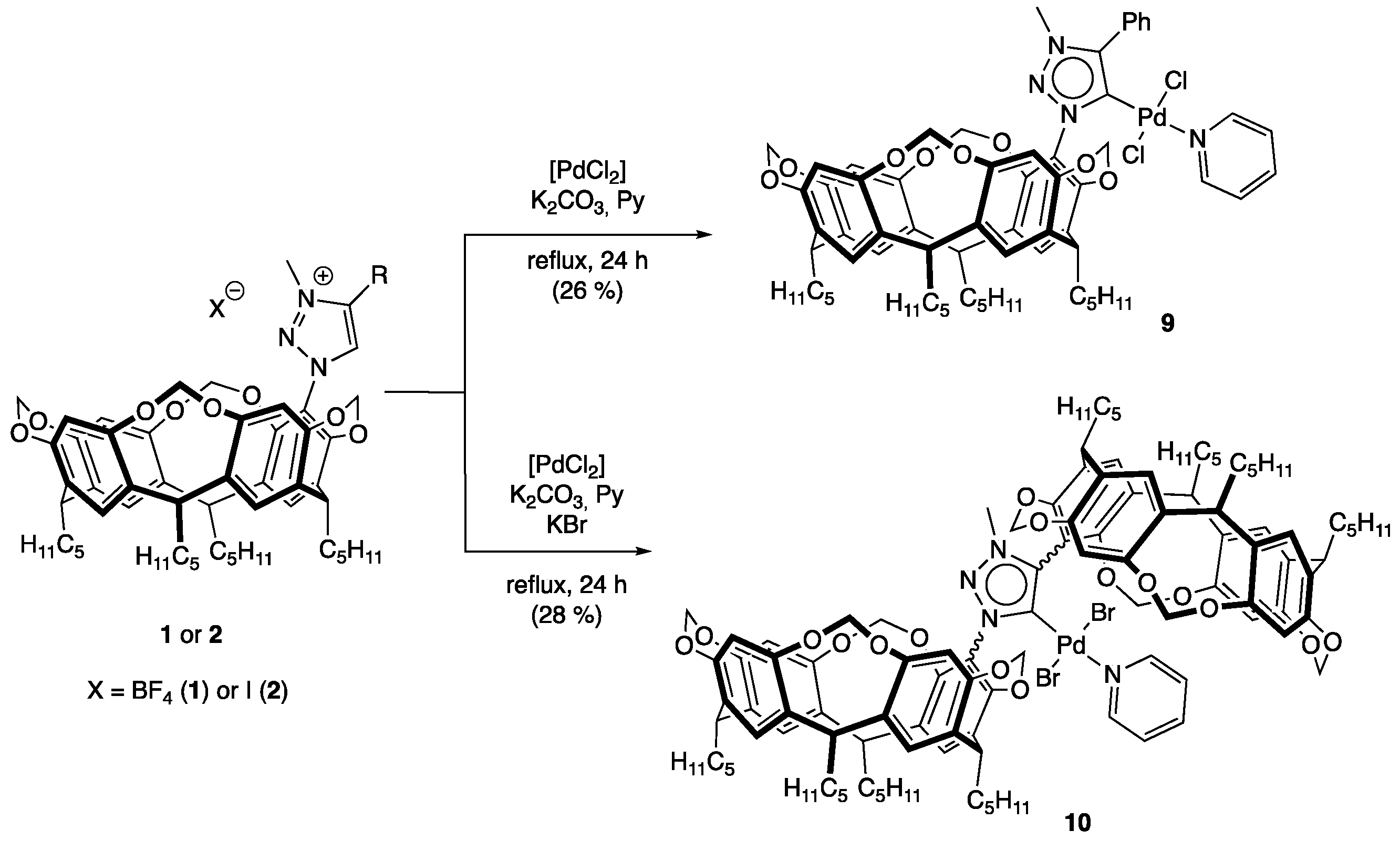
2.3. Synthesis of Palladium Complexes 9 and 10
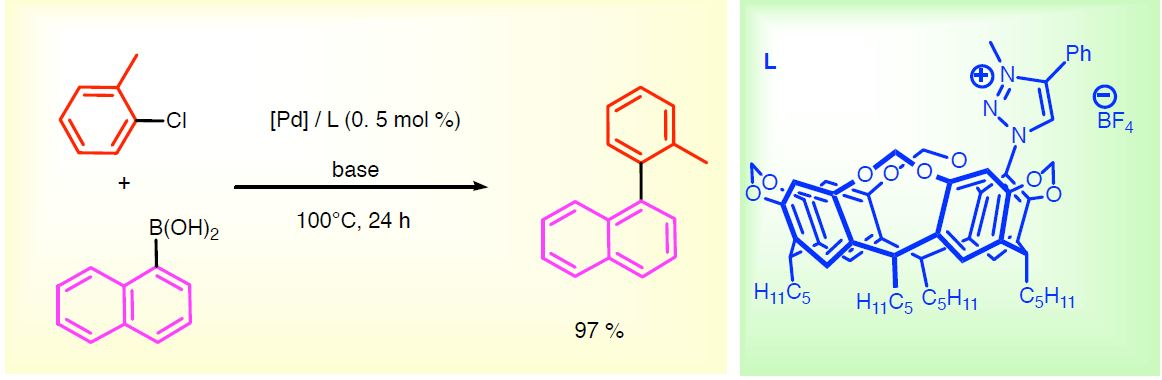
2.4. Catalytic Suzuki–Miyaura Cross-Coupling Reactions with Triazolium Salts 1 and 2
3. Materials and Methods
3.1. Experimental Section
3.2. Synthesis of 5-(Trimethylsilyl)ethynyl-4(24),6(10),12(16),18(22)-tetramethylenedioxy-2,8,14,20-tetra pentylresorcin[4]arene (4)
3.3. Synthesis of 5-Ethynyl-4(24),6(10),12(16),18(22)-tetramethylenedioxy-2,8,14,20-tetrapentylresorcin[4] arene (5)
3.4. Synthesis of 1-{4(24),6(10),12(16),18(22)-Tetramethylenedioxy-2,8,14,20-tetrapentylresorcin[4]arene-5-yl-4-phenyl-1H-1,2,3-triazole (7)
3.5. Synthesis of 1,4-bis{4(24),6(10),12(16),18(22)-Tetramethylenedioxy-2,8,14,20-tetrapentylresorcin[4]arene-5-yl}-1H-1,2,3-triazole (8)
3.6. Synthesis of 1-{4(24),6(10),12(16),18(22)-Tetramethylenedioxy-2,8,14,20-tetrapentylresorcin[4]arene-5-yl}-3-methyl-4-phenyl-1H-1,2,3-triazolium tetrafluoroborate (1)
3.7. Synthesis of 1,4-bis{4(24),6(10),12(16),18(22)-Tetramethylenedioxy-2,8,14,20-tetrapentylresorcin[4]arene-5-yl}-3-methyl-1H-1,2,3-triazolium iodide (2)
3.8. Synthesis of Trans-dichloro-{1-[4(24),6(10),12(16),18(22)-tetramethylenedioxy-2,8,14,20-tetrapentyl resorcin[4]arene-5-yl]-3-methyl-4-phenyl-1H-1,2,3-triazol-5-yliden}pyridine palladium(II) (9)
3.9. Synthesis of Trans-dibromo-{1,4-bis[4(24),6(10),12(16),18(22)-tetramethylenedioxy-2,8,14,20-tetrapentyl resorcin[4]arene-5-yl]-3-methyl-1H-1,2,3-triazol-5-yliden}pyridine palladium(II) (10)
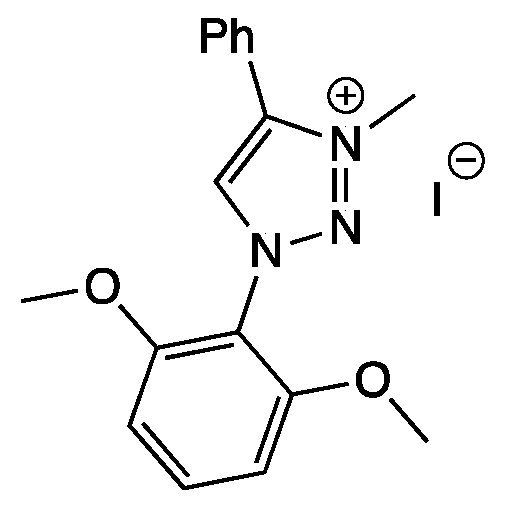
3.10. Synthesis of 1-(2,6-Dimethoxyphenyl)-3-methyl-4-phenyl-1H-1,2,3-triazolium iodide (11)
3.10.1. Step 1: Synthesis of 1-(2,6-Dimethoxyphenyl)-4-phenyl-1H-1,2,3-triazole
3.10.2. Step 2: Synthesis of 1-(2,6-Dimethoxyphenyl)-3-methyl-4-phenyl-1H-1,2,3-triazolium iodide (11)
3.11. Typical Procedure for the Palladium-Catalyzed Suzuki–Miyaura Cross-Coupling Reactions
3.12. X-ray Crystal Structure Analysis of Triazole 8
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Öfele, K. 1,3-Dimethyl-4-imidazolinyliden-(2)-pentacarbonylchrom ein neuer Übergangsmetall-Carben-Komplex. J. Organomet. Chem. 1968, 3, P42–P43. [Google Scholar] [CrossRef]
- Wanzlick, H.-W.; Schönherr, H.-J. Direct synthesis of a mercury salt-carbene complex. Angew. Chem. Int. Ed. 1968, 7, 141–142. [Google Scholar] [CrossRef]
- Teci, M.; Brenner, E.; Matt, D.; Toupet, L. N-Heterocyclic carbenes functioning as monoligating clamps. Eur. J. Inorg. Chem. 2013, 2013, 2841–2848. [Google Scholar] [CrossRef]
- Mathew, P.; Neels, A.; Albrecht, M. 1,2,3-Triazolylidenes as versatile abnormal carbene ligands for late transition metals. J. Am. Chem. Soc. 2008, 130, 13534–13535. [Google Scholar] [CrossRef]
- Guisado-Barrios, G.; Bouffard, J.; Donnadieu, B. Bertrand, G. Crystalline 1H-1,2,3-triazol-5-ylidenes: New stable mesoionic carbenes (MICs). Angew. Chem. Int. Ed. 2010, 49, 4759–4762. [Google Scholar] [CrossRef]
- Rostovtsev, V.V.; Green, L.G.; Fokin, V.V.; Sharpless, K.B. A stepwise Huisgen cycloaddition process: Copper(I)-catalyzed regioselective “ligation” of azides and terminal alkynes. Angew. Chem. Int. Ed. 2002, 41, 2596–2599. [Google Scholar] [CrossRef]
- Tornøe, C.W.; Christensen, C.; Meldal, M. Peptidotriazoles on solid phase: [1,2,3]-triazoles by regiospecific copper(I)-catalyzed 1,3-dipolar cycloadditions of terminal alkynes to azides. J. Org. Chem. 2002, 67, 3057–3064. [Google Scholar] [CrossRef] [PubMed]
- Terashima, T.; Inomata, S.; Ogata, K.; Fukuzawa, S.-I. Synthetic, structural, and catalytic studies of well-defined allyl 1,2,3-triazol-5-ylidene (tzNHC) palladium complexes. Eur. J. Inorg. Chem. 2012, 1387–1393. [Google Scholar] [CrossRef]
- Huang, J.; Hong, J.-T.; Hong, S.H. Suzuki–Miyaura cross-coupling reaction catalyzed by PEPPSI-type 1,4-di(2,6-diisopropylphenyl)-1,2,3-triazol-5-ylidene (tzIPr) palladium complex. Eur. J. Org. Chem. 2012, 2012, 6630–6635. [Google Scholar] [CrossRef]
- Bolje, A.; Košmrlj, J. A selective approach to pyridine appended 1,2,3-triazolium salts. Org. Lett. 2013, 15, 5084–5087. [Google Scholar] [CrossRef]
- Shaik, J.B.; Ramkumar, V.; Varghese, B.; Sankararaman, S. Synthesis and structure of trans-bis(1,4-dimesityl-3-methyl-1,2,3-triazol-5-ylidene)palladium(II) dichloride and diacetate. Suzuki–Miyaura coupling of polybromoarenes with high catalytic turnover efficiencies. Beilstein J. Org. Chem. 2013, 9, 698–704. [Google Scholar] [CrossRef]
- Wei, Y.; Petronilho, A.; Mueller-Bunz, H. Albrecht, M. Mesoionic triazolylidene nickel complexes: Synthesis, ligand lability, and catalytic C–C bond formation activity. Organometallics 2014, 33, 5834–5844. [Google Scholar] [CrossRef]
- Mendoza-Espinosa, D.; González-Olvera, R.; Negrón-Silva, G.E.; Angeles-Beltrán, D.; Suárez-Castillo, O.R.; Álvarez-Hernández, A.; Santillan, R. Phenoxy-linked mesoionic triazol-5-ylidenes as platforms for multinuclear transition metal complexes. Organometallics 2015, 34, 4529–4542. [Google Scholar] [CrossRef]
- Mohan, A.; Ramkumar, V.; Sankararaman, S. Synthesis and structures of (−)menthyl and (+)neomenthyl substituted enantio pure bis(1,2,3-triazol-5-ylidene)PdI2 complexes and PEPPSI type (1,2,3-triazol-5-ylidene)(pyridine)PdI2 complexes. Comparison of catalytic activities for C–C coupling. J. Organomet. Chem. 2015, 799–800, 115–121. [Google Scholar] [CrossRef]
- Sureshbabu, B.; Ramkumar, V.; Sankararaman, S. A mild and efficient method for the synthesis of structurally diverse 1,2,3-triazolylidene palladium(II) diiodo complexes. Comparison of catalytic activities for Suzuki–Miyaura coupling. J. Organomet. Chem. 2015, 799–800, 232–238. [Google Scholar] [CrossRef]
- Mitsui, T.; Sugihara, M.; Tokoro, Y.; Fukuzawa, S.-I. Synthesis of adamantyl substituted 1,2,3-triazol-5-ylidene ligands and their PEPPSI-type palladium complexes. Tetrahedron 2015, 71, 1509–1514. [Google Scholar] [CrossRef]
- Hettmanczyk, L.; Schmid, B.; Hohloch, S.; Sarkar, B. Palladium(II)-acetylacetonato complexes with mesoionic carbenes: Synthesis, structures and their application in the Suzuki–Miyaura cross coupling reaction. Molecules 2016, 21, 1561. [Google Scholar] [CrossRef]
- Kumar, A.; Prakasham, A.P.; Kumar Gangwar, M.; Vishnoi, P.; Butcher, R.J.; Ghosh, P. An efficient synthetic approach to trans-(NHC)2Pd(R)Br type complexes and their use in Suzuki–Miyaura cross-coupling reactions. Eur. J. Inorg. Chem. 2017, 2017, 2144–2154. [Google Scholar] [CrossRef]
- Inomata, S.; Hiroki, H.; Terashima, T.; Ogata, K.; Fukuzawa, S.-I. 1,2,3-Triazol-5-ylidene–palladium complex catalyzed Mizoroki–Heck and Sonogashira coupling reactions. Tetrahedron 2011, 67, 7263–7267. [Google Scholar] [CrossRef]
- Keske, E.C.; Zenkina, O.V.; Wang, R.; Crudden, C.M. Synthesis and structure of palladium 1,2,3-triazol-5-ylidene mesoionic carbene PEPPSI complexes and their catalytic applications in the Mizoroki–Heck reaction. Organometallics 2012, 31, 6215–6221. [Google Scholar] [CrossRef]
- Gazvoda, M.; Virant, M.; Pevec, A.; Urankar, D.; Bolje, A.; Kocevar, M.; Kosmrlj, J. A mesoionic bis(Py-tzNHC)palladium(II) complex catalyses “green” Sonogashira reaction through an unprecedented mechanism. Chem. Commun. 2016, 52, 1571–1574. [Google Scholar] [CrossRef]
- Ros, A.; Alcarazo, M.; Iglesias-Sigüenza, J.; Díez, E.; Álvarez, E.; Fernández, R.; Lassaletta, J.M. Stereoselective synthesis of rhodium(I) 4-(dialkylamino)triazol-5-ylidene complexes. Organometallics 2008, 27, 4555–4564. [Google Scholar] [CrossRef]
- Hohloch, S.; Duecker, F.L.; van der Meer, M.; Sarkar, B. Copper(I) complexes of mesoionic carbene: Structural characterization and catalytic hydrosilylation reactions. Molecules 2015, 20, 7379–7395. [Google Scholar] [CrossRef]
- Maity, R.; van der Meer, M.; Hohloch, S.; Sarkar, B. Di- and trinuclear iridium(III) complexes with poly-mesoionic carbenes synthesized through selective base-dependent metalation. Organometallics 2015, 34, 3090–3096. [Google Scholar] [CrossRef]
- Sabater, S.; Müller-Bunz, H.; Albrecht, M. Carboxylate-functionalized mesoionic carbene precursors: Decarboxylation, ruthenium bonding, and catalytic activity in hydrogen transfer reactions. Organometallics 2016, 35, 2256–2266. [Google Scholar] [CrossRef]
- Wei, Y.; Liu, S.-X.; Mueller-Bunz, H.; Albrecht, M. Application in selective aldehyde hydrosilylation. ACS Catal. 2016, 6, 8192–8200. [Google Scholar] [CrossRef]
- Bolje, A.; Hohloch, S.; Košmrlj, J.; Sarkar, B. RuII, IrIII and OsII mesoionic carbene complexes: Efficient catalysts for transfer hydrogenation of selected functionalities. Dalton Trans. 2016, 45, 15983–15993. [Google Scholar] [CrossRef] [PubMed]
- Bheeter, L.P.; Wei, D.; Dorcet, V.; Roisnel, T.; Ghosh, P.; Sortais, J.-B.; Darcel, C. 1,2,4-Triazole-based N-heterocyclic carbene nickel complexes—Synthesis and catalytic application. Eur. J. Inorg. Chem. 2015, 2015, 5226–5231. [Google Scholar] [CrossRef]
- Schöffler, A.L.; Makarem, A.; Rominger, F.; Straub, B.F. Dinuclear thiazolylidene copper complex as highly active catalyst for azid-alkyne cycloadditions. Beilstein J. Org. Chem. 2016, 12, 1566–1572. [Google Scholar] [CrossRef] [PubMed]
- Mendoza-Espinosa, D.; González-Olvera, R.; Osornio, C.; Negrón-Silva, G.E.; Álvarez-Hernández, A.; Bautista-Hernández, C.I.; Suárez-Castillo, O.R. Structural diversity of phenoxy functionalized triazol-5-ylidene palladium(II) complexes and their application in C-N bond formation. J. Organomet. Chem. 2016, 803, 142–149. [Google Scholar] [CrossRef]
- Guo, F.-J.; Sun, J.; Xu, Z.-Q.; Kühn, F.E.; Zang, S.-L.; Zhou, M.-D. C-S cross-coupling of aryl halides with alkyl thiols catalyzed by in-situ generated nickel(II) N-heterocyclic carbene complexes. Catal. Commun. 2017, 96, 11–14. [Google Scholar] [CrossRef]
- Strydom, I.; Guisado-Barrios, G.; Fernández, I.; Liles, D.C.; Peris, E.; Bezuidenhout, D.I. A hemilabile and cooperative N-donor-functionalized 1,2,3-triazol-5-ylidene ligand for alkyne hydrothiolation reactions. Chem. Eur. J. 2017, 23, 1393–1401. [Google Scholar] [CrossRef]
- Şahin, N.; Sémeril, D.; Brenner, E.; Matt, D.; Özdemir, İ.; Kaya, C.; Toupet, L. Resorcinarene-functionalised imidazolium salts as ligand precursors for palladium-catalysed Suzuki–Miyaura cross-couplings. ChemCatChem 2013, 5, 1116–1125. [Google Scholar] [CrossRef]
- Şahin, N.; Sémeril, D.; Brenner, E.; Matt, D.; Özdemir, İ.; Kaya, C.; Toupet, L. Subtle steric effects in nickel-catalysed kumada-Tamao-Corriu cross-coupling using resorcinarenyl-imidazolium salts. Eur. J. Org. Chem. 2013, 2013, 4443–4449. [Google Scholar] [CrossRef]
- Şahin, N.; Sémeril, D.; Brenner, E.; Matt, D.; Kaya, C.; Toupet, L. Palladium-catalysed Suzuki–Miyaura cross-coupling with imidazolylidene ligands substituted by crowded resorcinarenyl and calixarenyl units. Turk. J. Chem. 2015, 39, 1171–1179. [Google Scholar] [CrossRef]
- Kaloğlu, M.; Sémeril, D.; Brenner, E.; Matt, D.; Özdemir, İ.; Toupet, L. The influence of imidazolylidene ligands with bulky resorcinarenyl substituents on catalysts for Suzuki–Miyaura coupling. Eur. J. Inorg. Chem. 2016, 2016, 1115–1120. [Google Scholar] [CrossRef]
- Kaloğlu, M.; Şahin, N.; Sémeril, D.; Brenner, E.; Matt, D.; Özdemir, İ.; Kaya, C.; Toupet, L. Copper-catalysed allylic substitution using 2,8,14,20-tetrapentylresorcinarenyl-substituted imidazolium salts. Eur. J. Org. Chem. 2015, 2015, 7310–7316. [Google Scholar] [CrossRef]
- Natarajan, N.; Chavagnan, T.; Sémeril, D.; Brenner, E.; Matt, D.; Ramesh, R.; Toupet, L. Cavitand chemistry: Nickel half-sandwich complexes with imidazolylidene ligands bearing one or two resorcinarenyl substituents. Eur. J. Inorg. Chem. 2018, 2018, 890–896. [Google Scholar] [CrossRef]
- Kantchev, E.A.B.; O’Brien, C.J.; Organ, M.G. Palladium complexes of N-heterocyclic carbenes as catalysts for cross-coupling reactions—A synthetic chemist’s perspective. Angew. Chem. Int. Ed. 2007, 46, 2768–2813. [Google Scholar] [CrossRef]
- Valente, C.; Çalimsiz, S.; Hoi, K.H.; Mallik, D.; Sayah, M.; Organ, M.G. The development of bulky palladium NHC complexes for the most-challenging cross-coupling reactions. Angew. Chem. Int. Ed. 2012, 51, 3314–3332. [Google Scholar] [CrossRef]
- Campeau, L.-C.; Hazari, N. Cross-coupling and related reactions: Connecting past success to the development of new reactions for the future. Organometallics 2019, 38, 3–35. [Google Scholar] [CrossRef]
- Praetorius, J.M.; Crudden, C.M. N-Heterocyclic carbene complexes of rhodium: Structure, stability and reactivity. Dalton Trans. 2008, 4079–4094. [Google Scholar] [CrossRef] [PubMed]
- Chavagnan, T.; Sémeril, D.; Matt, D.; Toupet, L. Substrate-selective olefin hydrogenation with a cavitand-based bis(N-anisyl iminophosphorane). Eur. J. Org. Chem. 2017, 2017, 70–76. [Google Scholar] [CrossRef]
- Chavagnan, T.; Sémeril, D.; Matt, D.; Toupet, L. Cavitand chemistry—Towards metallocapsular catalysts. Eur. J. Org. Chem. 2017, 2017, 313–323. [Google Scholar] [CrossRef]
- Elaieb, F.; Sémeril, D.; Matt, D. Catalytic behaviour of calixarenylphosphanes in nickel-catalysed Suzuki–Miyaura cross-coupling. Eur. J. Inorg. Chem. 2017, 2017, 685–693. [Google Scholar] [CrossRef]
- Nasielski, J.; Hadei, N.; Achonduh, G.; Kantchev, E.A.B.; O’Brien, C.J.; Lough, A.; Organ, M.G. Structure-activity relationship analysis of Pd–PEPPSI complexes in cross-couplings: A close inspection of the catalytic cycle and the precatalyst activation model. Chem. Eur. J. 2010, 16, 10844–10853. [Google Scholar] [CrossRef] [PubMed]
- Buchowicz, W.; Kozioł, A.; Jerzykiewicz, L.B.; Lis, T.; Pasynkiewicz, S.; Pecherzewska, A.; Pietrzykowski, A. N-Heterocyclic carbene complexes of cyclopentadienylnickel(II): Synthesis, structure and catalytic activity in styrene polymerization. J. Mol. Catal. A 2006, 257, 118–123. [Google Scholar] [CrossRef]
- El Moll, H.; Sémeril, D.; Matt, D.; Toupet, L. Regioselective grafting of two -CH2P(X)Ph2 Units (X = O, Lone Pair) onto a resorcin[4]arene-derived cavitand. Eur. J. Org. Chem. 2010, 2010, 1158–1168. [Google Scholar] [CrossRef]
- Hwang, H.; Kim, J.; Jeong, J.; Chang, S. Regioselective introduction of heteroatoms at the C-8 position of quinoline N-oxides: Remote C-H activation using N-oxide as a stepping stone. J. Am. Chem. Soc. 2014, 136, 10770–10776. [Google Scholar] [CrossRef]
- Zhao, Y.; van Nguyen, H.; Male, L.; Craven, P.; Buckley, B.R.; Fossey, J.S. Phosphino-triazole ligands for palladium-catalyzed cross-coupling. Organometallics 2018, 37, 4224–4241. [Google Scholar] [CrossRef] [PubMed]
- Sheldrick, G.M. A short history of SHELX. Acta Cryst. 2008, A64, 112–122. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
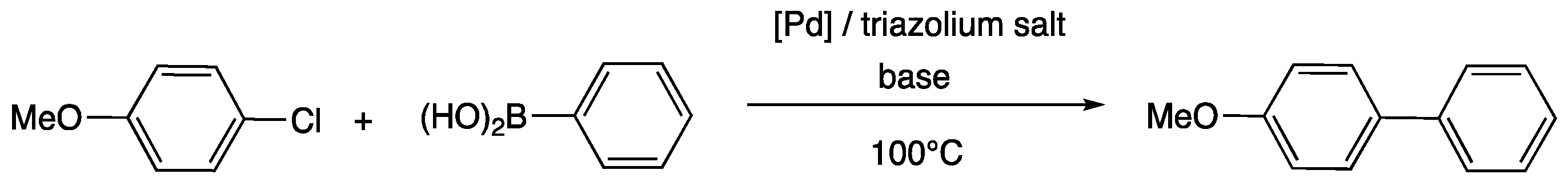{kind=link}
| Entry | Triazolium Salt | [Pd] | Base | Solvent | Conversion (%) |
|---|---|---|---|---|---|
| 1 | 1 | [Pd(OAc)2] | Cs2CO3 | DMF | 3 |
| 2 | 2 | 8 | |||
| 3 | 1 | [Pd(OAc)2] | K2CO3 | DMF | traces |
| 4 | 2 | traces | |||
| 5 | 1 | [Pd(OAc)2] | NaH | DMF | 1 |
| 6 | 2 | 18 | |||
| 7 | 1 | [Pd(OAc)2] | K3PO4 | DMF | traces |
| 8 | 2 | traces | |||
| 9 | 1 | [Pd(OAc)2] | tBuOK | DMF | 30 |
| 10 | 1 | dioxane | 47 | ||
| 11 | 2 | DMF | 25 | ||
| 12 | 1 | [PdCl(η3-C3H5)]2 | tBuOK | DMF | traces |
| 13 | 2 | 19 | |||
| 14 | 1 | [PdCl2(PhCN)2] | tBuOK | DMF | 28 |
| 15 | 2 | DMF | 29 | ||
| 16 | 2 | dioxane | 43 | ||
| 17 | 1 | [PdCl2(cod)] | tBuOK | DMF | 31 |
| 18 | 1 | dioxane | 36 | ||
| 19 | 2 | DMF | 23 | ||
| 20 | 1 | [Pd2(dba)3] | tBuOK | DMF | 9 |
| 21 | 2 | 17 | |||
| 22 | / | Complex 9 | tBuOK | dioxane | 46 |
| 23 | / | [Pd(OAc)2] | tBuOK | dioxane | 8 |
| 24 | / | [PdCl2(PhCN)2] | tBuOK | dioxane | 6 |
| Entry | ArCl | Triazolium Salt - Conditions |  |  |  |  | |
| 1 |  | 1 - A (5 h) | conv. (%) | 100 | 98 | 100 | 98 |
| 2 | 2 - B (5 h) | conv. (%) | 96 | 81 | 80 | 78 | |
| 3 | 11 - A (5 h) | conv. (%) | 31 | 41 | |||
| 4 | 11 - B (5 h) | conv. (%) | 18 | 19 | |||
| 5 |  | 1 - A (5 h) | conv. (%) | 95 | 77 | 80 | 99 |
| 6 | 2 - B (5 h) | conv. (%) | 100 | 85 | 83 | 100 | |
| 7 | 11 - A (5 h) | conv. (%) | 15 | 16 | |||
| 8 | 11 - B (5 h) | conv. (%) | 5 | 6 | |||
| 9 |  | 1 - A (24 h) | conv. (%) | 100 | 100 | 63 | 97 |
| 10 | 2 - B (24 h) | conv. (%) | 83 | 65 | 52 | 76 | |
| 11 |  | 1 - A (24 h) | conv. (%) | 100 | 99 | 95 | 100 |
| 12 | 2 - B (24 h) | conv. (%) | 98 | 99 | 83 | 99 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sémeril, D.; Matt, D.; Ramesh, R. Synthesis of the First Resorcin[4]arene-Functionalized Triazolium Salts and Their Use in Suzuki–Miyaura Cross-Coupling Reactions. Catalysts 2019, 9, 388. https://doi.org/10.3390/catal9040388
Sémeril D, Matt D, Ramesh R. Synthesis of the First Resorcin[4]arene-Functionalized Triazolium Salts and Their Use in Suzuki–Miyaura Cross-Coupling Reactions. Catalysts. 2019; 9(4):388. https://doi.org/10.3390/catal9040388
Chicago/Turabian StyleSémeril, David, Dominique Matt, and Rengan Ramesh. 2019. "Synthesis of the First Resorcin[4]arene-Functionalized Triazolium Salts and Their Use in Suzuki–Miyaura Cross-Coupling Reactions" Catalysts 9, no. 4: 388. https://doi.org/10.3390/catal9040388
APA StyleSémeril, D., Matt, D., & Ramesh, R. (2019). Synthesis of the First Resorcin[4]arene-Functionalized Triazolium Salts and Their Use in Suzuki–Miyaura Cross-Coupling Reactions. Catalysts, 9(4), 388. https://doi.org/10.3390/catal9040388