Electrocatalytic Oxidation of Small Molecule Alcohols over Pt, Pd, and Au Catalysts: The Effect of Alcohol’s Hydrogen Bond Donation Ability and Molecular Structure Properties



Abstract
:1. Introduction
2. Results and Discussion
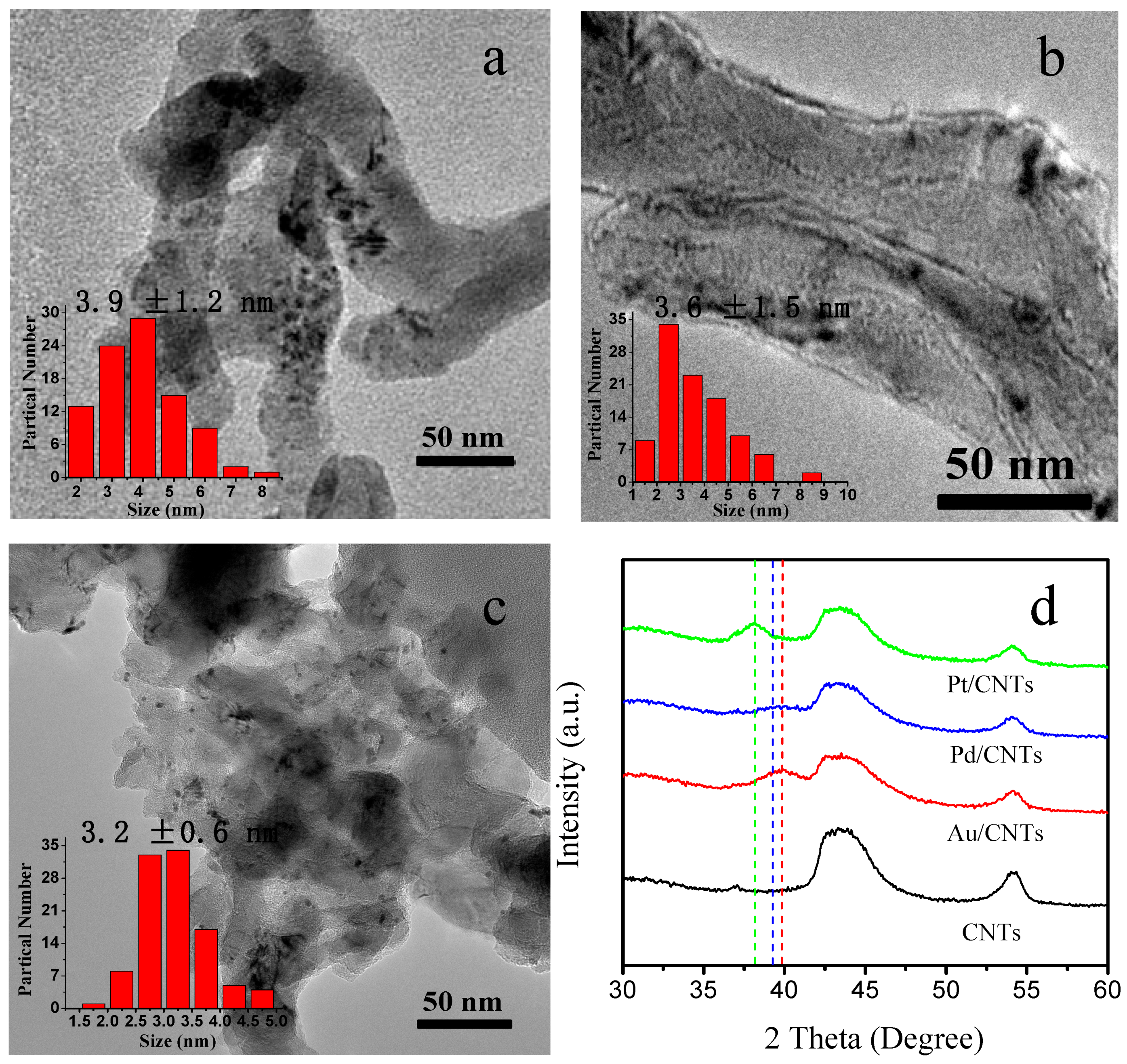
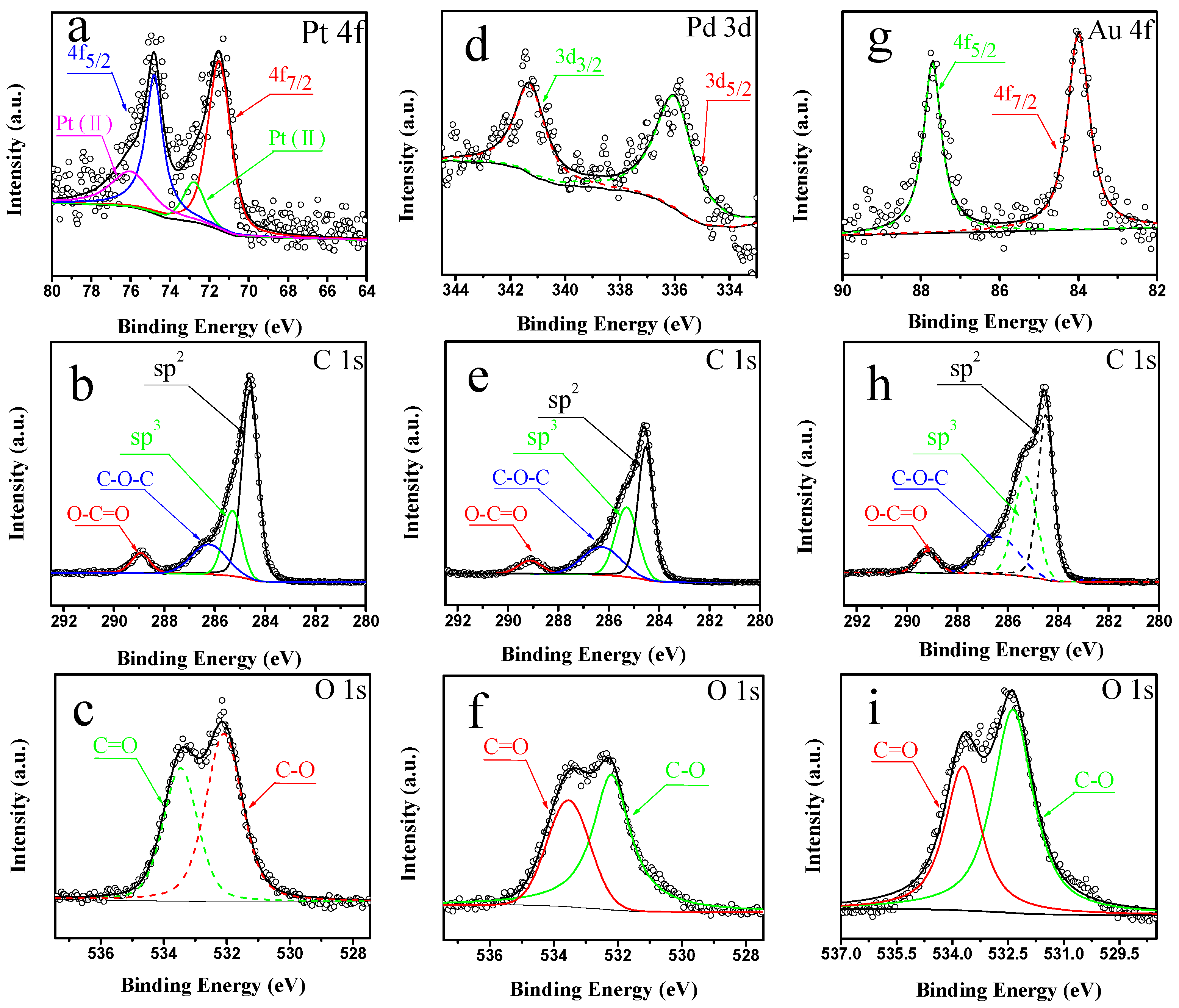
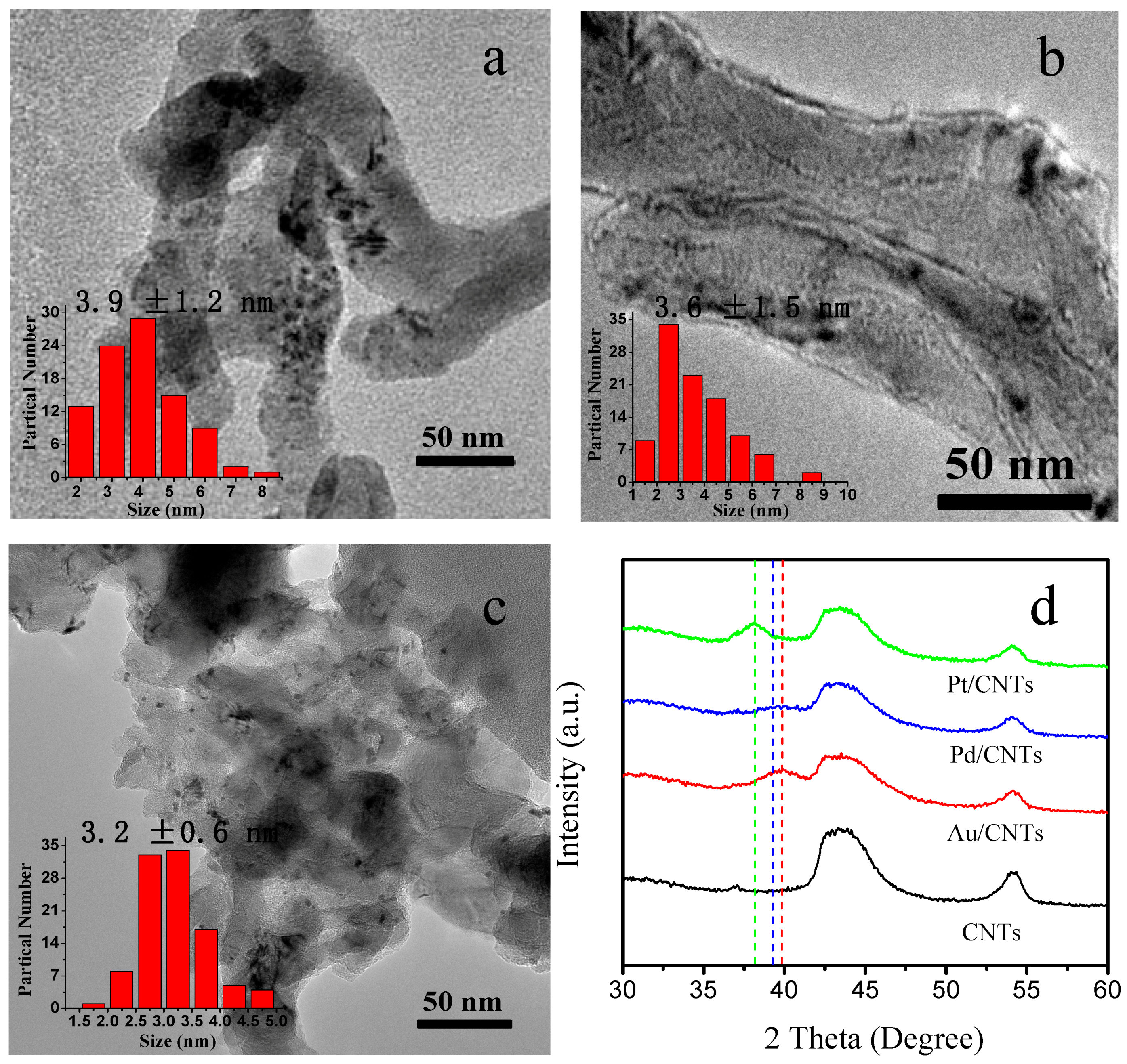
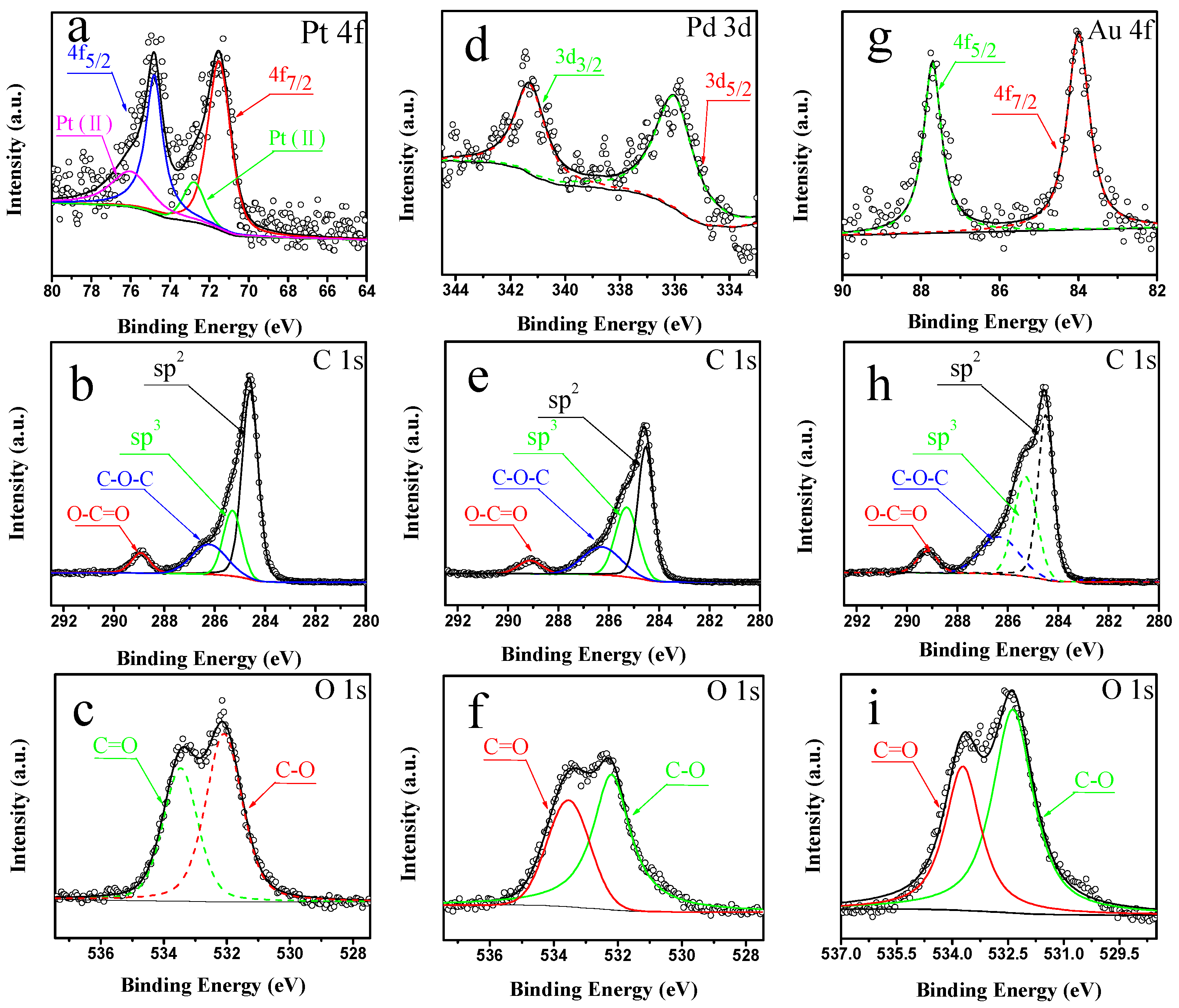
2.1. Characterization of Pt/CNTs, Pd/CNTs, and Au/CNTs Catalysts
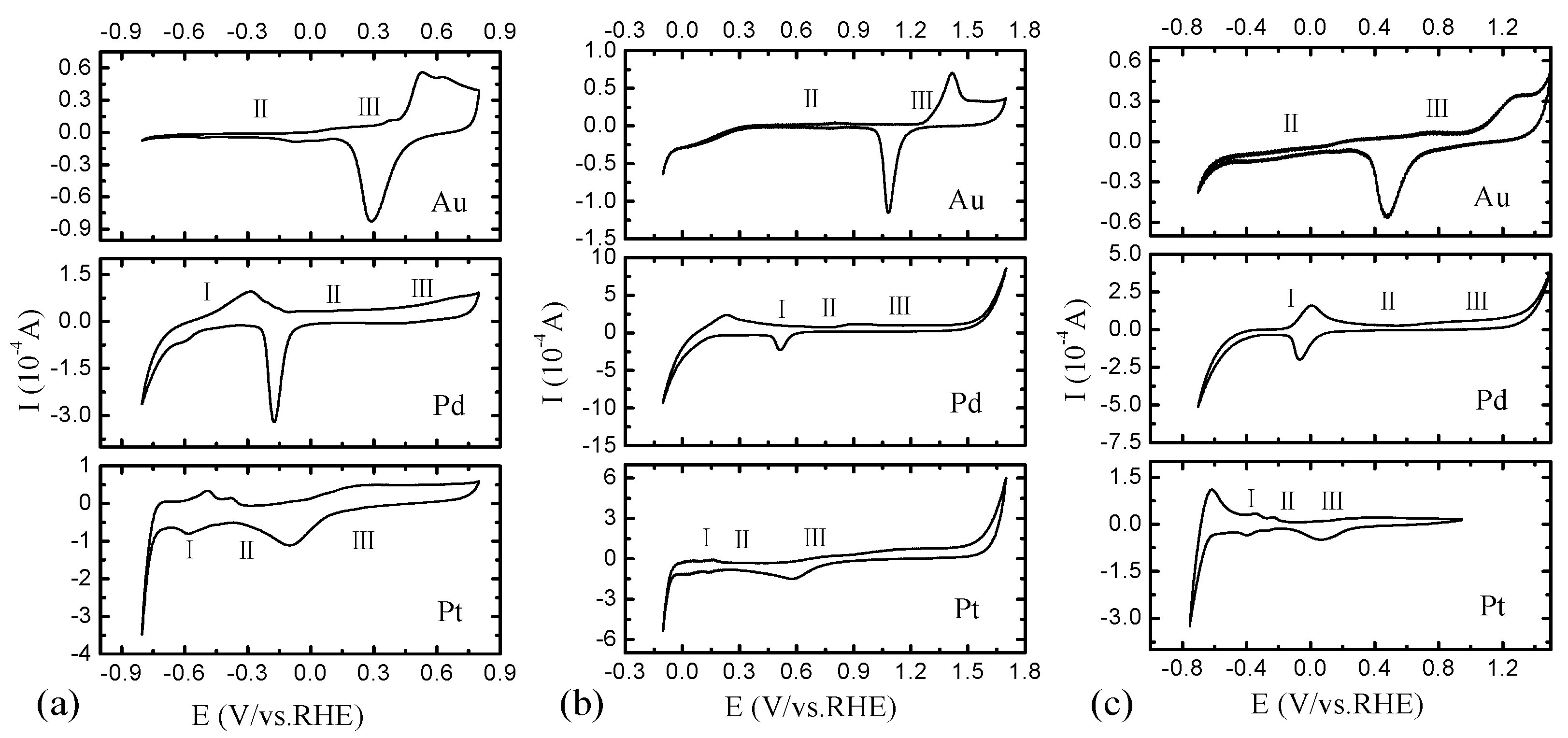
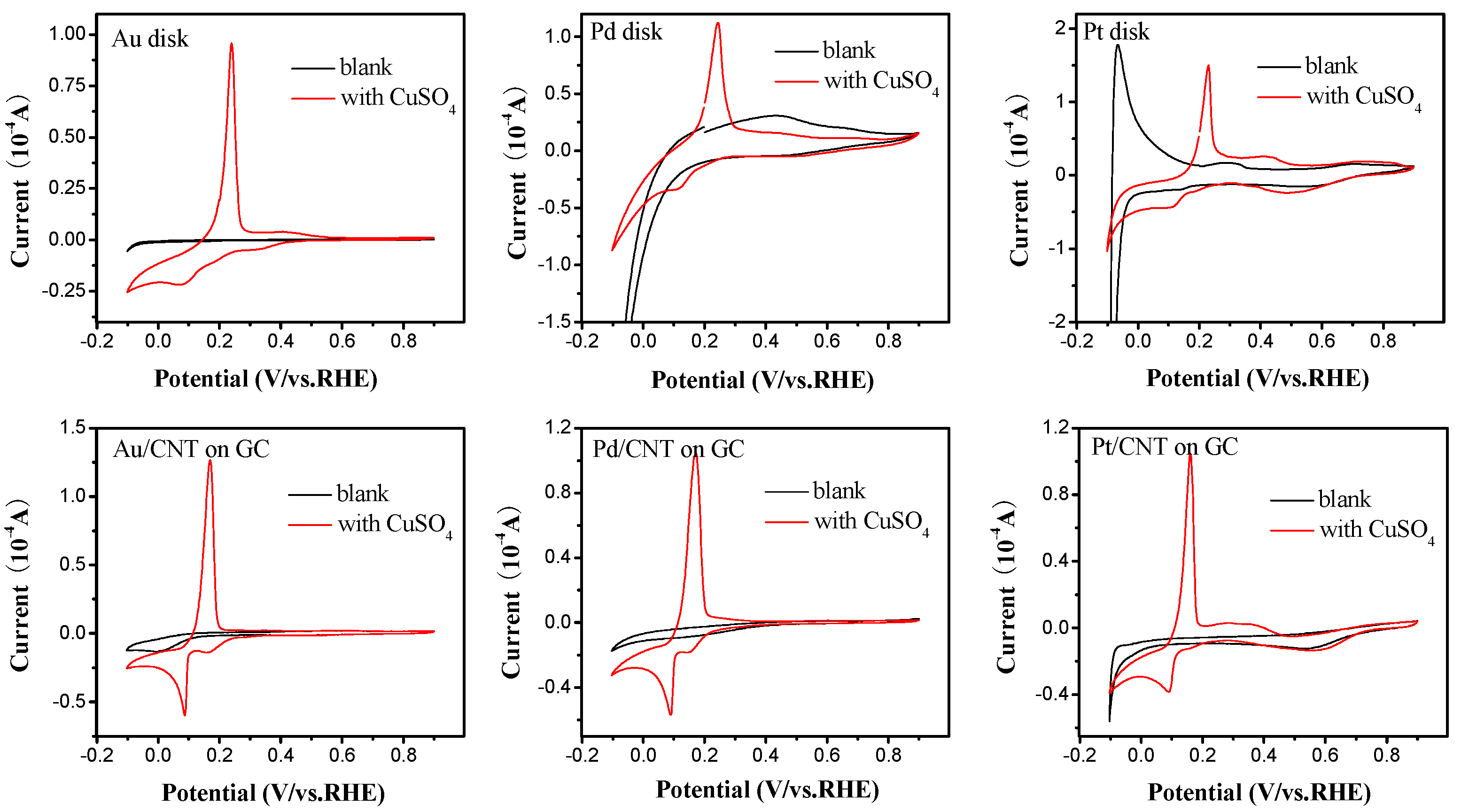
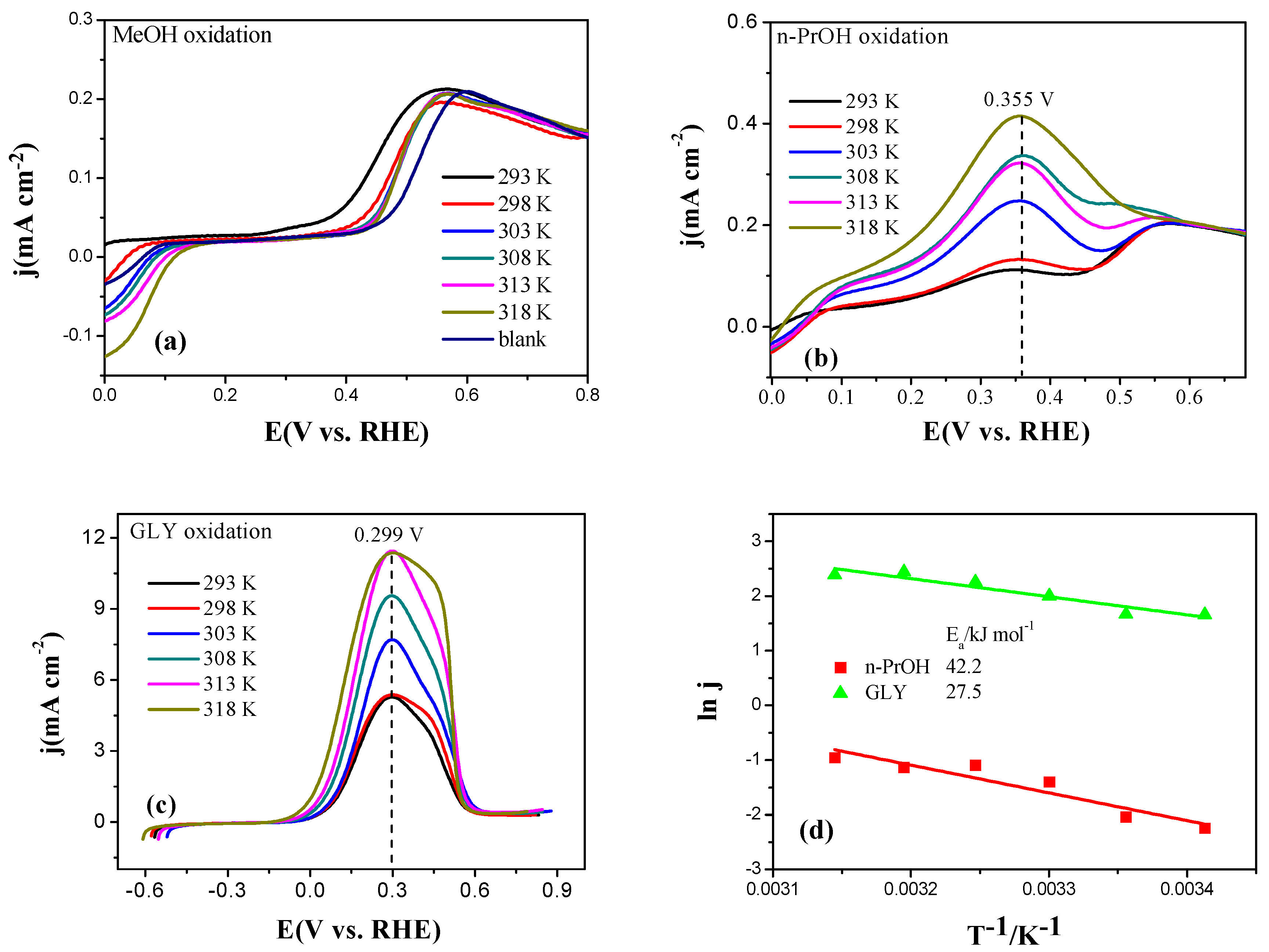
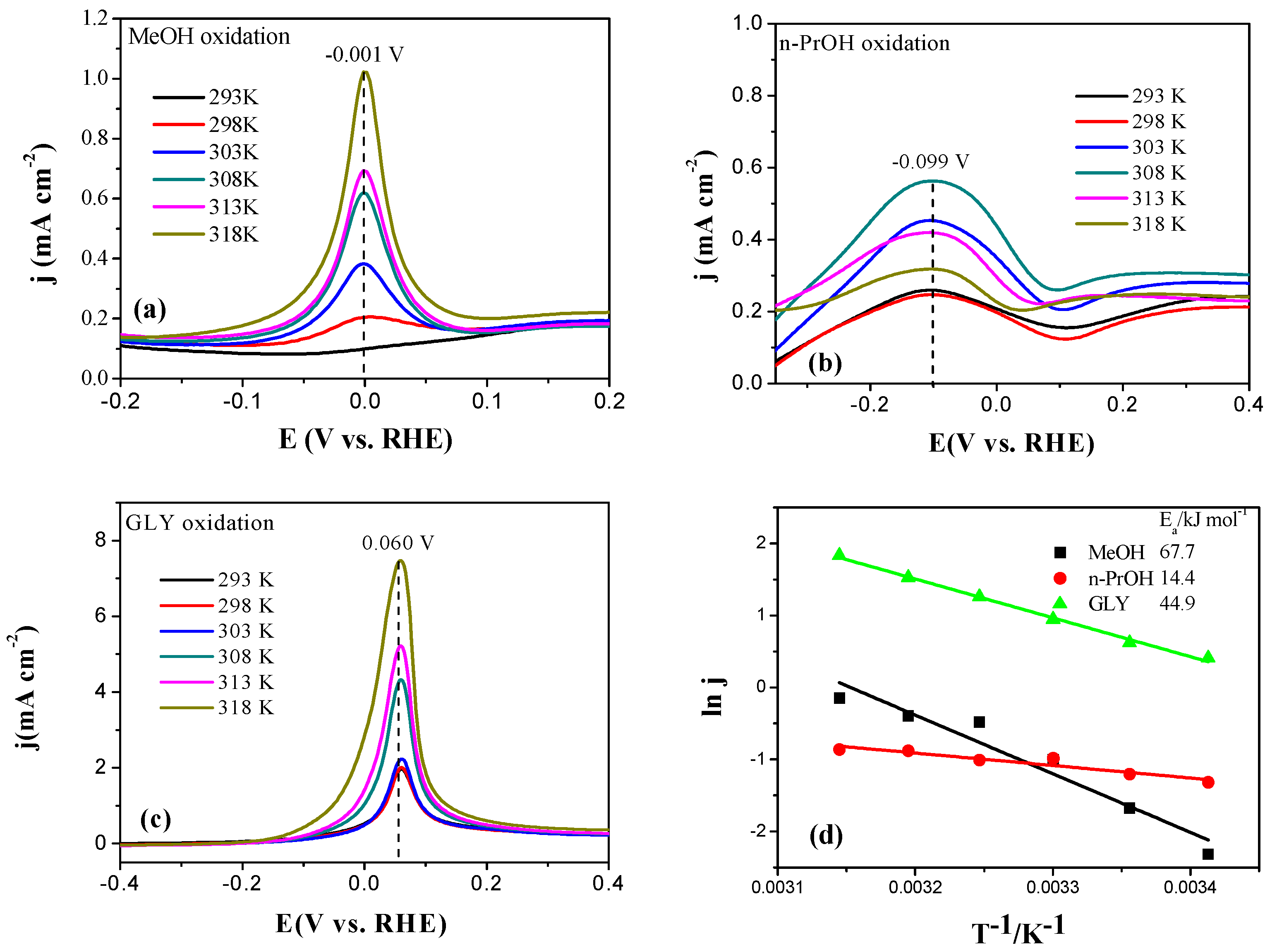
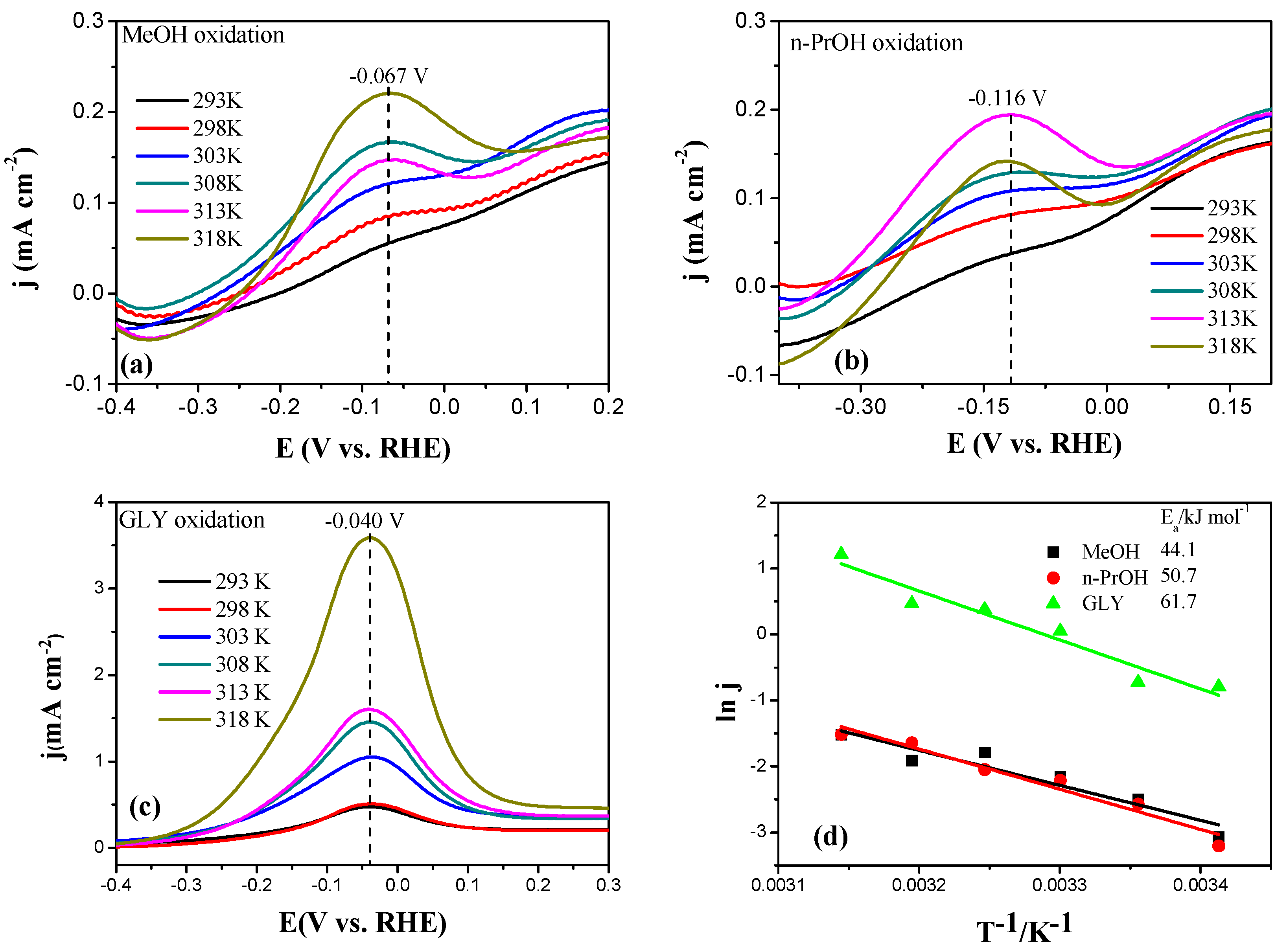
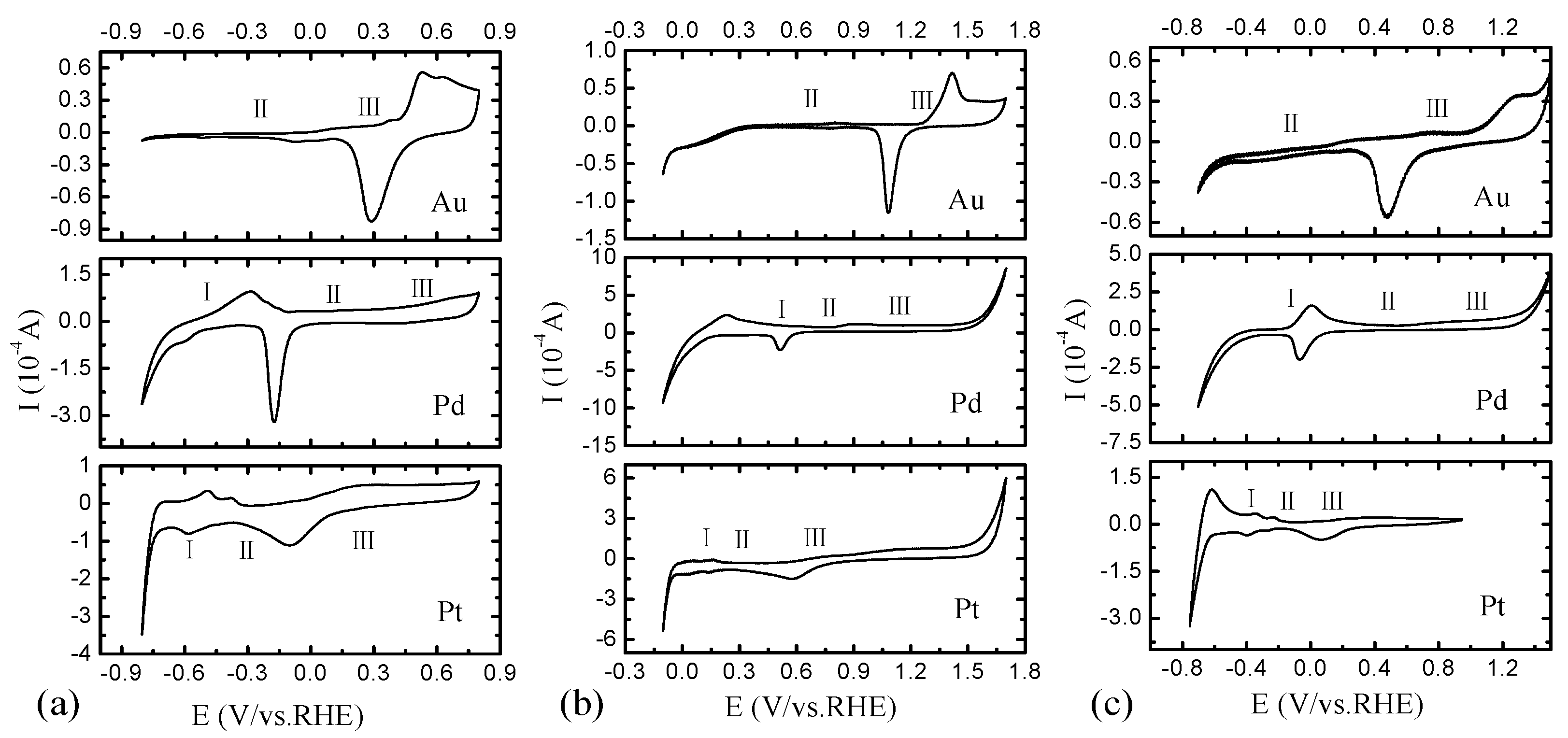
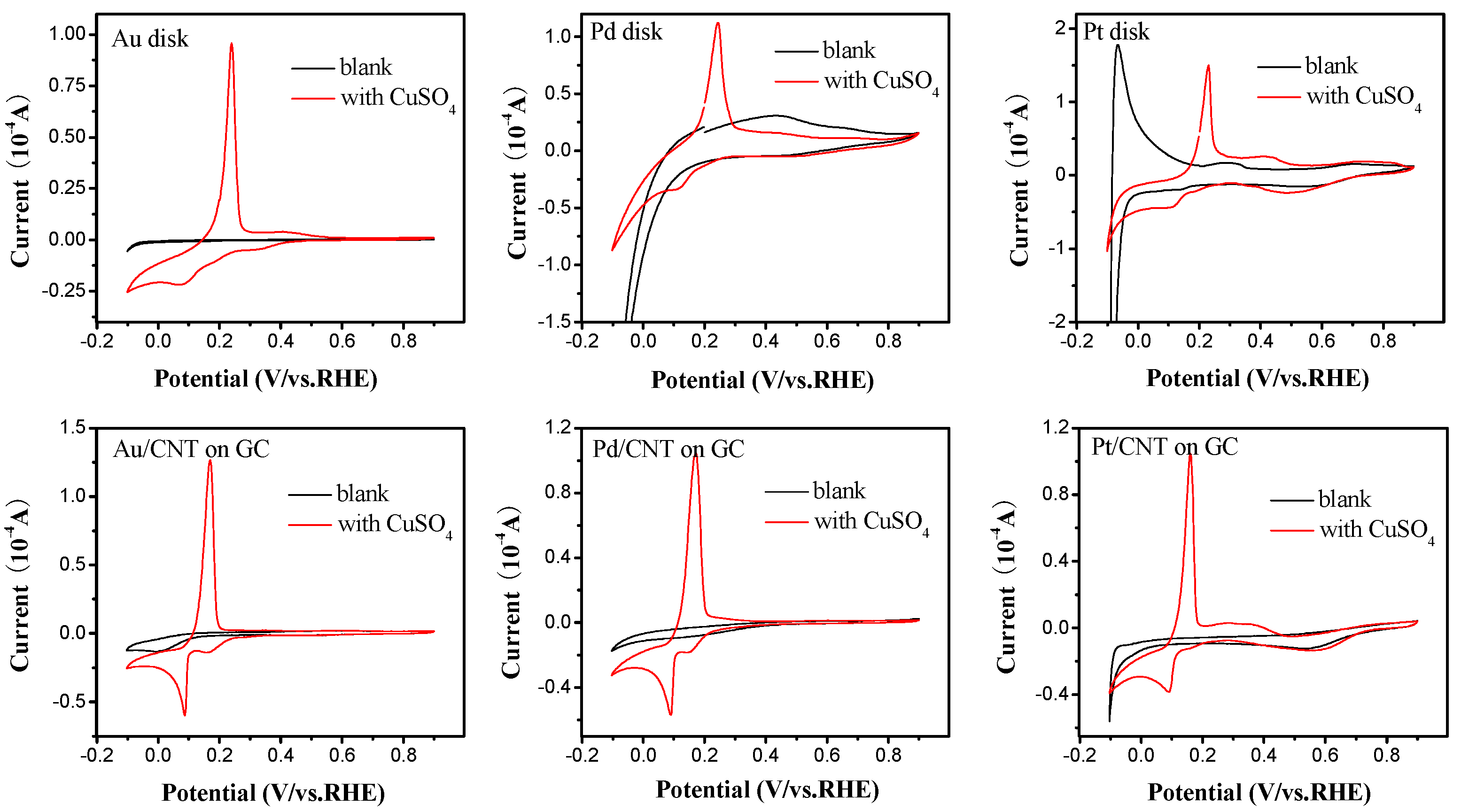
2.2. Electrochemical Characterization of Pt, Pd, and Au Disk Electrodes
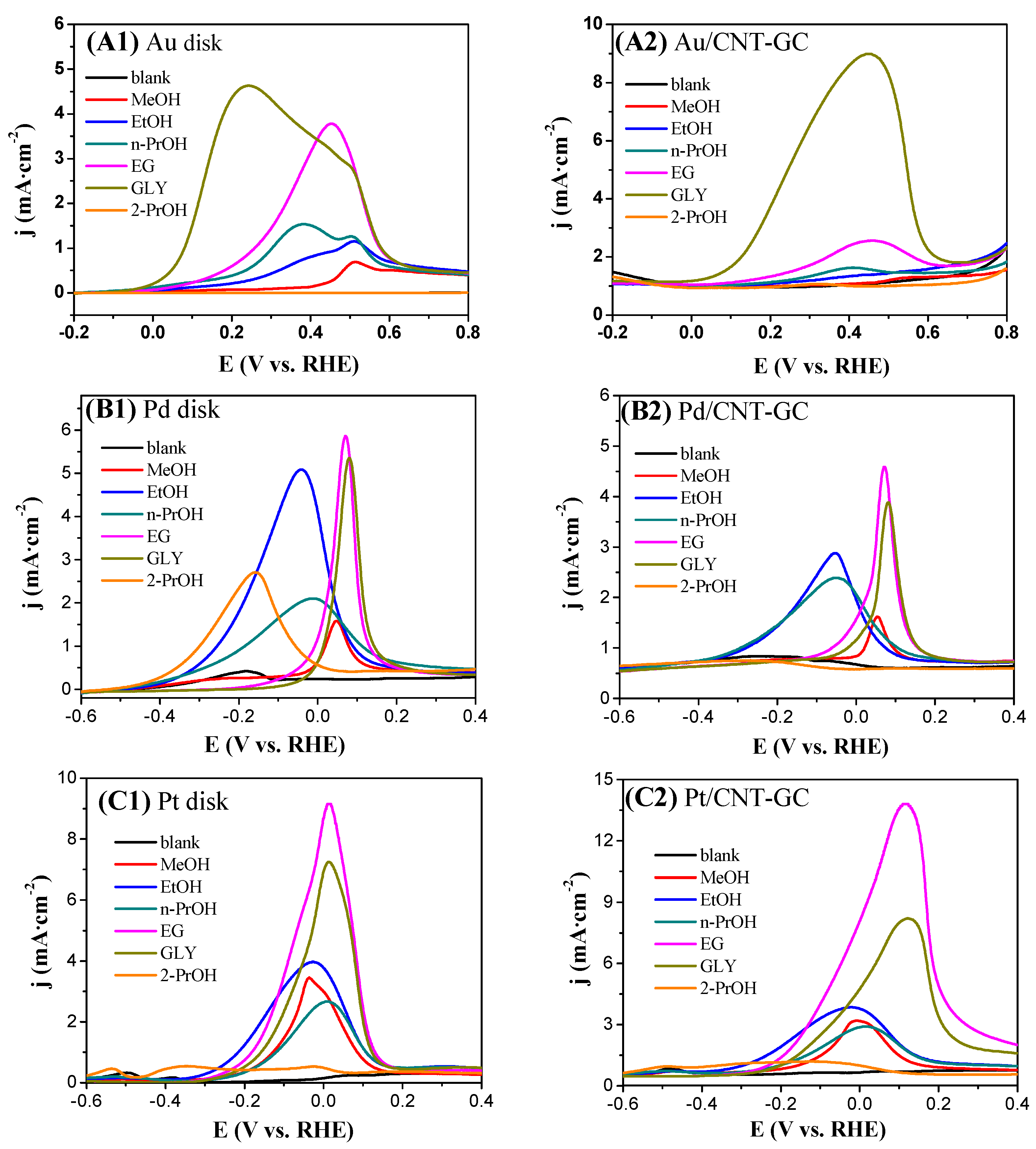
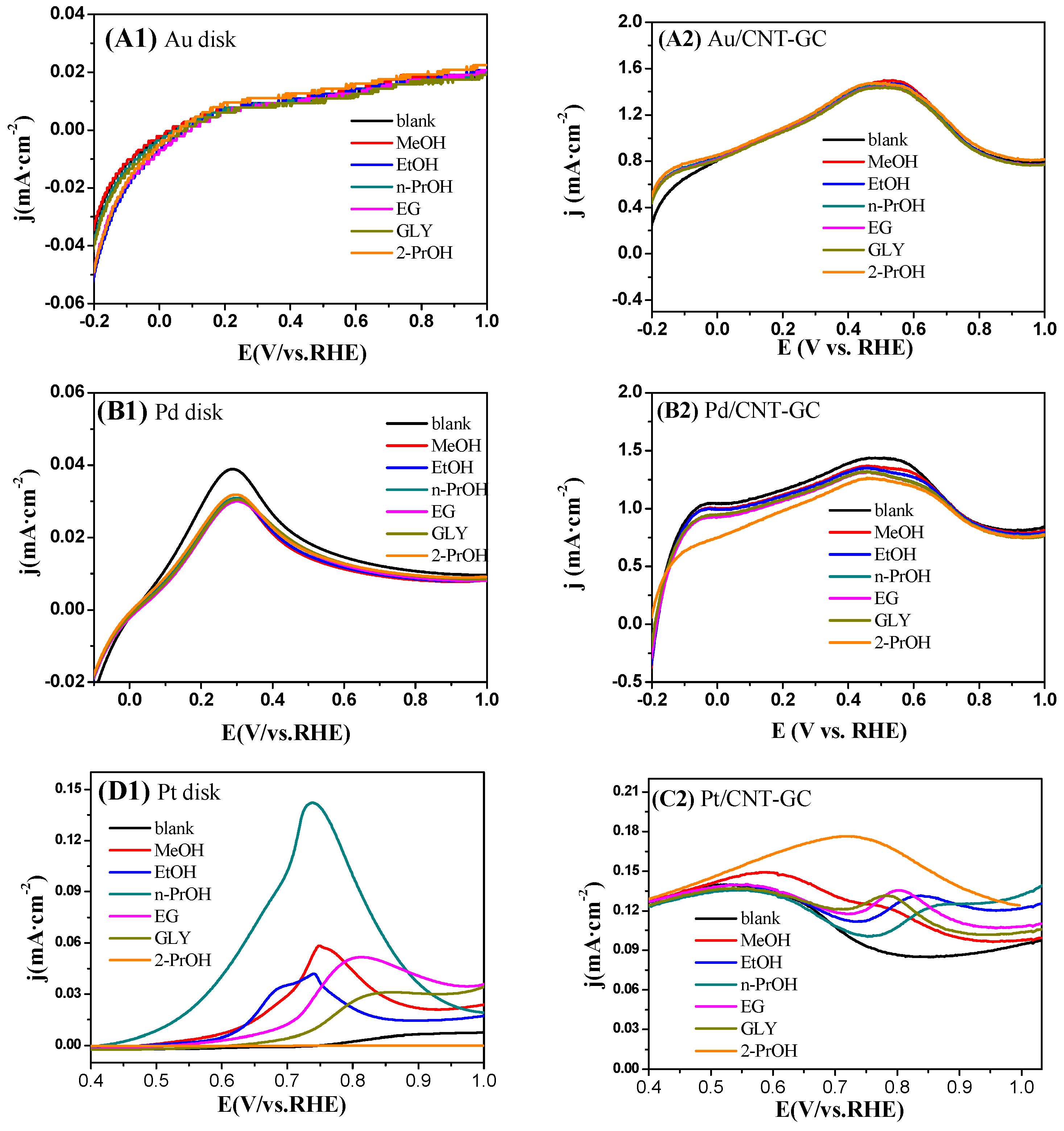
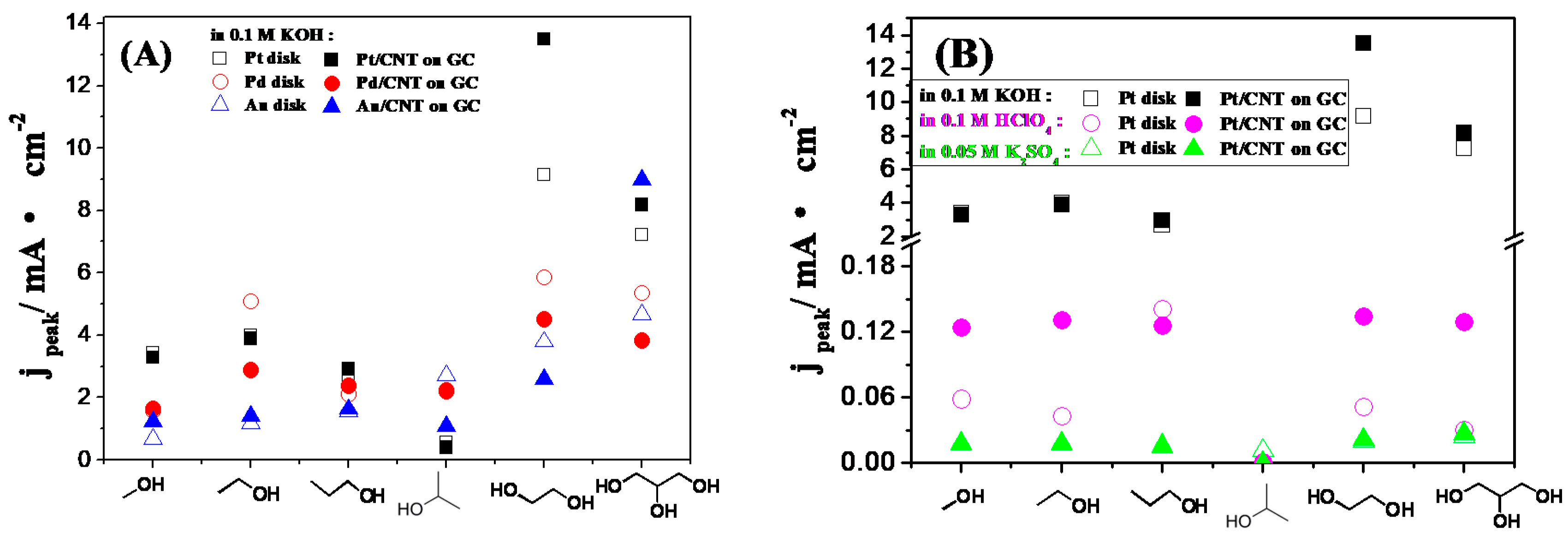
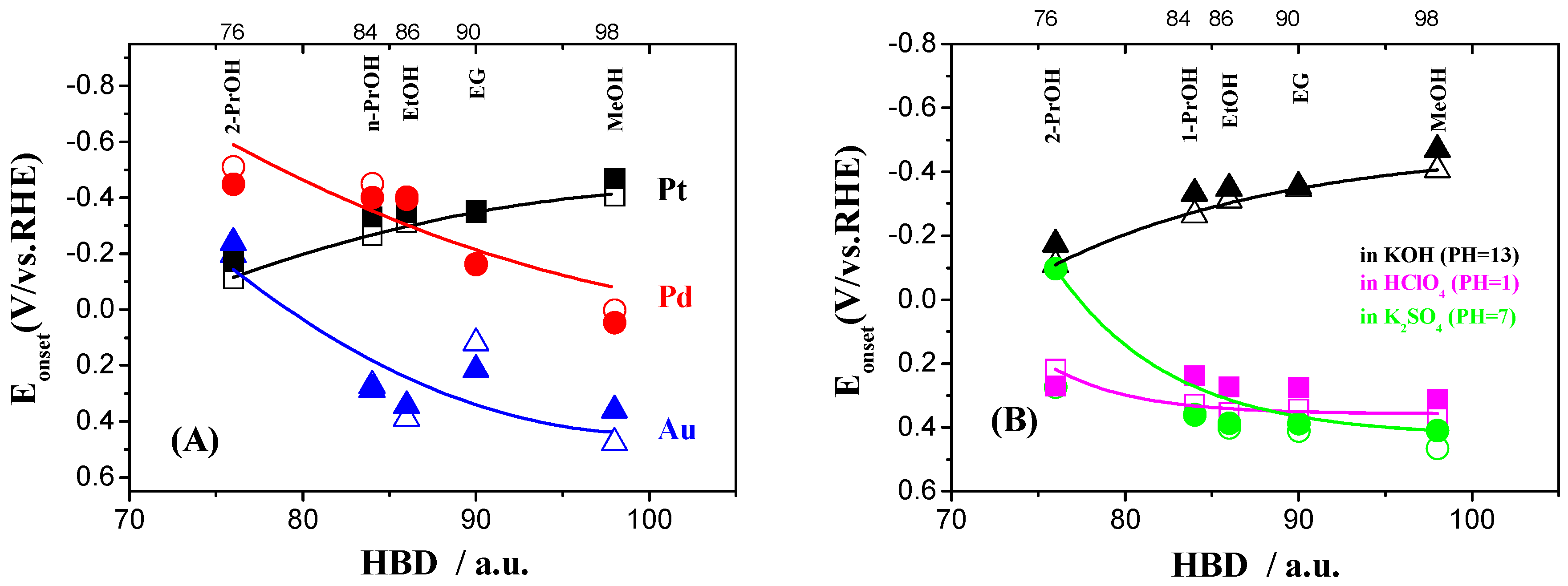
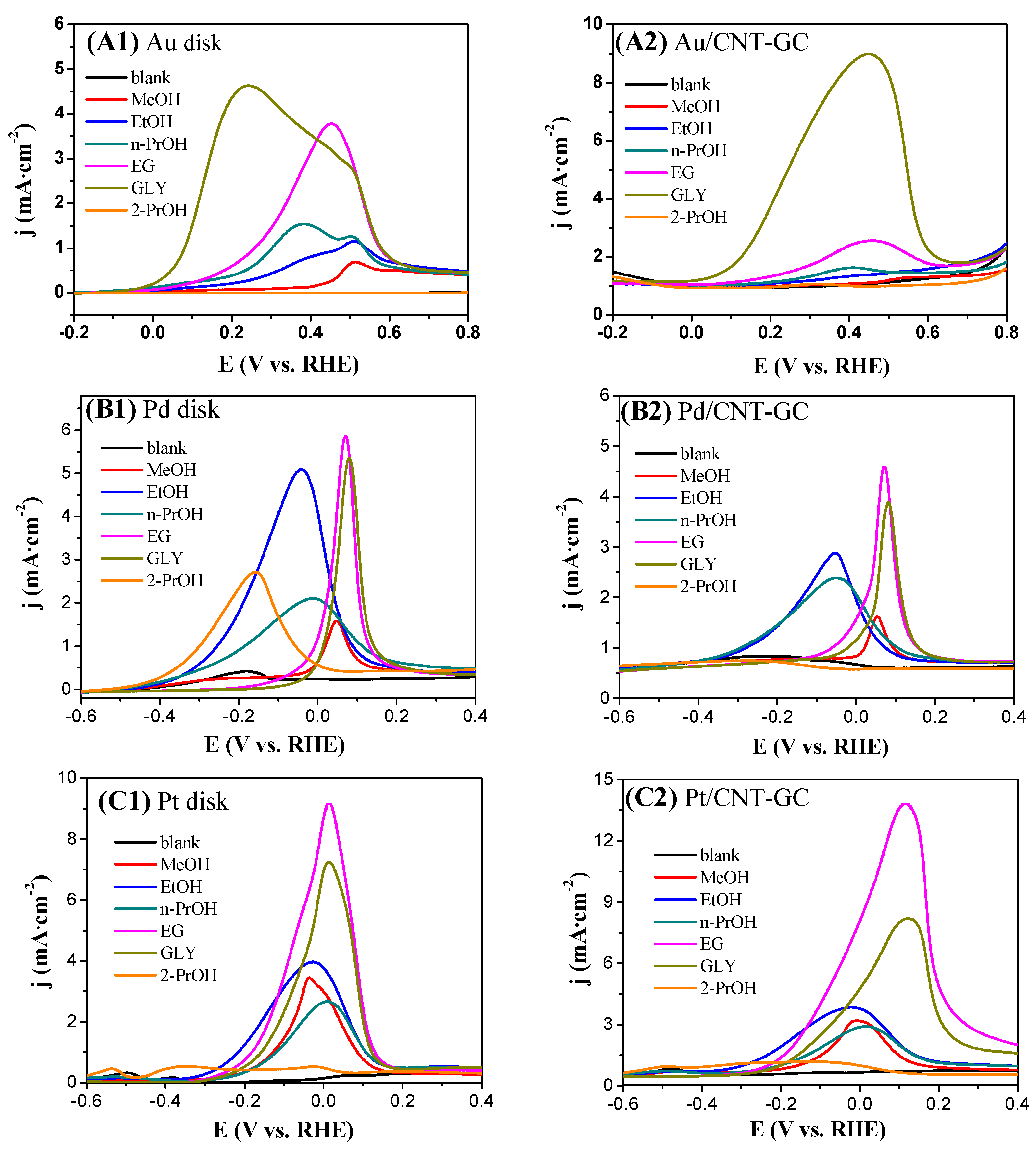
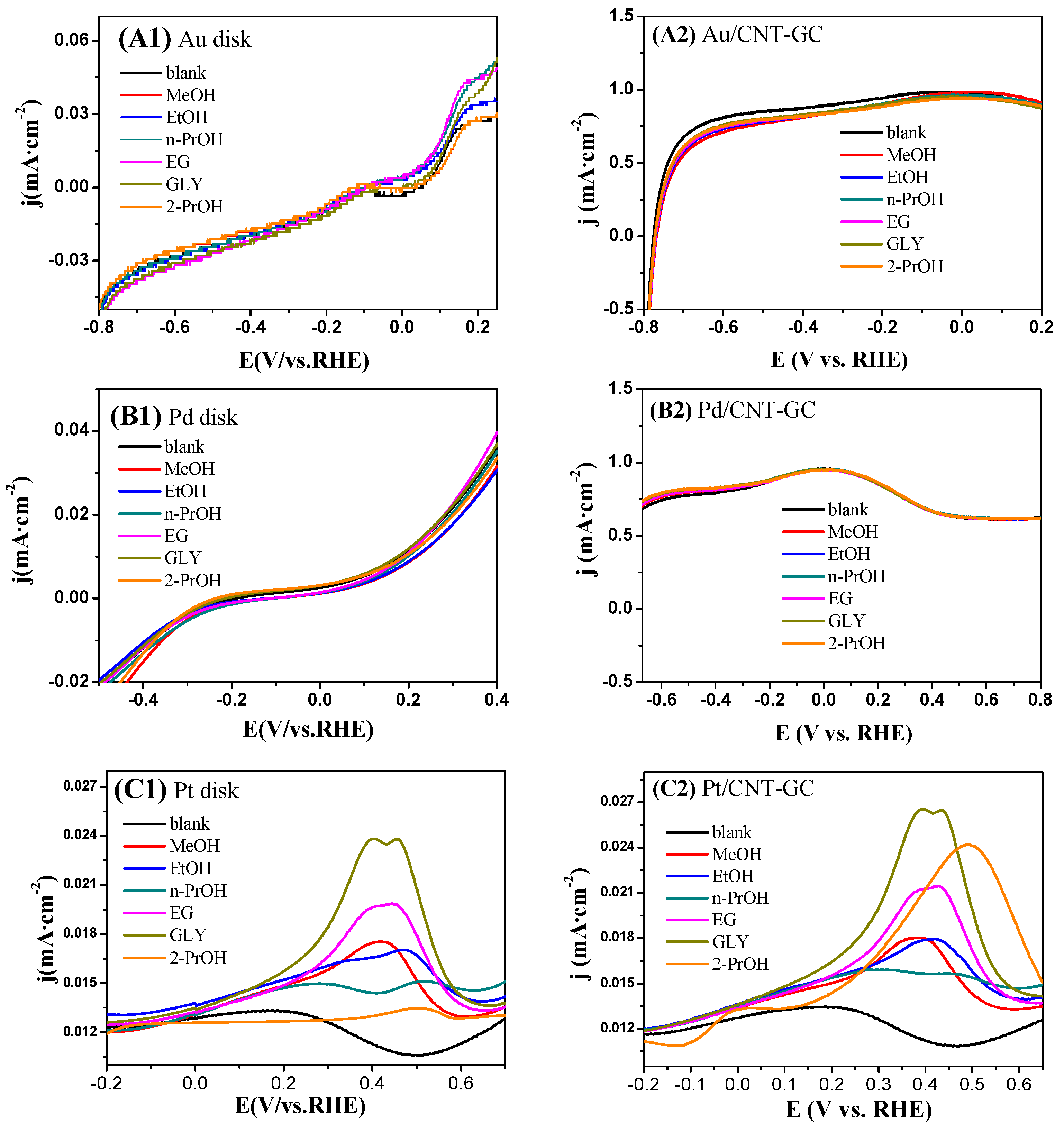
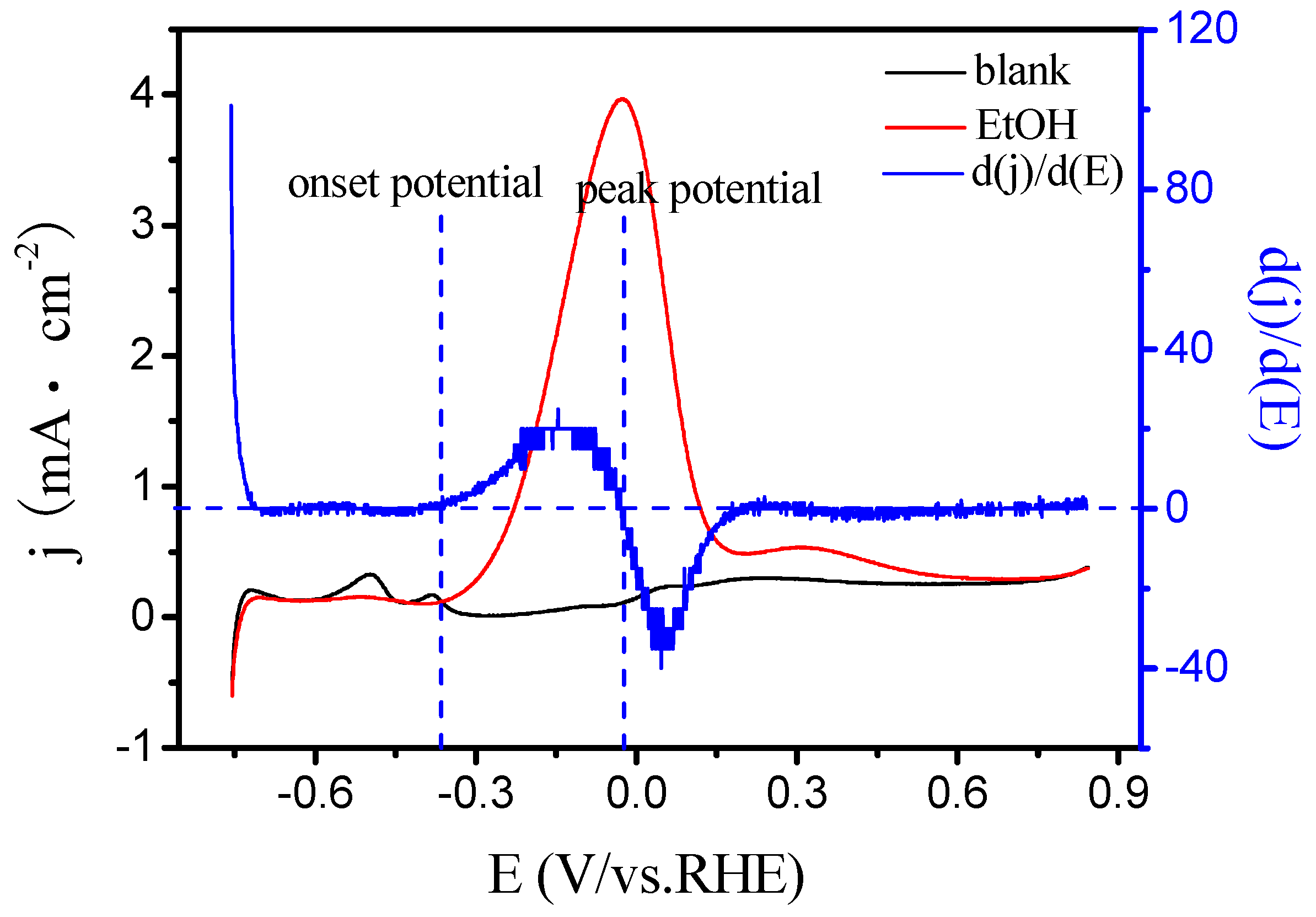
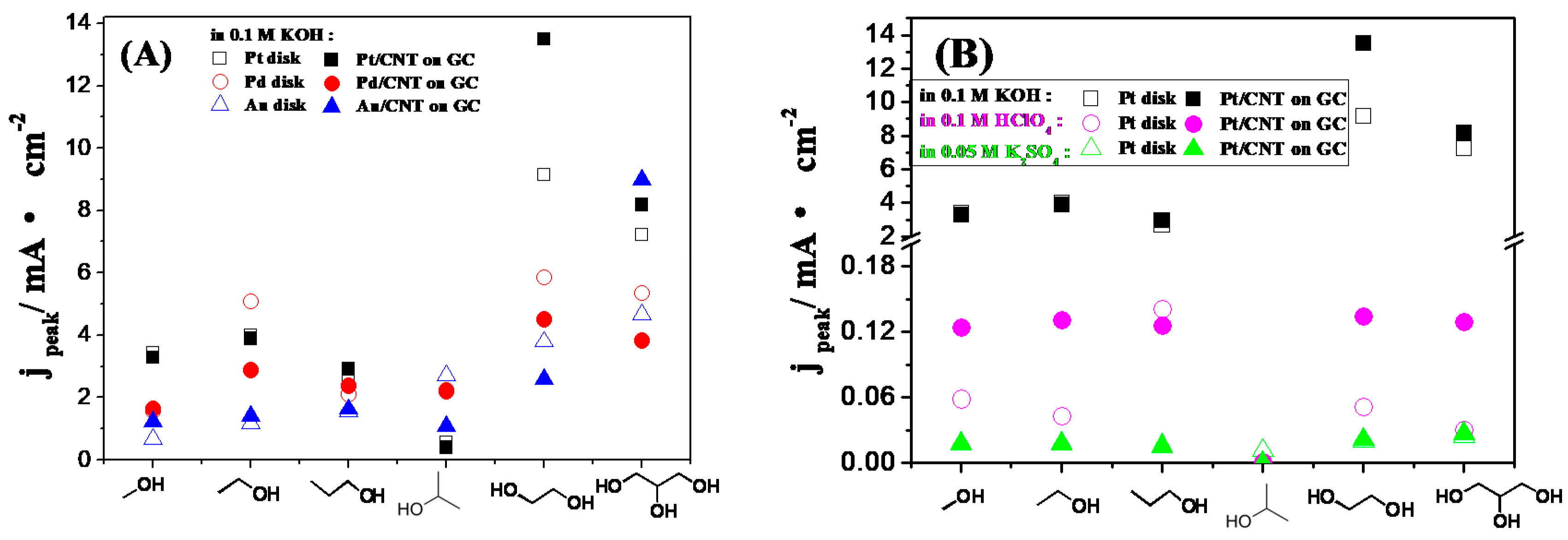
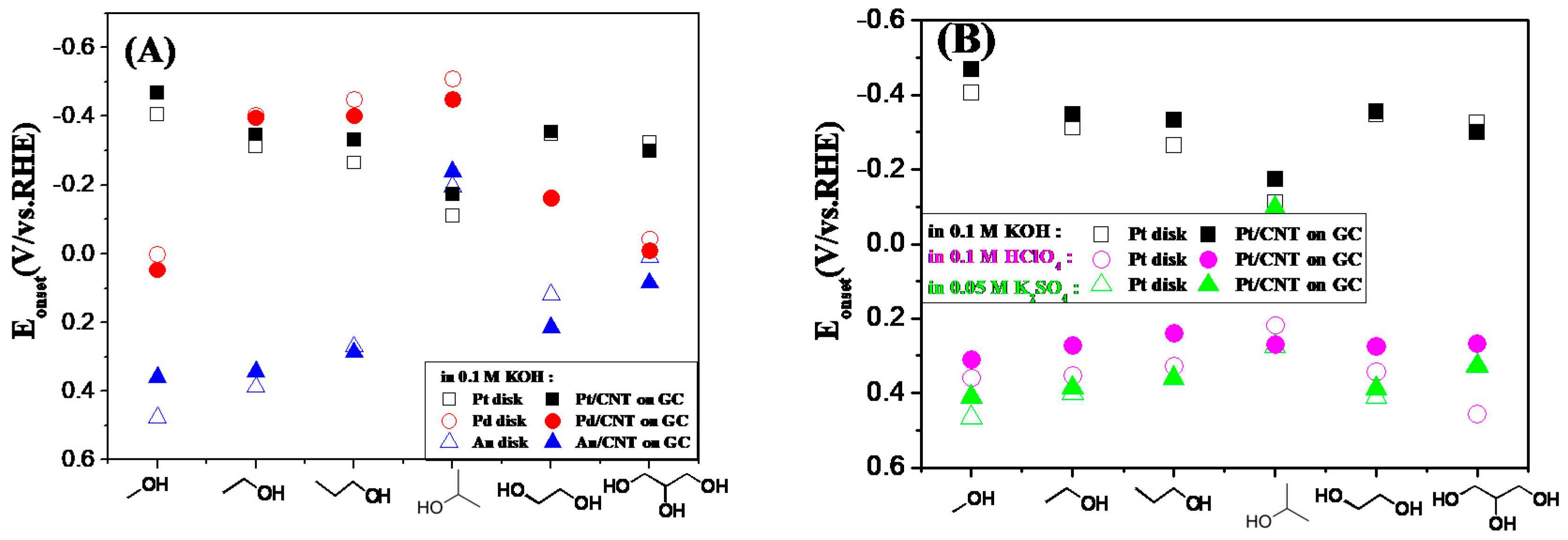
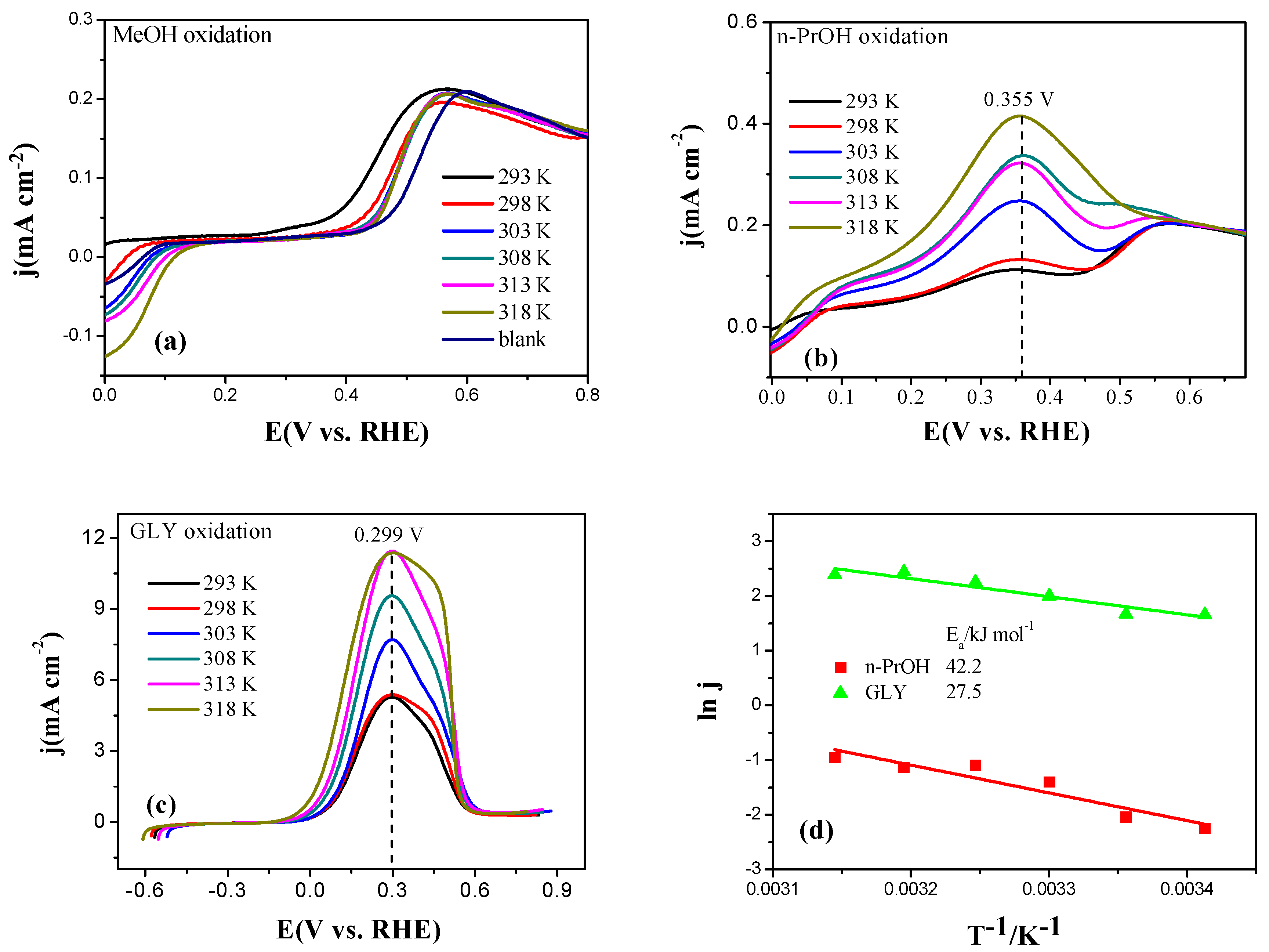
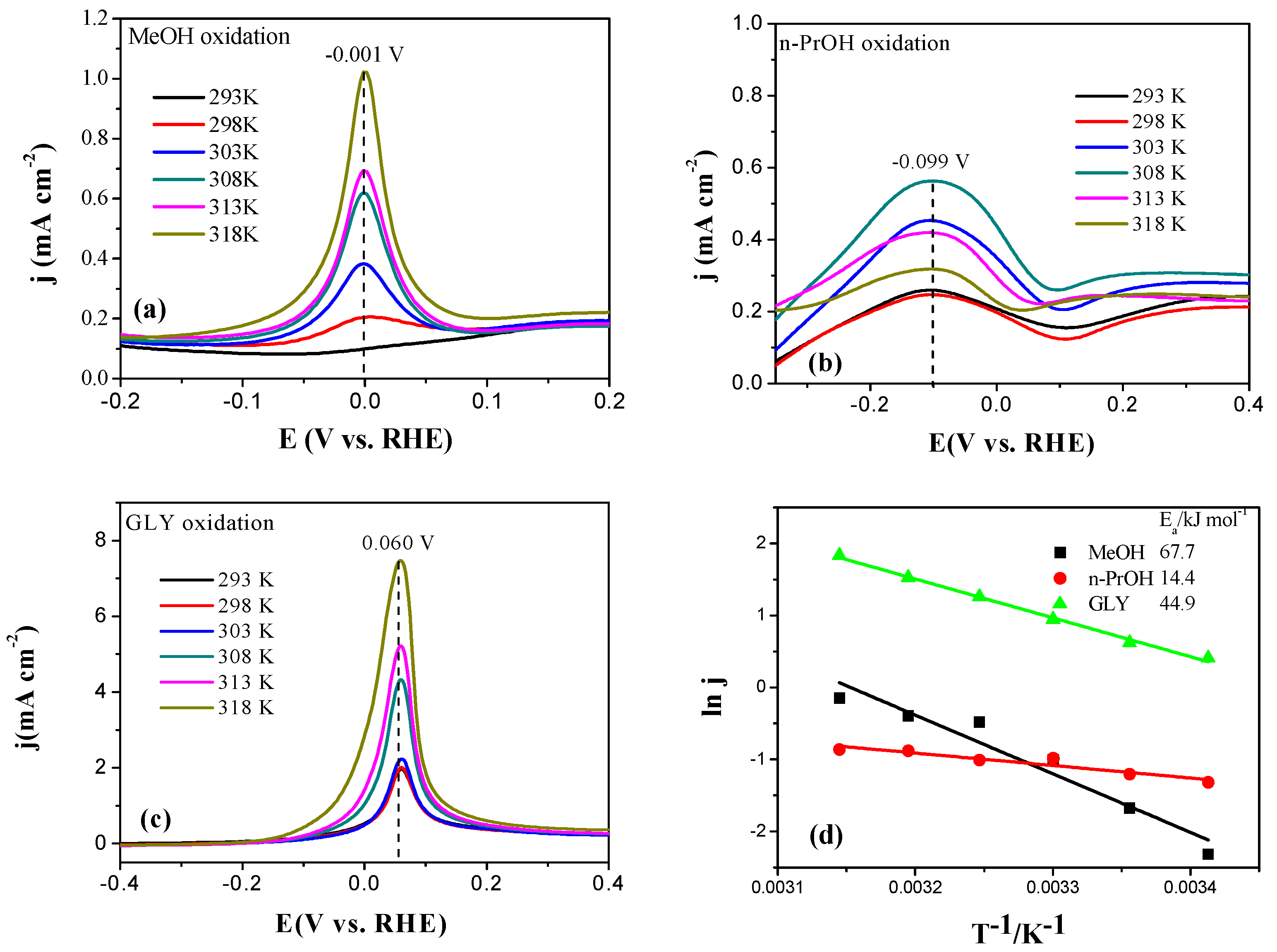
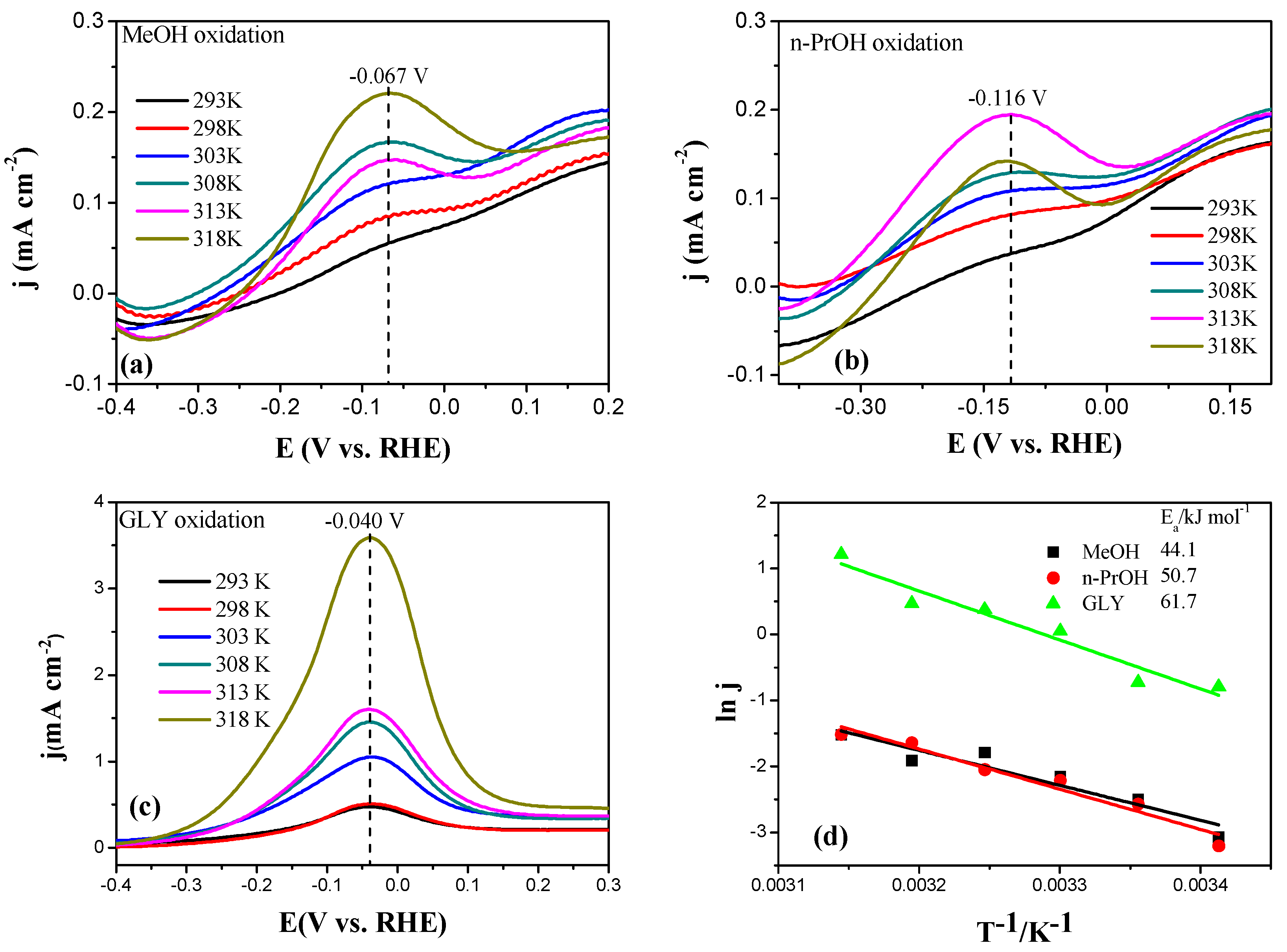
2.3. Electrocatalytic Oxidation Behaviors of Alcohols over Different Electrodes
3. Experimental
3.1. Materials and Reagents
3.2. Catalyst Synthesis and Physical Characterizations
3.3. Fabrication and Electrochemical Characterization of Electrodes
3.4. Apparent Activation Energy of Electrodes
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Ye, J.; Liu, J.; Xu, C.; Jiang, S.; Tong, Y. Electrooxidation of 2-propanol on Pt, Pd and Au in alkaline medium. Electrochem. Commun. 2007, 9, 2760–2763. [Google Scholar] [CrossRef]
- Winter, M.; Brodd, R.J. What Are Batteries, Fuel Cells, and Supercapacitors? Chem. Rev. 2004, 104, 4245–4269. [Google Scholar] [CrossRef] [PubMed]
- Xin, L.; Zhang, Z.; Wang, Z.; Li, W. Simultaneous Generation of Mesoxalic Acid and Electricity from Glycerol on a Gold Anode Catalyst in Anion-Exchange Membrane Fuel Cells. ChemCatChem 2012, 4, 1105–1114. [Google Scholar] [CrossRef]
- Zhang, Z.; Xin, L.; Sun, K.; Li, W. Pd–Ni electrocatalysts for efficient ethanol oxidation reaction in alkaline electrolyte. J. Hydrogen Energy 2011, 36, 12686–12697. [Google Scholar] [CrossRef]
- Zalineeva, A.; Serov, A.; Padilla, M.; Martinez, U.; Artyushkova, K.; Baranton, S.; Coutanceau, C.; Atanassov, P.B. Self-supported PdxBi catalysts for the electrooxidation of glycerol in alkaline media. J. Am. Chem. Soc. 2014, 136, 3937–3945. [Google Scholar] [CrossRef]
- Benipal, N.; Qi, J.; Liu, Q.; Li, W. Carbon nanotube supported PdAg nanoparticles for electrocatalytic oxidation of glycerol in anion exchange membrane fuel cells. Appl. Catal. B Environ. 2017, 210, 121–130. [Google Scholar] [CrossRef]
- Steele, B.C.H.; Heinzel, A. Materials for fuel-cell technologies. Nature 2001, 414, 345–352. [Google Scholar] [CrossRef]
- Carreue, L.; Friedrich, K.A.; Stimming, U. Fuel Cells—Fundamentals and Applications. Fuel Cells 2015, 1, 5–39. [Google Scholar] [CrossRef]
- Berger, D.J. Fuel Cells and Precious-Metal Catalysts. Science 1999, 286, 49. [Google Scholar] [CrossRef]
- Luo, J.; Maye, M.M.; Kariuki, N.N.; Wang, L.; Njoki, P.; Lin, Y.; Schadt, M.; Naslund, H.R.; Zhong, C.-J. Electrocatalytic oxidation of methanol: Carbon-supported gold–platinum nanoparticle catalysts prepared by two-phase protocol. Catal. Today 2005, 99, 291–297. [Google Scholar] [CrossRef]
- Samant, P.V.; Fernandes, J.B.; Rangel, C.M.; Figueiredo, J.L. Carbon xerogel supported Pt and Pt–Ni catalysts for electro-oxidation of methanol in basic medium. Catal. Today 2005, 102–103, 173–176. [Google Scholar] [CrossRef]
- Zhang, X.; Tsang, K.-Y.; Chan, K.-Y. Electrocatalytic properties of supported platinum–cobalt nanoparticles with uniform and controlled composition. J. Electroanal. Chem. 2004, 573, 1–9. [Google Scholar] [CrossRef]
- Kadirgan, F.; Beden, B.; Leger, J.M.; Lamy, C. Synergistic effect in the electrocatalytic oxidation of methanol on platinum+palladium alloy electrodes. J. Electroanal. Chem. 1981, 125, 89–103. [Google Scholar] [CrossRef]
- Horacio, R.C.; Ernesto, R.G. Direct Alcohol Fuel Cells; Springer: Dordrecht, The Netherlands, 2014; pp. 33–81. ISBN 978-94-007-7707-1. [Google Scholar]
- Camara, G.A.; de Lima, R.B.; Iwasita, T. Catalysis of ethanol electrooxidation by PtRu: The influence of catalyst composition. Electrochem. Commun. 2004, 6, 812–815. [Google Scholar] [CrossRef]
- Shen, P.K.; Xu, C. Alcohol oxidation on nanocrystalline oxide Pd/C promoted electrocatalysts. Electrochem. Commun. 2006, 8, 184–188. [Google Scholar] [CrossRef]
- Xu, C.; Cheng, L.; Shen, P.; Liu, Y. Methanol and ethanol electrooxidation on Pt and Pd supported on carbon microspheres in alkaline media. Electrochem. Commun. 2007, 9, 997–1001. [Google Scholar] [CrossRef]
- Bianchini, C.; Shen, P.K. Palladium-Based Electrocatalysts for Alcohol Oxidation in Half Cells and in Direct Alcohol Fuel Cells. Chem. Rev. 2009, 109, 4183–4206. [Google Scholar] [CrossRef] [PubMed]
- Kwon, Y.; Schouten, K.J.P.; Koper, M.T.M. Mechanism of the Catalytic Oxidation of Glycerol on Polycrystalline Gold and Platinum Electrodes. ChemCatChem 2011, 3, 1176–1185. [Google Scholar] [CrossRef]
- Simões, M.; Baranton, S.; Coutanceau, C. Electrochemical Valorisation of Glycerol. ChemSusChem 2012, 55, 2106–2124. [Google Scholar] [CrossRef] [PubMed]
- Nishimura, K.; Kunimatsu, K.; Enyo, M. Electrocatalysis on Pd + Au alloy electrodes: Part III. IR spectroscopic studies on the surface species derived from CO and CH3OH in NaOH solution. J. Electroanal. Chem. 1989, 260, 167–179. [Google Scholar] [CrossRef]
- Xu, C.; Tian, Z.; Chen, Z.; Jiang, S.P. Pd/C promoted by Au for 2-propanol electrooxidation in alkaline media. Electrochem. Commun. 2008, 10, 246–249. [Google Scholar] [CrossRef]
- Antolini, E.; Gonzalez, E.R. Alkaline direct alcohol fuel cells. J. Power Sources 2010, 195, 3431–3450. [Google Scholar] [CrossRef]
- Ago, H.; Kugler, T.; Cacialli, F.; Salaneck, W.R.; Shaffer, M.S.P.; Windle, A.H.; Friend, R.H. Work Functions and Surface Functional Groups of Multiwall Carbon Nanotubes. J. Phys. Chem. B 1999, 103, 8116–8121. [Google Scholar] [CrossRef]
- Yu, H.; Jin, Y.; Peng, F.; Wang, H.; Yang, J. Kinetically controlled side-wall functionalization of carbon nanotubes by nitric acid oxidation. J. Phys. Chem. C 2008, 112, 6758–6763. [Google Scholar] [CrossRef]
- Beden, B.; Çetin, I.; Kahyaoglu, A.; Takky, D.; Lamy, C. Electrocatalytic oxidation of saturated oxygenated compounds on gold electrodes. J. Catal. 1987, 104, 37–46. [Google Scholar] [CrossRef]
- Rieger, P.H. Electrochemistry; Springer: Dordrecht, The Netherlands, 1994; pp. 187–198. [Google Scholar] [CrossRef]
- Parker, V.D.; Tilset, M. Rapid, low activation energy, selective, direct reactions of cation radicals with nucleophiles. An experimental test of theory. J. Am. Chem. Soc. 1987, 109, 2521–2523. [Google Scholar] [CrossRef]
- Marcus, Y. The Properties of Organic Liquids that are Relevant to their Use as Solvating Solvents. Chem. Soc. Rev. 1993, 22, 409–416. [Google Scholar] [CrossRef]
- Pan, Z.F.; Chen, R.; An, L.; Li, Y.S. Alkaline anion exchange membrane fuel cells for cogeneration of electricity and valuable chemicals. J. Power Sources 2017, 365, 430–445. [Google Scholar] [CrossRef]
- Li, M.; Liu, P.; Adzic, R.R. Platinum monolayer electrocatalysts for anodic oxidation of alcohols. J. Phys. Chem. Lett. 2012, 3, 3480–3485. [Google Scholar] [CrossRef]
- Ozoemena, K.I.; Chen, S. Nanomaterials for Fuel Cell Catalysis; Springer: Dordrecht, The Netherlands, 2016; pp. 479–486. [Google Scholar] [CrossRef]
- Li, N.; Zhou, Q.; Tian, S.; Zhao, H.; Li, X.; Adkins, J.; Gu, Z.; Zhao, L.; Zheng, J. Electrocatalytic oxidation of alcohols on single gold particles in highly ordered SiO2 cavities. Electrochim. Acta 2013, 109, 546–553. [Google Scholar] [CrossRef]
- Michaelides, A.; Ranea, V.A.; de Andres, P.L.; King, D.A. General model for water monomer adsorption on close-packed transition and noble metal surfaces. Phys. Rev. Lett. 2003, 90, 216102-1–216102-4. [Google Scholar] [CrossRef]
- Costa-Amaral, R.; Da Silva, J.L. The adsorption of alcohols on strained Pt3Ni(111) substrates: A density functional investigation within the D3 van der Waals correction†. Phys. Chem. Chem. Phys. 2018, 20, 24210–24221. [Google Scholar] [CrossRef]
- Schennach, R.; Eichler, A.; Rendulic, K.D. Adsorption and Desorption of Methanol on Pd (111) and on a Pd/V Surface Alloy. J. Phys. Chem. B 2003, 107, 2552–2558. [Google Scholar] [CrossRef]
- Wang, L.; He, C.Z.; Zhang, W.H.; Li, Z.Y.; Yang, J.L. Methanol-selective oxidation pathways on Au surfaces: A first-principles study. J. Phys. Chem. C 2014, 118, 17511–17520. [Google Scholar] [CrossRef]
- Xu, Z.F.; Wang, Y.X. Effects of alloyed metal on the catalysis activity of Pt for ethanol partial oxidation: Adsorption and dehydrogenation on Pt3M (M = Pt, Ru, Sn, Re, Rh, And Pd). J. Phys. Chem. C 2011, 115, 20565–20571. [Google Scholar] [CrossRef]
- Sheng, T.; Lin, W.F.; Hardacre, C.; Hu, P. Role of water and adsorbed hydroxyls on ethanol electrochemistry on Pd: New mechanism, active centers, and energetics for direct ethanol fuel cell running in alkaline medium. J. Phys. Chem. C 2014, 118, 5762–5772. [Google Scholar] [CrossRef]
- Freire, R.L.; Kiejna, A.; Da Silva, J.L. Adsorption of water and ethanol on noble and transition-metal substrates: A density functional investigation within van der Waals corrections. Phys. Chem. Chem. Phys. 2016, 18, 29526–29536. [Google Scholar] [CrossRef]
- Iwasita, T.; Nart, F.C.; Vielstich, W. An FTIR Study of the Catalytic Activity of a 85: 15 Pt: Ru Alloy for Methanol Oxidation. Phys. Chem. 1990, 94, 1030–1034. [Google Scholar] [CrossRef]
- Pandey, R.K.; Lakshminarayanan, V. Ethanol electrocatalysis on gold and conducting polymer nanocomposites: A study of the kinetic parameters. Appl. Catal. B Environ. 2012, 125, 271–281. [Google Scholar] [CrossRef]
- Jia, C.; Yin, H.M.; Ma, R.Y.; Ge, X.B.; Zhou, A.Q.; Xu, X.H.; Ding, Y. Enhanced photoelectrocatalytic activity of methanol oxidation on TiO2-decorated nanoporous gold. J. Phys. Chem. C 2009, 113, 16138–16143. [Google Scholar] [CrossRef]
- Gupta, S.S.; Datta, J. A comparative study on ethanol oxidation behavior at Pt and PtRh electrodeposits. J. Electroanal. Chem. 2006, 594, 65–72. [Google Scholar] [CrossRef]
- Su, Y.Z.; Xu, C.W.; Liu, J.P.; Liu, Z.Q. Electrooxidation of 2-propanol compared ethanol on Pd electrode in alkaline medium. J. Power Sources 2009, 194, 295–297. [Google Scholar] [CrossRef]
- Liang, Z.X.; Zhao, T.S.; Xu, J.B.; Zhu, L.D. Mechanism study of the ethanol oxidation reaction on palladium in alkaline media. Electrochim. Acta 2009, 54, 2203–2208. [Google Scholar] [CrossRef]
- Roychowdhury, C.; Matsumoto, F.; Mutolo, P.F.; Abruna, H.D.; DiSalvo, F.J. Synthesis, Characterization, and Electrocatalytic Activity of PtBi Nanoparticles Prepared by the Polyol Process. Chem. Mater. 2005, 17, 5871–5876. [Google Scholar] [CrossRef]
- Lopez-Sanchez, J.A.; Dimitratos, N.; Hammond, C.; Brett, G.L.; Kesavan, L.; White, S.; Miedziak, P.; Tiruvalam, R.; Jenkins, R.L.; Carley, A.F.; et al. Facile removal of stabilizer-ligands from supported gold nanoparticles. Nat. Chem. 2011, 3, 551–556. [Google Scholar] [CrossRef] [PubMed]
- Green, C.L.; Kucernak, A. Determination of the Platinum and Ruthenium Surface Areas in Platinum-Ruthenium Alloy Electrocatalysts by Underpotential Deposition of Copper. I. Unsupported Catalysts. J. Phys. Chem. B 2002, 106, 1036–1047. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Catalysts | Metal Loading a (wt%) | Particle Size b (nm) | Grain Size c (nm) | 2θ d (°) | EAS e (m2/g) |
|---|---|---|---|---|---|
| Pt/CNTs | 0.85 | 3.2 | 2.2 | 38.2 | 110.0 |
| Pd/CNTs | 0.75 | 3.6 | 2.3 | 39.1 | 123.0 |
| Au/CNTs | 0.80 | 3.9 | 2.4 | 40 | 100.0 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, B.; Tao, L.; Cheng, Y.; Yang, F.; Jin, Y.; Zhou, C.; Yu, H.; Yang, Y. Electrocatalytic Oxidation of Small Molecule Alcohols over Pt, Pd, and Au Catalysts: The Effect of Alcohol’s Hydrogen Bond Donation Ability and Molecular Structure Properties. Catalysts 2019, 9, 387. https://doi.org/10.3390/catal9040387
Wang B, Tao L, Cheng Y, Yang F, Jin Y, Zhou C, Yu H, Yang Y. Electrocatalytic Oxidation of Small Molecule Alcohols over Pt, Pd, and Au Catalysts: The Effect of Alcohol’s Hydrogen Bond Donation Ability and Molecular Structure Properties. Catalysts. 2019; 9(4):387. https://doi.org/10.3390/catal9040387
Chicago/Turabian StyleWang, Bei, Liu Tao, Yu Cheng, Fang Yang, Yuguang Jin, Chunmei Zhou, Hao Yu, and Yanhui Yang. 2019. "Electrocatalytic Oxidation of Small Molecule Alcohols over Pt, Pd, and Au Catalysts: The Effect of Alcohol’s Hydrogen Bond Donation Ability and Molecular Structure Properties" Catalysts 9, no. 4: 387. https://doi.org/10.3390/catal9040387
APA StyleWang, B., Tao, L., Cheng, Y., Yang, F., Jin, Y., Zhou, C., Yu, H., & Yang, Y. (2019). Electrocatalytic Oxidation of Small Molecule Alcohols over Pt, Pd, and Au Catalysts: The Effect of Alcohol’s Hydrogen Bond Donation Ability and Molecular Structure Properties. Catalysts, 9(4), 387. https://doi.org/10.3390/catal9040387