On the High Structural Heterogeneity of Fe-Impregnated Graphitic-Carbon Catalysts from Fe Nitrate Precursor



Abstract
1. Introduction
2. Results
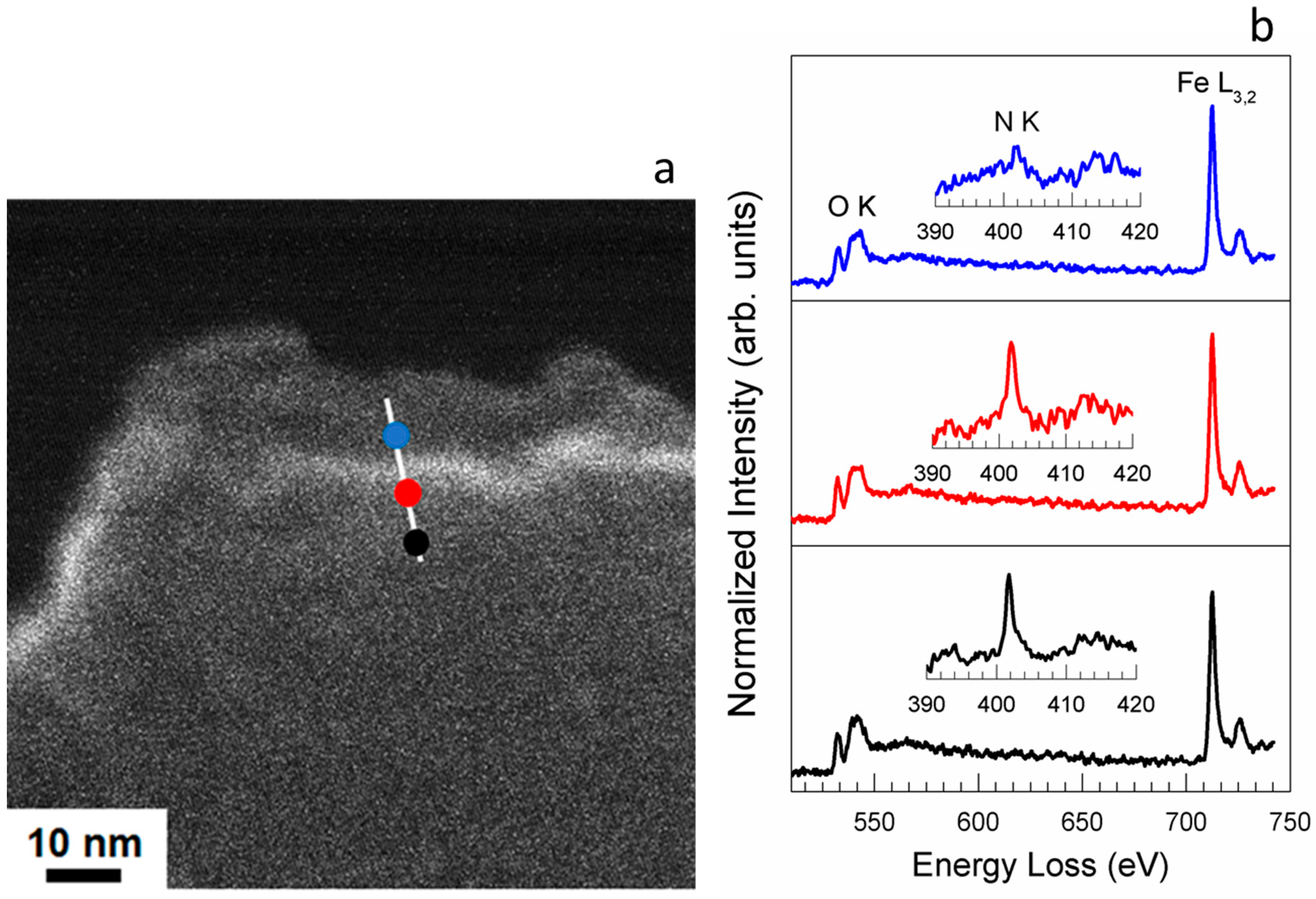
2.1. Electron Microscopy Structural Analysis
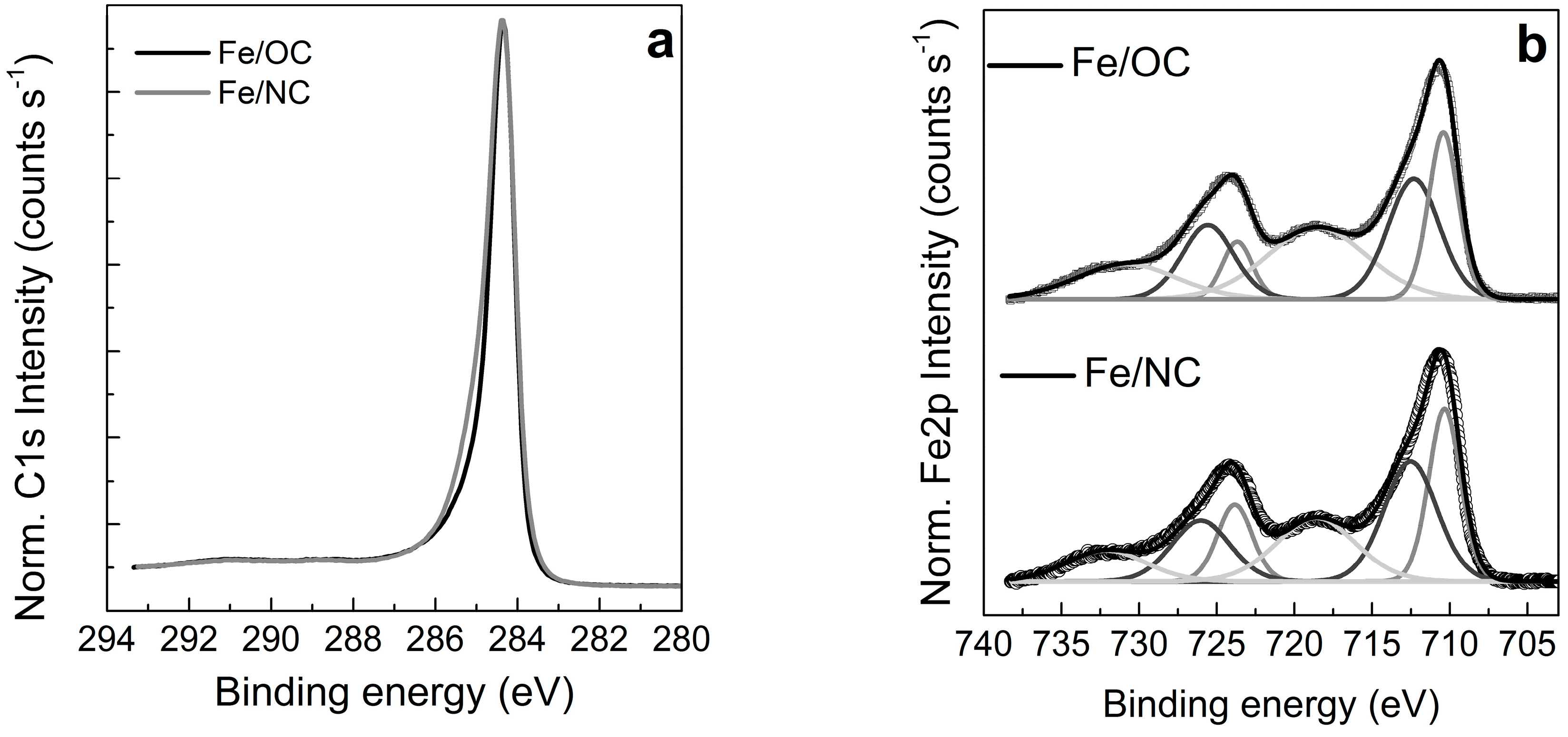
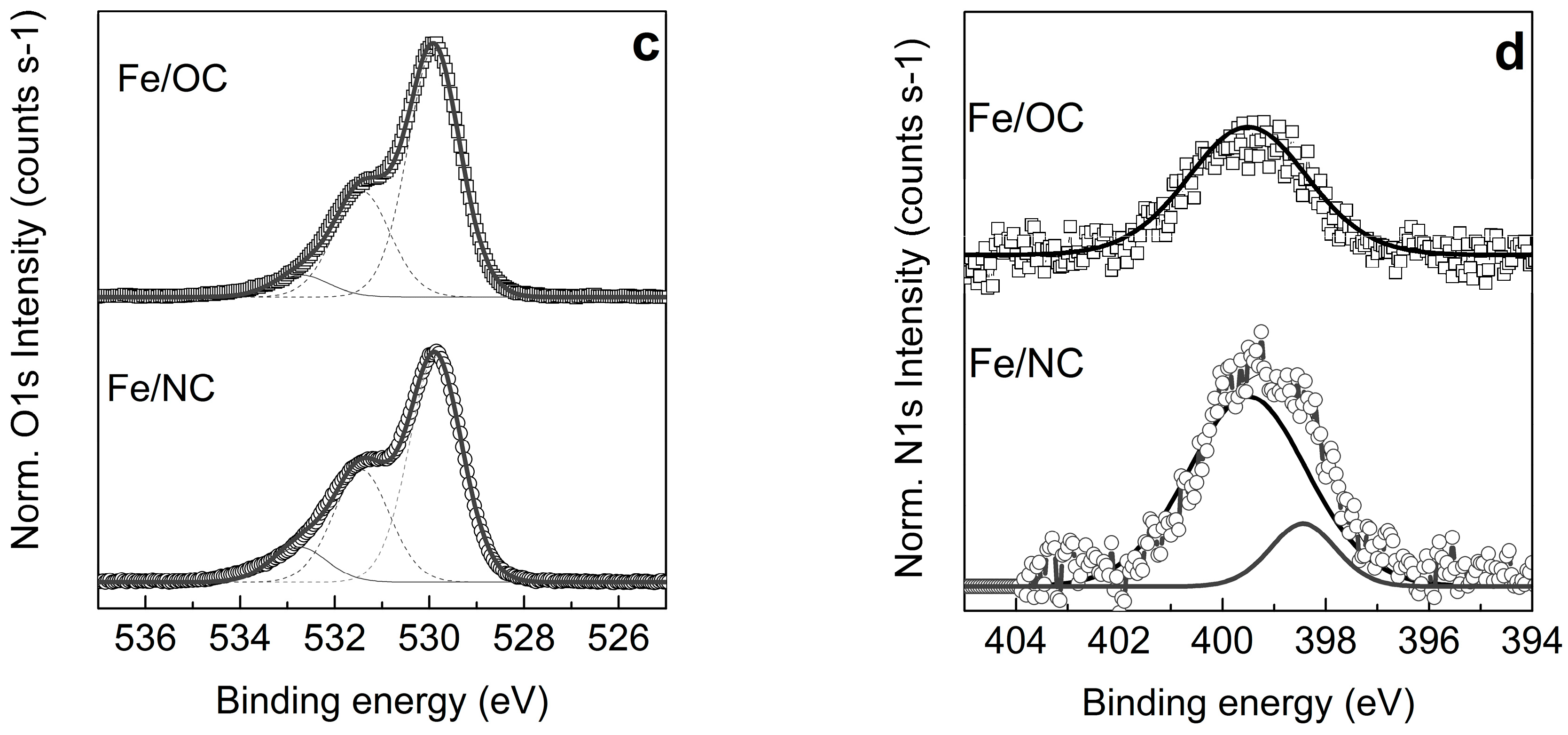
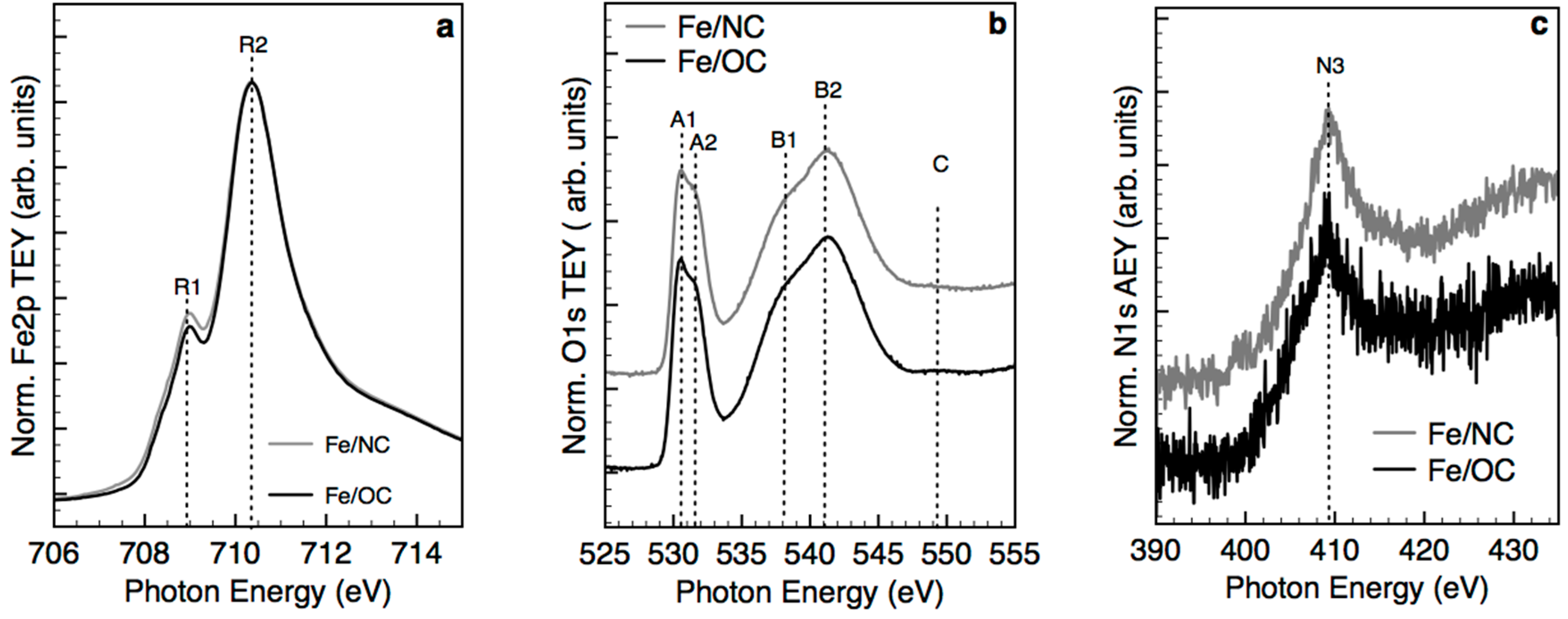
2.2. X-ray Photoemission Spectroscopy and Absorption Spectroscopy in Ultra High Vacuum (UHV)
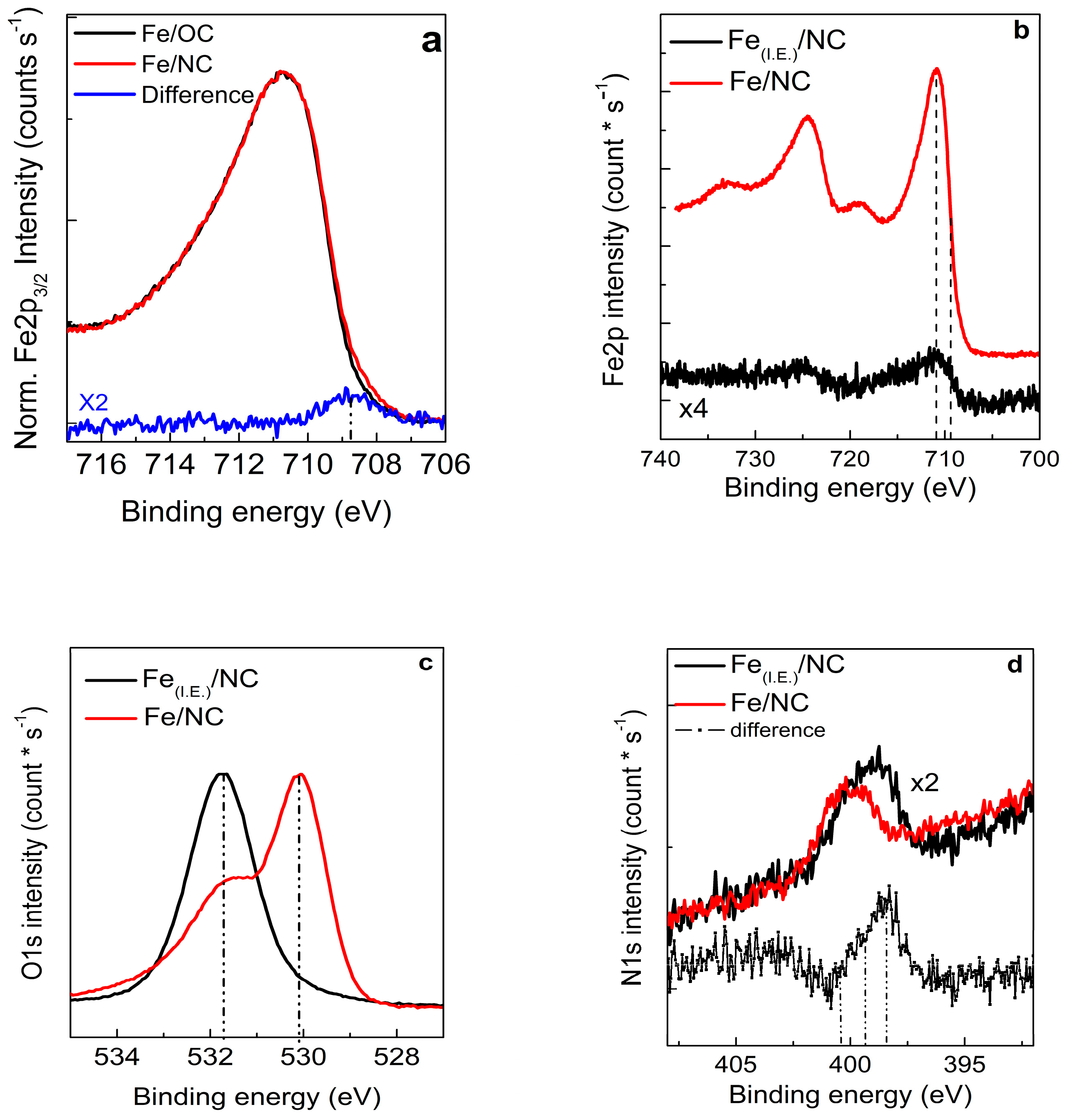
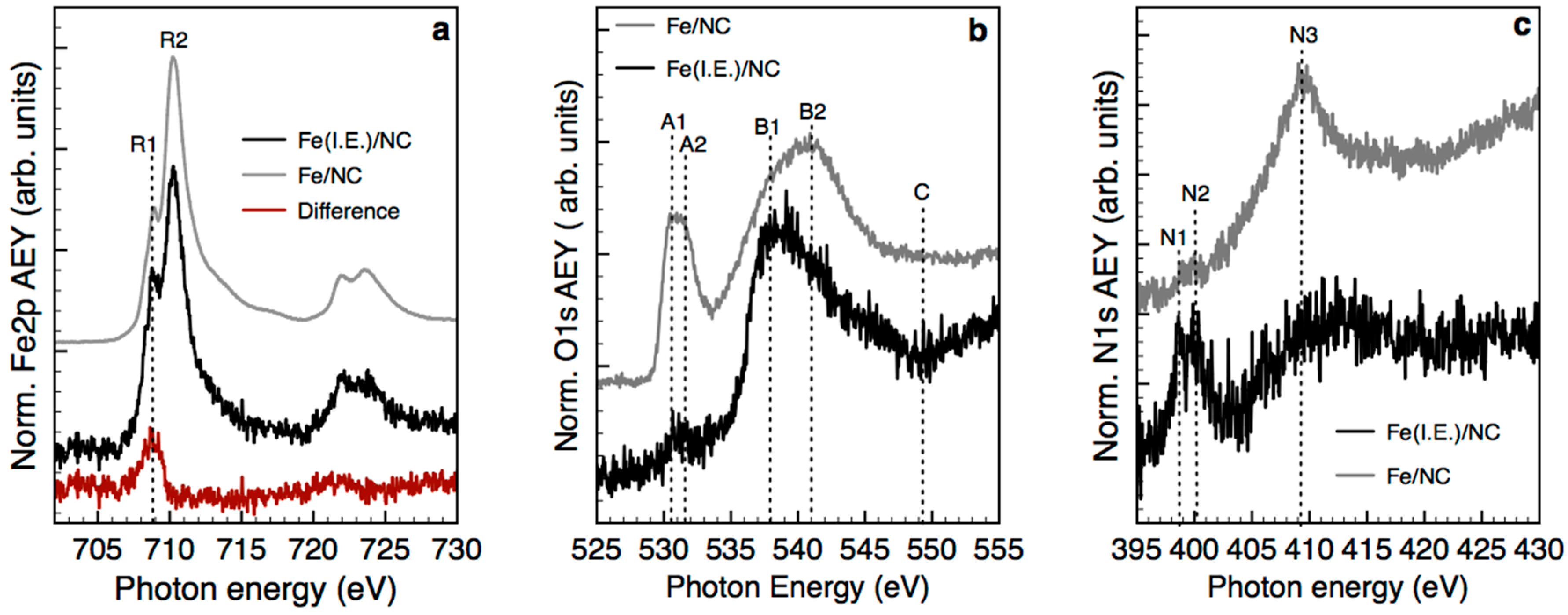
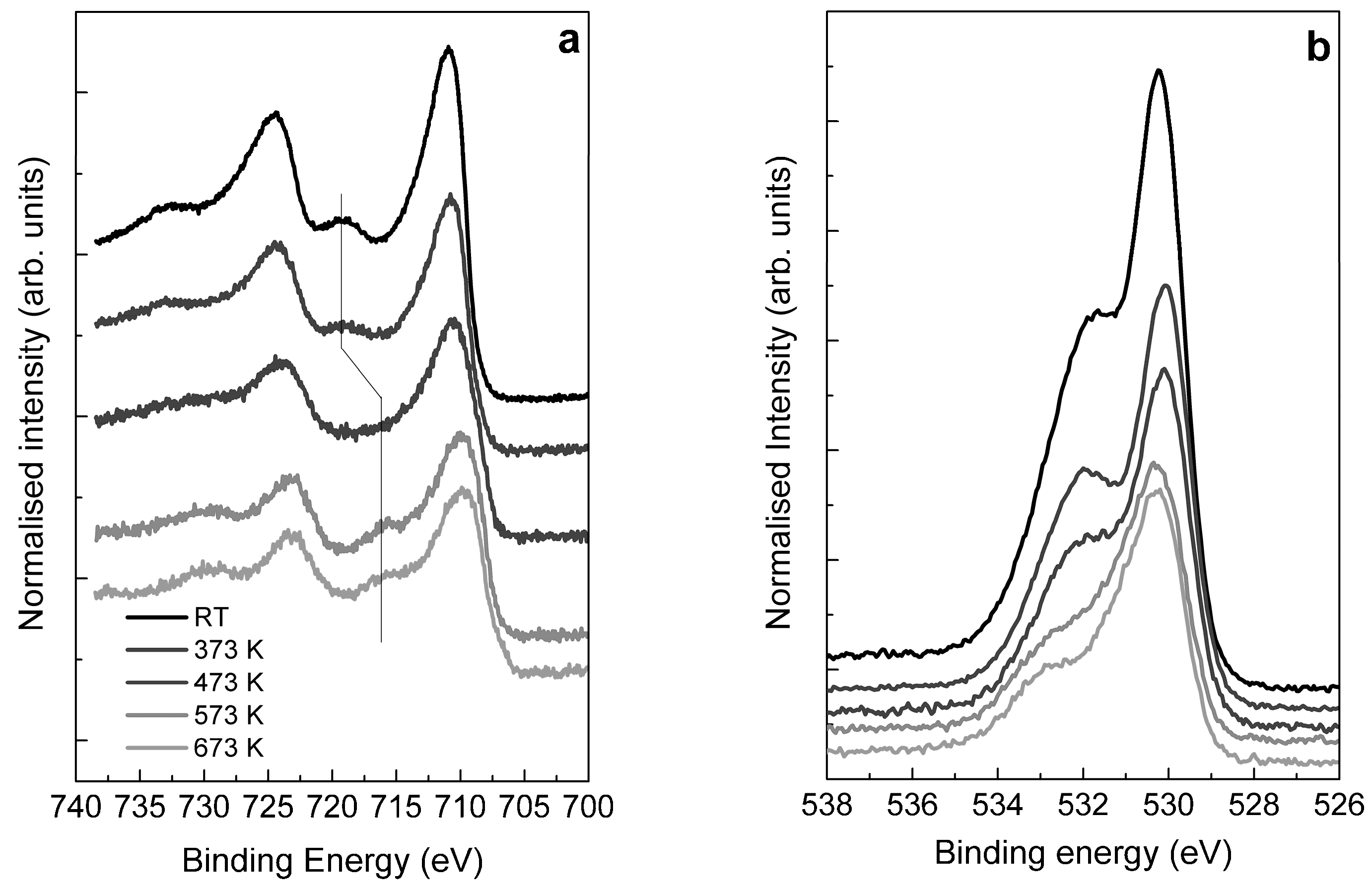
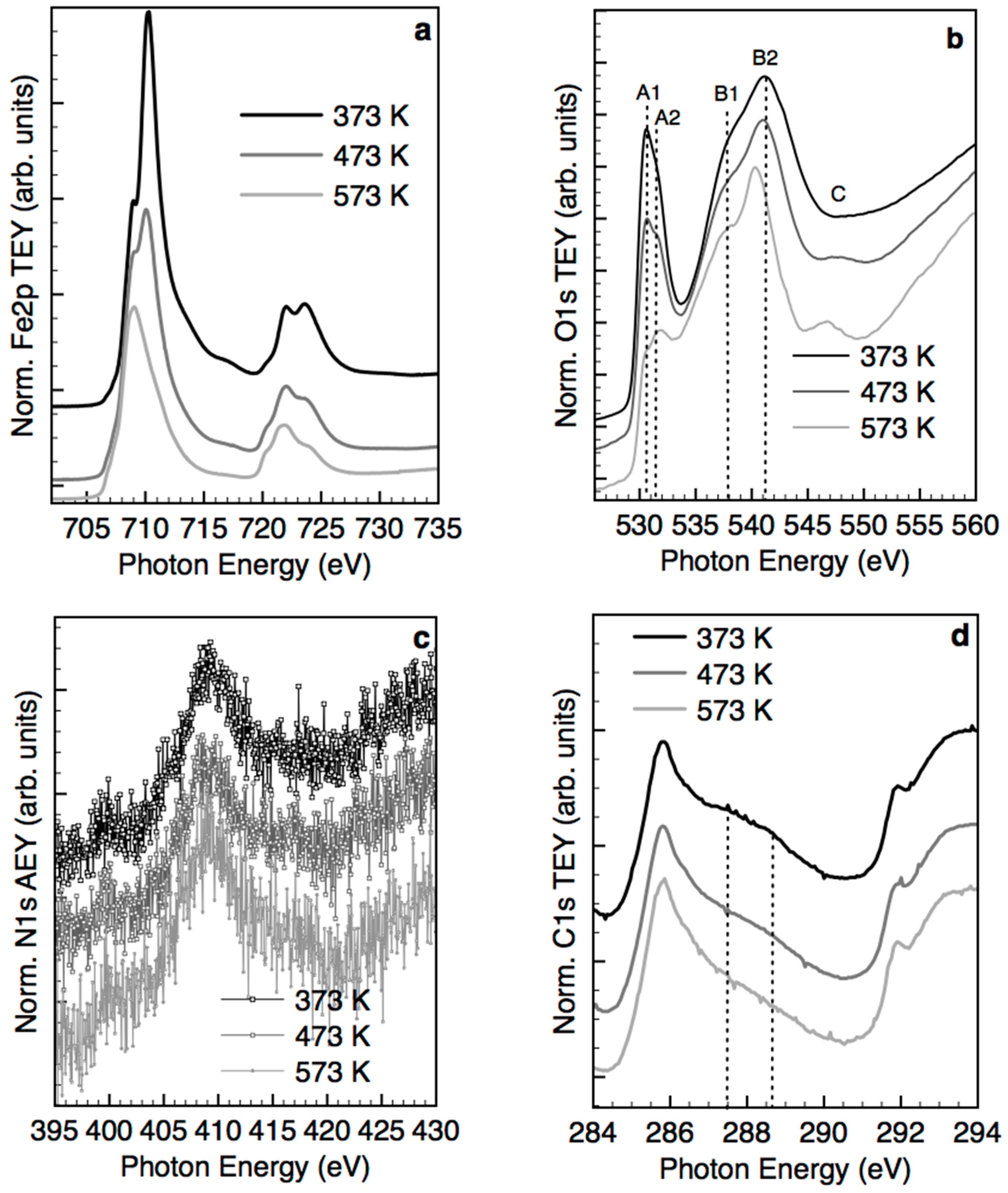
2.3. Structural Transformation upon Annealing in UHV by X-ray Absorption Spectroscopy, X-ray Photoemission Spectroscopy and TEM
3. Discussion
4. Materials and Methods
4.1. Synthesis of Impregnated Fe/OC, Fe/NC and Fe(I.E.)/NC
4.2. X-ray Photoelectron and Absorption Spectroscopy
4.3. Electron Microscopy Techniques
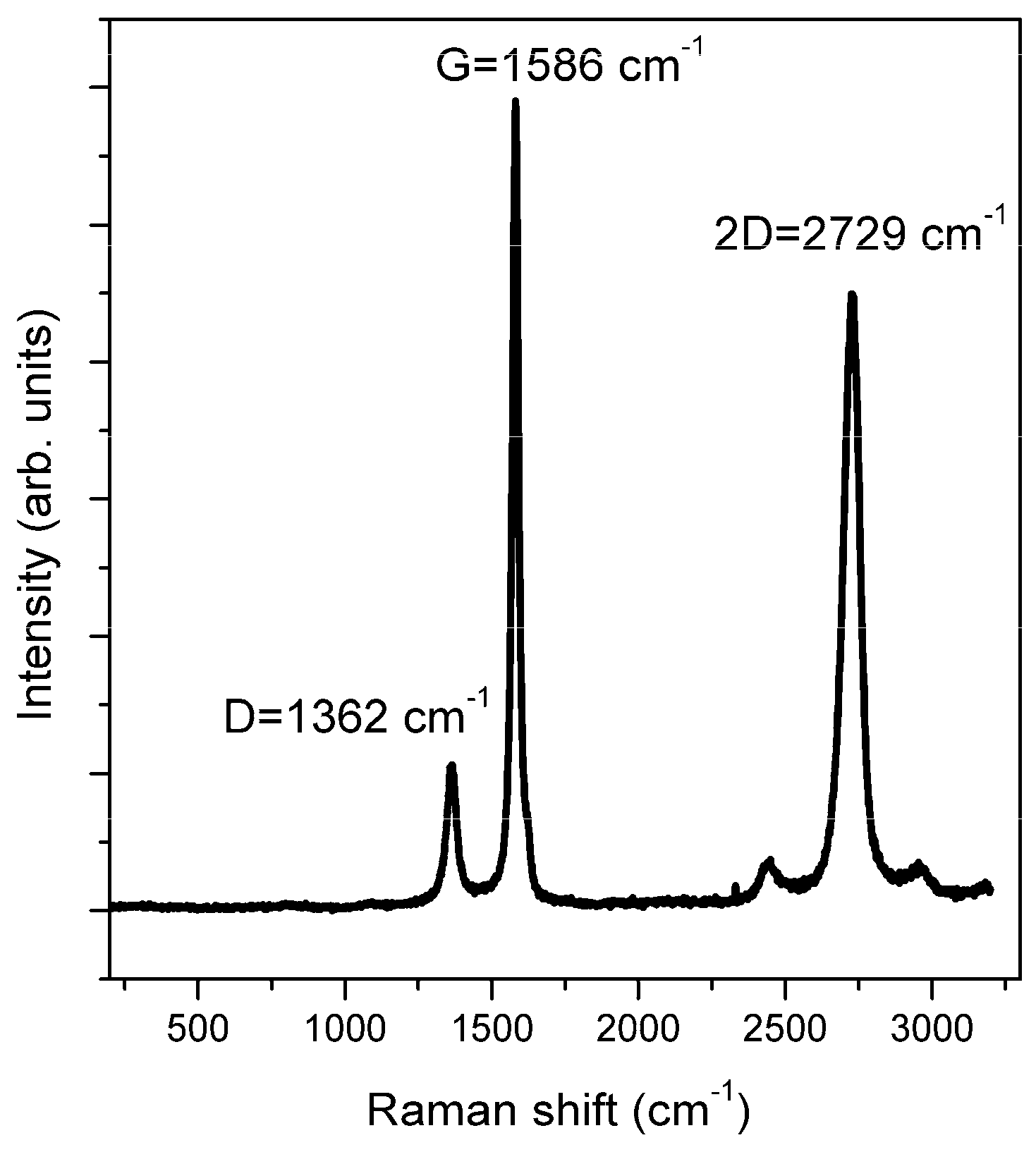
4.4. Raman
5. Conclusions
- (a)
- Single atoms and clusters are formed during the first impregnation step from the chemisorption of soluble Fe(III) species on the heteroatoms at the graphite edges. In the specific case of N-functionalized graphitic carbon, the high local pH in proximity to the functional groups induces clustering of the Fe species in parallel to their chemisorption on the carbon surface. Specifically, anionic Fe(III) species interact with the pyridine N functional groups on the C surface (or few basic and thermally stable O species on the OC support) and are stabilized in an Fe(II) oxidation state. Moreover, more abundant cationic Fe(III) species interact with carboxylates species on the OC surface.
- (b)
- The majority of the Fe(III) species in solution slowly polymerize during the drying process, forming Fe(III)-oxyhydroxy-nitrate NPs, which deposit on the carbon surface with no specific interaction with it. The thermal annealing in N2 transforms these Fe(III)-oxyhydroxy-nitrate NPs into big agglomerates of ferrihydrite NPs containing N impurities as Fe–N–O bonds in sp3 configuration but with a different spectroscopic fingerprint than nitrate moieties.
Author Contributions
Acknowledgments
Conflicts of Interest
References
- Turishchev, S.Y.; Antipov, S.S.; Novolokina, N.V.; Chuvenkova, O.A.; Melekhov, V.V.; Ovsyannikov, R.; Senkovskii, B.V.; Timchenko, A.A.; Ozoline, O.N.; Domashevskaya, E.P. A soft X-ray synchrotron study of the charge state of iron ions in the ferrihydrite core of the ferritin Dps protein in Escherichia coli. Biophysics 2016, 61, 705–710. [Google Scholar] [CrossRef]
- Tang, C.; Zhang, Q. Can metal–nitrogen–carbon catalysts satisfy oxygen electrochemistry? J. Mater. Chem. A 2016, 14, 4998–5001. [Google Scholar] [CrossRef]
- Zhu, Z.; Zhang, B.; Liu, X.; Wang, D.-W.; Su, D.S. Unravelling the Structure of Electrocatalytically Active Fe–N Complexes in Carbon for the Oxygen Reduction Reaction. Angew. Chem. Int. Ed. 2014, 53, 10673–10677. [Google Scholar] [CrossRef]
- Gangeri, M.; Caudo, S.; Perathoner, S.; Centi, G.; Amadou, J.; Bégin, D.; Pham-Huu, C.; Ledoux, M.J.; Tessonnier, J.P.; Su, D.S.; et al. Fe and Pt carbon nanotubes for the electrocatalytic conversion of carbon dioxide to oxygenates. Catal. Today 2009, 143, 57–63. [Google Scholar] [CrossRef]
- Ferrero, G.A.; Preuss, K.; Marinovic, A.; Jorge, A.B.; Mansor, N.; Brett, J.L.D.; Fuertes, A.B.; Sevilla, M.; Titirici, M.M. Fe–N-Doped Carbon Capsules with Outstanding Electrochemical Performance and Stability for the Oxygen Reduction Reaction in Both Acid and Alkaline Conditions. ACS Nano 2016, 10, 5922–5932. [Google Scholar] [CrossRef]
- Genovese, C.; Schuster, M.E.; Gibson, E.K.; Gianolio, D.; Posligua, V.; Grau-Crespo, R.; Cibin, G.; Wells, P.P.; Garai, D.; Solokha, V.; et al. Operando spectroscopy study of the carbon dioxide electro-reduction by iron species on nitrogen-doped carbon. Nat. Comms 2018, 9, 1–12. [Google Scholar] [CrossRef]
- Arrigo, R.; Schuster, M.E.; Wrabetz, S.; Girgsdies, F.; Tessonnier, J.P.; Centi, G.; Perathoner, S.; Su, D.S.; Schlogl, R. New Insights from Microcalorimetry on the FeOx/CNT-Based Electrocatalysts Active in the Conversion of CO2 to Fuels. ChemSusChem 2012, 5, 577–586. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Perathoner, S.; Ampelli, C.; Mebrahtu, C.; Su, D.S.; Centi, G. Room-Temperature Electrocatalytic Synthesis of NH3 from H2O and N2 in a Gas–Liquid–Solid Three-Phase Reactor. ACS Sustainable Chem. Eng. 2017, 5, 7393–7400. [Google Scholar] [CrossRef]
- Bahome, M.C.; Jewell, L.L.; Hildebrandt, D.; Glasser, D.; Coville, N.J. Fischer–Tropsch synthesis over iron catalysts supported on carbon nanotubes. App. Catal. A: General 2005, 287, 60–67. [Google Scholar] [CrossRef]
- Aluha, J.; Boahene, P.; Dalai, A.; Hu, Y.; Bere, K.; Braidy, N.; Abatzoglou, N. Synthesis and Characterization of Co/C and Fe/C Nanocatalysts for Fischer–Tropsch Synthesis: A Comparative Study Using a Fixed-Bed Reactor. Ind. Eng. Chem. Res. 2015, 54, 10661–10674. [Google Scholar] [CrossRef]
- Torres Galvis, H.M.; Bitter, J.H.; Khare, C.B.; Ruitenbeek, M.; Dugulan, A.I.; de Jong, K.P. Supported Iron Nanoparticles as Catalysts for Sustainable Production of Lower Olefins. Science 2012, 335, 835–838. [Google Scholar] [CrossRef] [PubMed]
- Gallios, G.P.; Tolkou, A.K.; Katsoyiannis, I.A.; Stefusova, K.; Vaclavikova, M.; Deliyanni, E.A. Adsorption of Arsenate by Nano Scaled Activated Carbon Modified by Iron and Manganese Oxides. Sustainability 2017, 9, 1684. [Google Scholar] [CrossRef]
- Munnik, P.; de Jongh, P.E.; de Jong, K.P. Recent Developments in the Synthesis of Supported Catalysts. Chem. Rev. 2015, 115, 6687–6718. [Google Scholar] [CrossRef]
- Bockris, J.O.; Khan, S.U.M. Surface Electrochemistry: A molecular Level Approach; Springer Science: New York, NY, USA, 1993. [Google Scholar]
- Arrigo, R.; Haevecker, M.; Wrabetz, S.; Blume, R.; Lerch, M.; McGregor, J.; Parrott, E.P.J.; Zeitler, J.A.; Gladden, L.F.; Knop-Gericke, A.; et al. Tuning the acid base properties of nanocarbon by functionalization via amination. JACS 2010, 132, 9616–9630. [Google Scholar] [CrossRef] [PubMed]
- Arrigo, R. Nitrogen Funstionalization of Carbon Nanofibers and Application in Heterogeneous Catalysis. Ph.D. Thesis, TU Berlin, Berlin, Germany, 2009. [Google Scholar]
- Jolivet, J.-P.; Chaneac, C.; Tronc, E. Iron oxide chemistry. From molecular clusters to extended solid networks. Chem. Commun. 2004, 481–487. [Google Scholar]
- Arrigo, R.; Schuster, M.E.; Xie, Z.; Yi, Y.; Wowsnick, G.; Sun, L.L.; Hermann, K.E.; Friedrich, M.; Kast, P.; Hävecker, M.; et al. Nature of the N–Pd Interaction in Nitrogen-Doped Carbon Nanotube Catalysts. ACS Catalysis 2015, 5, 2740–2756. [Google Scholar] [CrossRef]
- Arrigo, R.; Wrabetz, S.; Schuster, M.E.; Wang, D.; Villa, A.; Rosenthal, D.; Girsgdies, F.; Weinberg, G.; Prati, L.; Schlogl, R.; et al. Tailoring the morphology of Pd nanoparticles on CNTs by nitrogen and oxygen functionalization. Phys. Chem. Chem. Phys. 2012, 14, 10523–10532. [Google Scholar] [CrossRef] [PubMed]
- Arrigo, R.; Schuster, M.E.; Abate, S.; Wrabetz, S.; Amakawa, K.; Teschner, D.; Freni, M.; Centi, G.; Perathoner, S.; Havecker, M.; et al. Dynamics of Palladium on Nanocarbon in the Direct Synthesis of H2O2. ChemSusChem 2014, 7, 179–194. [Google Scholar] [CrossRef] [PubMed]
- Schuster, M.E.; Havecker, M.; Arrigo, R.; Blume, R.; Knauer, M.; Ivleva, N.P.; Su, D.S.; Niessner, R.; Schlogl, R. Surface Sensitive Study To Determine the Reactivity of Soot with the Focus on the European Emission Standards IV and VI. J. Phys. chem. A 2011, 115, 2568–2580. [Google Scholar] [CrossRef] [PubMed]
- Knauer, M.; Schuster, M.E.; Su, D.S.; Schlögl, R.; Niessner, R.; Ivleva, N.P. Soot Structure and Reactivity Analysis by Raman Microspectroscopy, Temperature-Programmed Oxidation, and High-Resolution Transmission Electron Microscopy. J. Phys. Chem. A 2009, 113, 13871–13880. [Google Scholar] [CrossRef]
- Michel, F.M.; Ehm, L.; Antao, S.M.; Lee, P.L.; Chupas, P.J.; Liu, G.; Strongin, D.R.; Schoonen, M.A.A.; Phillips, B.L.; Parise, J.B. The Structure of Ferrihydrite, a Nanocrystalline Material. Science 2007, 316, 1726–1729. [Google Scholar] [CrossRef]
- Weatherill, J.S.; Morris, K.; Bots, P.; Stawski, T.M.; Janssen, A.; Abrahamsen, L.; Blackham, R.; Shaw, S. Ferrihydrite formation: the role of Fe13 Keggin clusters. Environ. Sci. Technol. 2016, 50, 9333–9342. [Google Scholar] [CrossRef] [PubMed]
- Tanuma, S.; Powell, C.J.; Penn, D.R. Calculations of electron inelastic mean free paths. V. Data for 14 organic compounds over the 50–2000 eV range. Surf. Interface Anal. 1993, 21, 165–176. [Google Scholar] [CrossRef]
- Blume, R.; Rosenthal, D.; Li, H.; Tessonnier, J.P.; Knop-Gericke, A.; Schloegl, R. Characterizing Graphitic Carbon with X-ray Photoelectron Spectroscopy: A Step-by-Step Approach. ChemCatChem 2017, 7, 2871–2881. [Google Scholar] [CrossRef]
- Yamashita, T.; Hayes, P. Analysis of XPS spectra of Fe2+ and Fe3+ ions in oxide materials. Appl. Surf. Sci. 2008, 254, 2441–2449. [Google Scholar] [CrossRef]
- Grosvenor, A.P.; Kobe, B.A.; Biesinger, M.C.; McIntyre, N.S. Investigation of multiplet splitting of Fe 2p XPS spectra and bonding in iron compounds. Surf. Interface Anal. 2004, 36, 1564–1574. [Google Scholar] [CrossRef]
- Fujii, T.; de Groot, F.M.F.; Sawatzky, G.A.; Voogt, F.C.; Hibma, T.; Okada, K. In situ XPS analysis of various iron oxide films grown by NO2-assisted molecular-beam epitaxy. Phys. Rev. B 1999, 59, 3195–3202. [Google Scholar] [CrossRef]
- Li, H.B.; Gao, Y.Q.; Yang, G.W. Electrochemical route for accessing amorphous mixed-metal hydroxide nanospheres and magnetism. RSC Adv. 2015, 5, 45359–45367. [Google Scholar] [CrossRef]
- Rocha Costa Rizuti, T.; Oestereich, A.; Demidov, D.V.; Havecker, M.; Zafeiratos, S.; Weinberg, G.; Bukhtiyarov, V.I.; Knop-Gericke, A.; Schlogl, R. The silver-oxygen system in catalysis: new insights by near ambient pressure x-ray photoelectron spectroscopy. Phys. Chem. Chem. Phys. 2012, 14, 4554–4564. [Google Scholar] [CrossRef]
- Arrigo, R.; Schuster, M.E.; Abate, S.; Giorgianni, G.; Centi, G.; Perathoner, S.; Wrabetz, S.; Pfeifer, V.; Antonietti, M.; Schlögl, R. Pd supported on Carbon Nitride Boosts the Direct Hydrogen Peroxide Synthesis. ACS Catalysis 2016, 6, 6959–6966. [Google Scholar] [CrossRef]
- Purans, J.; Forest, B.; Cannes, C.; Sladkov, V.; David, F.; Venault, L.; Lecomte, M. Structural investigation of Pd(II) in concentrated nitric and perchloric acid solutions by XAFS. J. Phys. Chem: B 2005, 109, 11074–11082. [Google Scholar] [CrossRef]
- Peak, D.; Regier, T. Direct Observation of Tetrahedrally Coordinated Fe(III) in Ferrihydrite. Environ. Sci. Technol. 2012, 46, 3163–3168. [Google Scholar] [CrossRef] [PubMed]
- Giovannini, S.; Boscerini, F.; Carboni, R.; Signorini, L.; Pasquini, L.; Mahne, N.; Giglia, A.; Pedio, M.; Nannarone, S.; Benfatto, M.; et al. Multiple scattering analysis of O K-edge NEXAFS in iron oxides. Phys. Scr. 2005, 115, 424–427. [Google Scholar] [CrossRef]
- De Groot, F.M.F.; Grioni, M.; Fuggle, J.C.; Ghijsen, J.; Sawatzky, G.A.; Petersen, H. Oxygen 1s x-ray absorption of transition metal oxides. Phys. Rev. B 1989, 40, 5715–5722. [Google Scholar] [CrossRef]
- Wu, Y.Z.; Gota, S.; Jollet, F.; Pollak, M.; Gautier-Soyer, M.; Natoli, C.R. Characterization of iron oxides by X-ray absorption at the oxygen K edge using a full multiple-scattering approach. Phys. Rev. B 1997, 55, 2570–2577. [Google Scholar] [CrossRef]
- Bhattacharyya, A.; Stavitski, E.; Dvorak, J.; .Martinez, C.E. Redox interactions between Fe and cysteine: Spectroscopic studies and multiplet calculations. Geochim. Cosmochim. Acta 2013, 122, 89–100. [Google Scholar] [CrossRef]
- McDonald, T.M.; Mason, J.A.; Kong, X.; Bloch, E.D.; Gygi, D.; Dani, A.; Crocellà, V.; Giordanino, F.; Odoh, S.O.; Drisdell, W.S.; et al. Cooperative insertion of CO2 in diamine-appended metal-organic frameworks. Nature 2015, 519, 303–308. [Google Scholar] [CrossRef]
- Khyzhun, O.Y.; Strunskus, T.; Wöll, C.; Gies, H.; Staemmler, V. Comparison of the O Kα x-ray emission bands in micro- and mesoporous silica materials and in α-quartz. J. Chem. Phys. 2008, 129, 084711-1. [Google Scholar]
- Narioka, S.; Ishii, H.; Ouchi, Y.; Yokoyama, T.; Ohta, T.; Seki, K. XANES Spectroscopic Studies of Evaporated Porphyrin Films: Molecular Orientation and Electronic Structure. J. Phys. Chem. 1995, 99, 1332–1337. [Google Scholar] [CrossRef]
- Chen, J.G. NEXAFS investigations of transition metal oxides, nitrides, carbides, sulfides and other interstitial compounds. Surf. Sci. Rep. 1997, 30, 1–152. [Google Scholar] [CrossRef]
- Schuster, M.E.; Teschner, D.; Popovic, J.; Ohmer, N.; Girgsdies, F.; Tornow, J.; Willinger, M.G.; Samuelis, D.; Titirici, M.-M.; Maier, J.; et al. Charging and Discharging Behavior of Solvothermal LiFePO4 Cathode Material Investigated by Combined EELS/NEXAFS Study. Chem. Mater. 2014, 26, 1040–1047. [Google Scholar] [CrossRef]
- Miedema, P.S.; de Groot, F.M.F. The iron L edges: Fe 2p X-ray absorption and electron energy loss spectroscopy. J. Elect. Spectros. Rel. Phen. 2013, 187, 32–48. [Google Scholar] [CrossRef]
- Gautier-Soyer, M. X-ray absorption spectroscopy: A tool to study the local atomic and electronic structure of ceramics. J. Eu. Ceram. Soc. 1998, 18, 2253–2261. [Google Scholar] [CrossRef]
- Janney, D.E.; Cowley, J.M.; Buseck, P.R. Transmission electron microscopy of synthetic 2-and 6-line ferrihydrite. Clays Clay Miner. 2000, 48, 111–119. [Google Scholar] [CrossRef]
- Campbell, A.S.; Schwertmann, U.; Campbell, P.A. Formation of Cubic Phases on Heating Ferrihydrite. Clay Minerals 1997, 32, 615–622. [Google Scholar] [CrossRef]
- Leveneur, J.; Waterhouse, G.I.N.; Kennedy, J.; Metson, J.B.; Mitchell, D.R.G. Nucleation and Growth of Fe Nanoparticles in SiO2: A TEM, XPS and FE L-edge XANES Investigation. J. Phys. Chem. C 2011, 115, 20978–20985. [Google Scholar] [CrossRef]
- Stöhr, J. NEXAFS Spectroscopy; Springer: Berlin, Germany, 1992. [Google Scholar]
- Schwertmann, U.; Friedl, J.; Stanjek, H. From Fe(III) Ions to Ferrihydrite and then Hematite. J. Col. Interf. Sci. 1999, 209, 215–223. [Google Scholar] [CrossRef] [PubMed]
- Melnikov, P.; Nascimento, V.A.; Arkhangelsky, I.V.; Zanoni Consolo, L.Z.; de Oliveira, L.C.S. Thermal decomposition mechanism of iron(III) nitrate and characterization of intermediate products by the technique of computerized modeling. J. Therm. Anal. Calorim. 2014, 114, 141–151. [Google Scholar] [CrossRef]














{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | O | N | Fe | C | Si |
|---|---|---|---|---|---|
| Fe/NC | 9.3 | 0.5 | 2.4 | 87.3 | 0.5 |
| Fe/OC | 13.6 | 0.4 | 3.6 | 81.9 | 0.5 |
| Fe(I.E.)/NC b | 8.5 | 0.44 | 0.06 | 86.5 | 4.5 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Arrigo, R.; Schuster, M.E. On the High Structural Heterogeneity of Fe-Impregnated Graphitic-Carbon Catalysts from Fe Nitrate Precursor. Catalysts 2019, 9, 303. https://doi.org/10.3390/catal9040303
Arrigo R, Schuster ME. On the High Structural Heterogeneity of Fe-Impregnated Graphitic-Carbon Catalysts from Fe Nitrate Precursor. Catalysts. 2019; 9(4):303. https://doi.org/10.3390/catal9040303
Chicago/Turabian StyleArrigo, Rosa, and Manfred Erwin Schuster. 2019. "On the High Structural Heterogeneity of Fe-Impregnated Graphitic-Carbon Catalysts from Fe Nitrate Precursor" Catalysts 9, no. 4: 303. https://doi.org/10.3390/catal9040303
APA StyleArrigo, R., & Schuster, M. E. (2019). On the High Structural Heterogeneity of Fe-Impregnated Graphitic-Carbon Catalysts from Fe Nitrate Precursor. Catalysts, 9(4), 303. https://doi.org/10.3390/catal9040303