Hydrogenation of Bio-Oil Model Compounds over Raney-Ni at Ambient Pressure



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
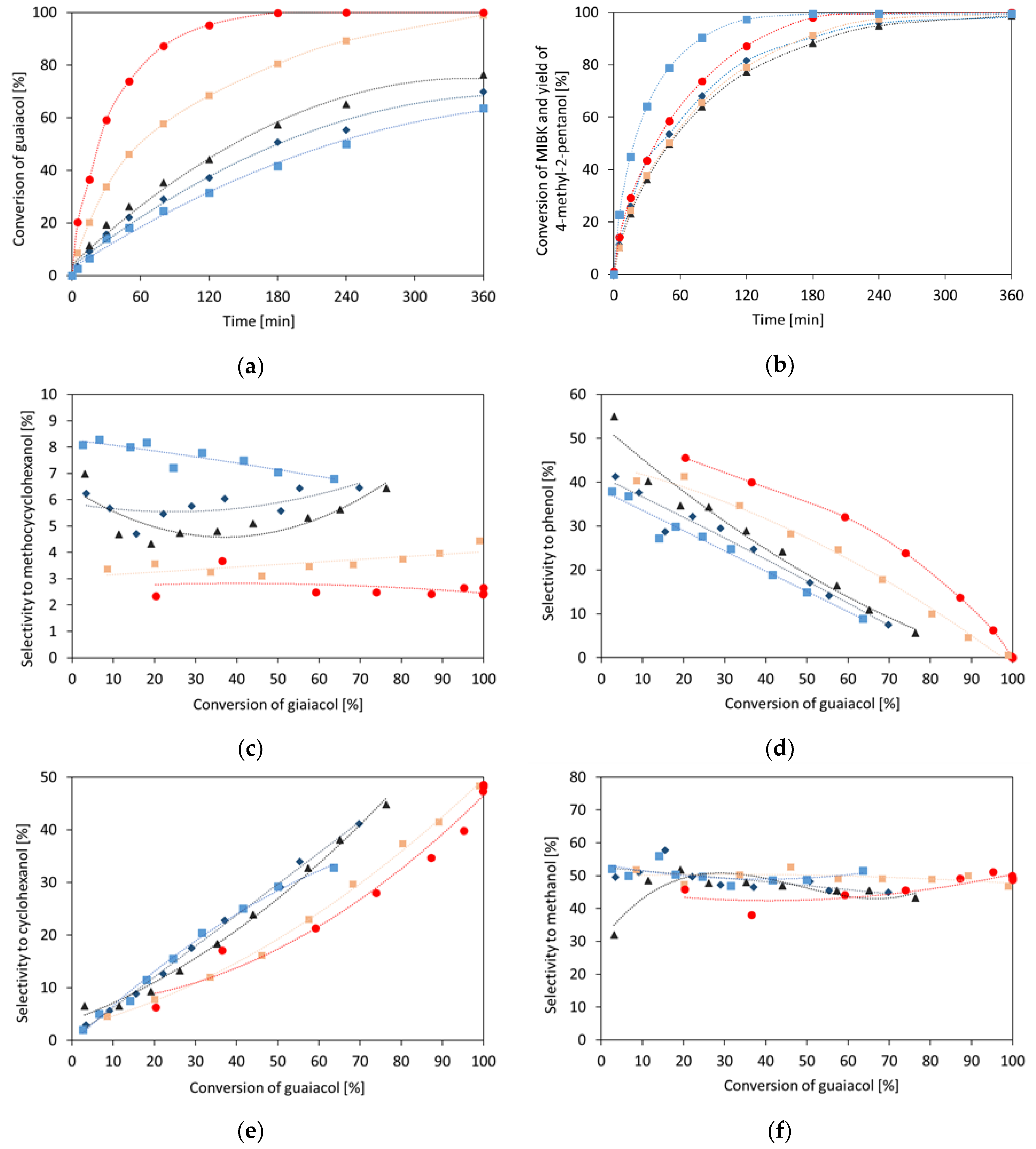
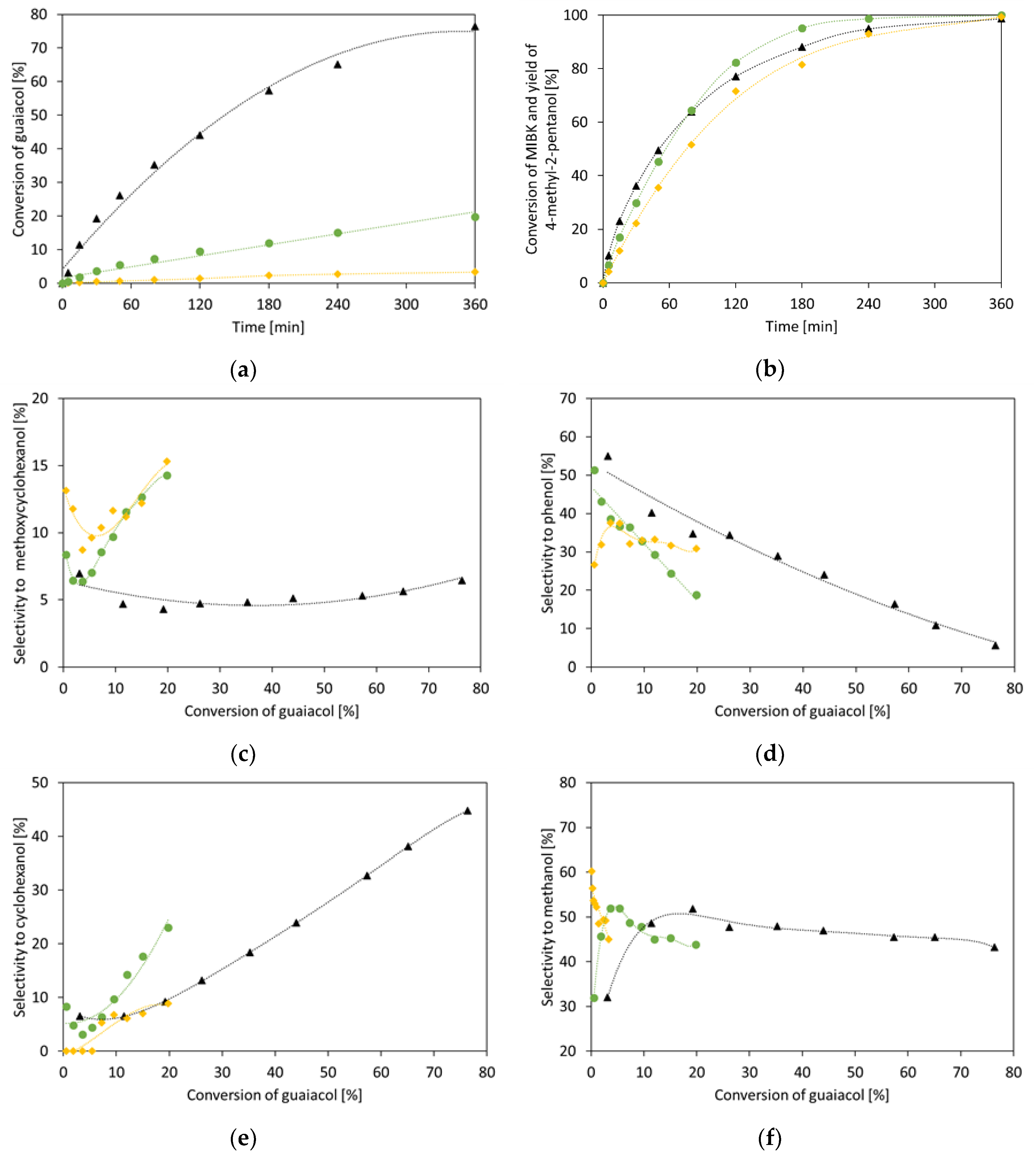
2. Results and Discussion
2.1. Single Compounds Hydrogenation
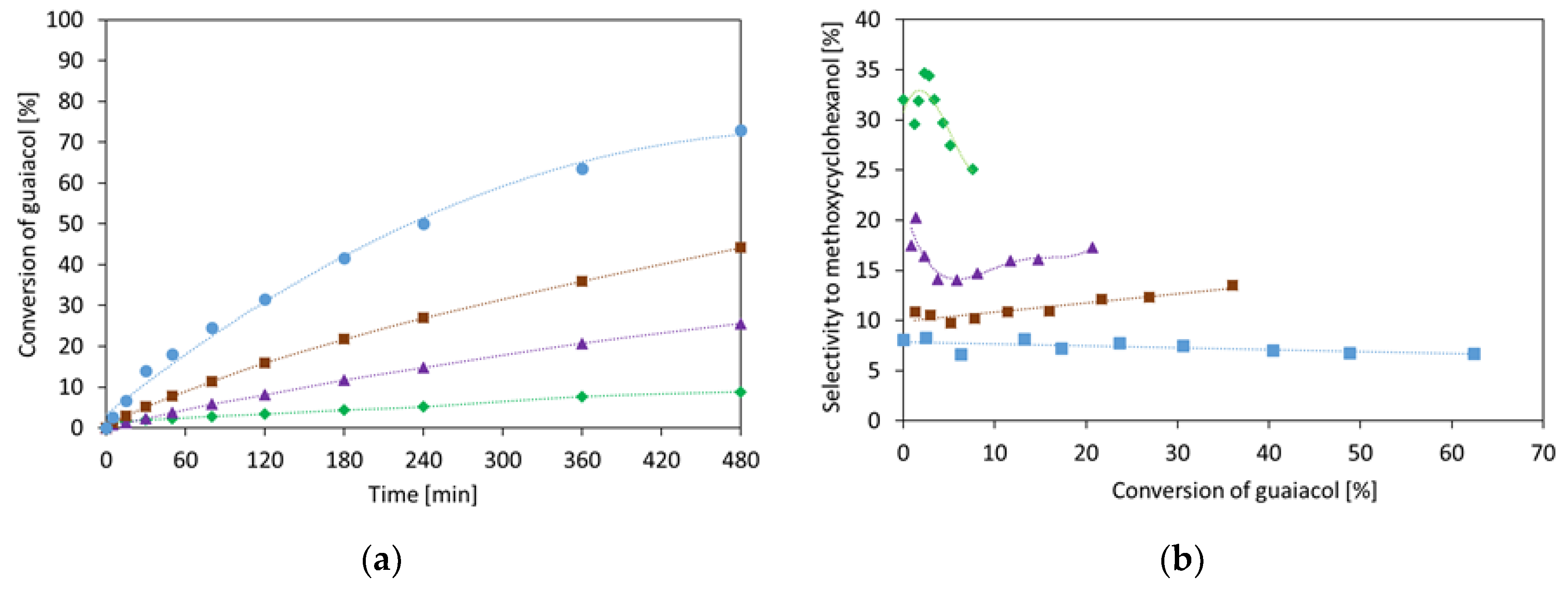
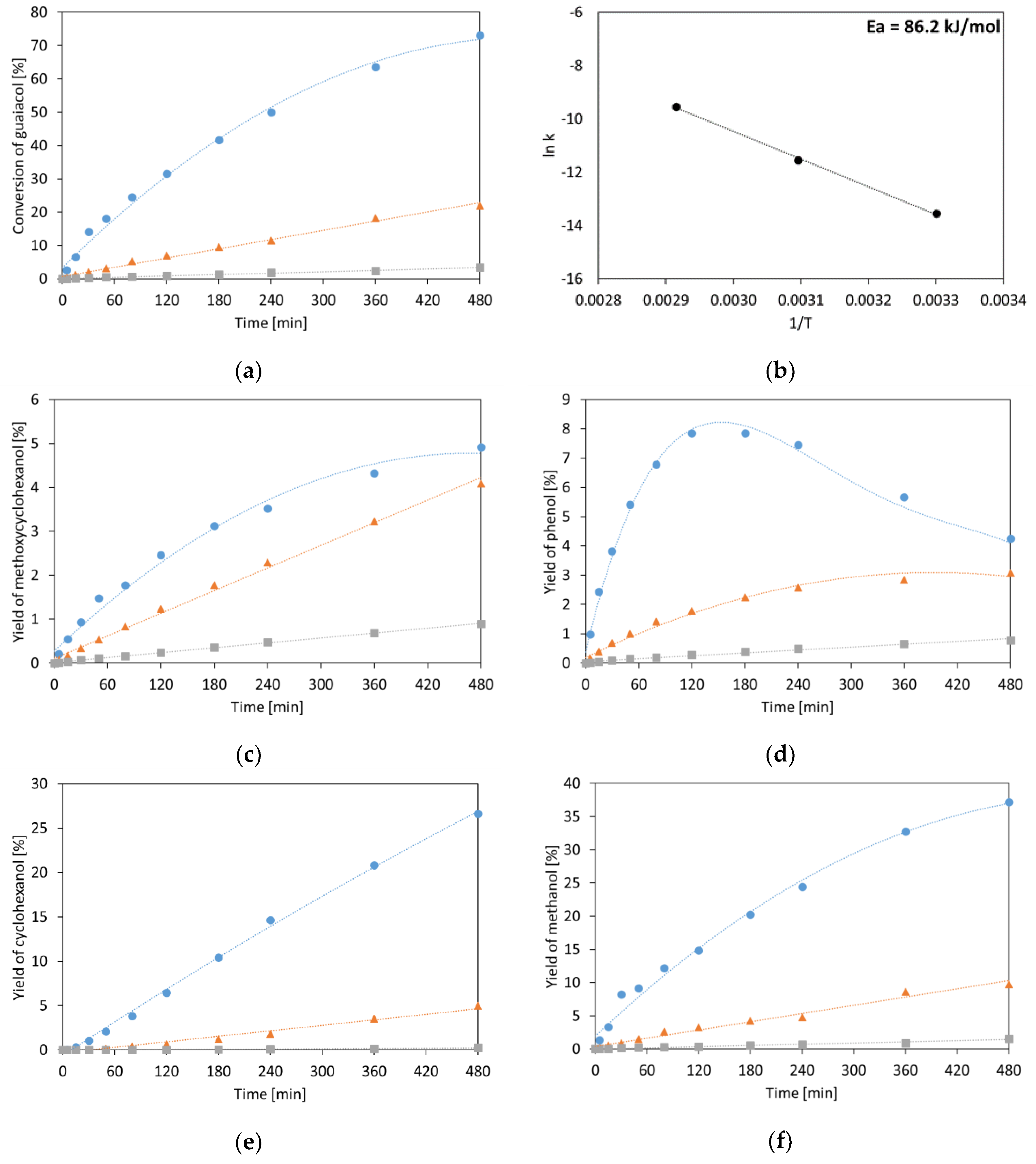
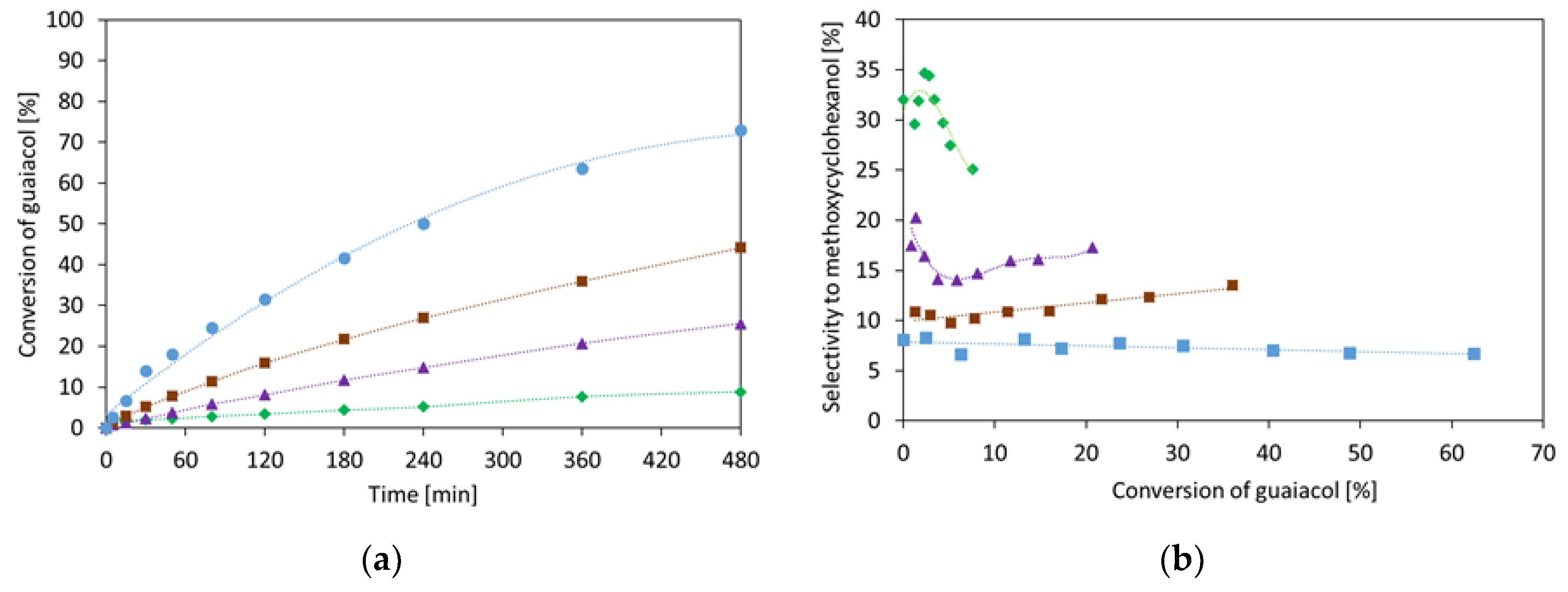
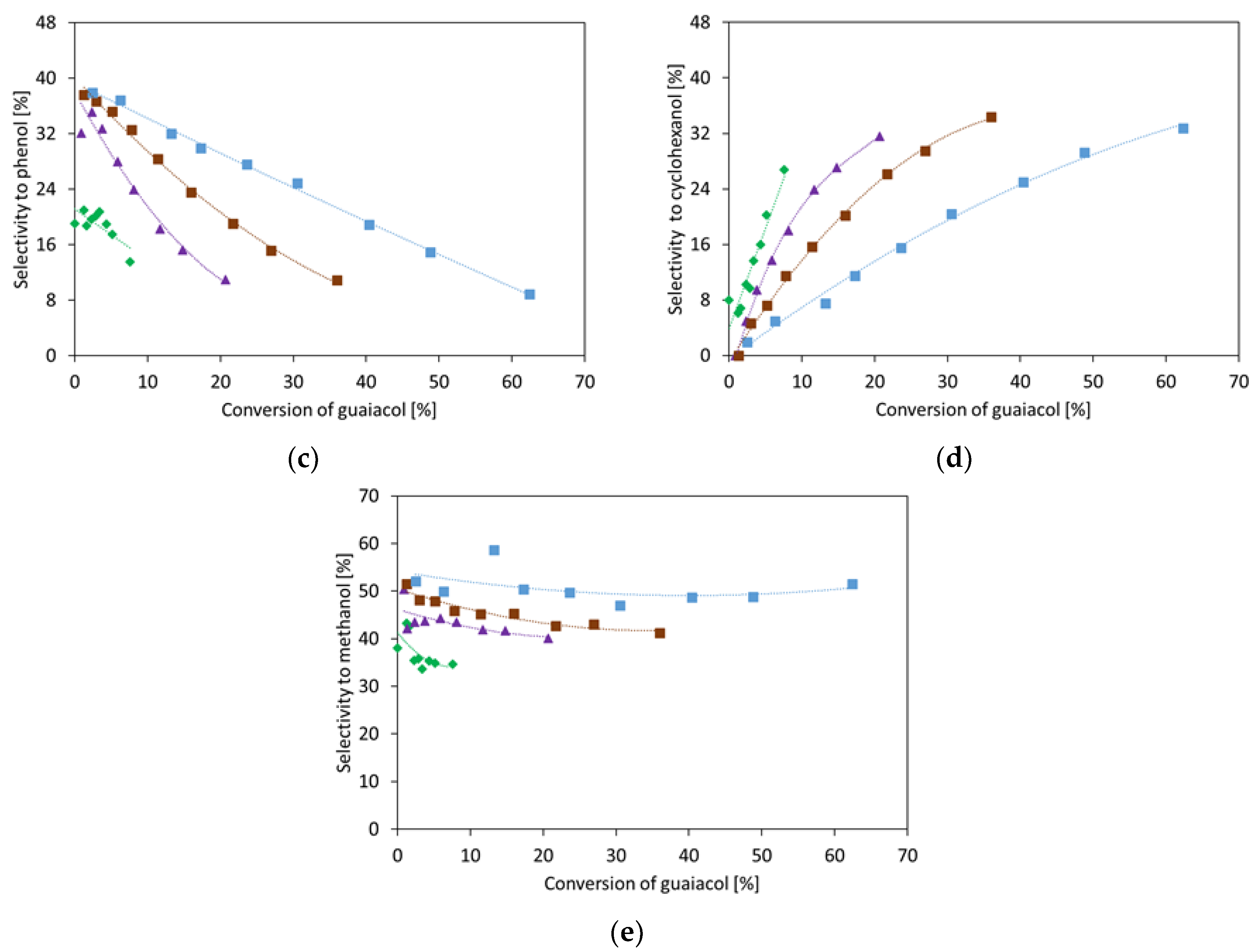
2.1.1. Guaiacol
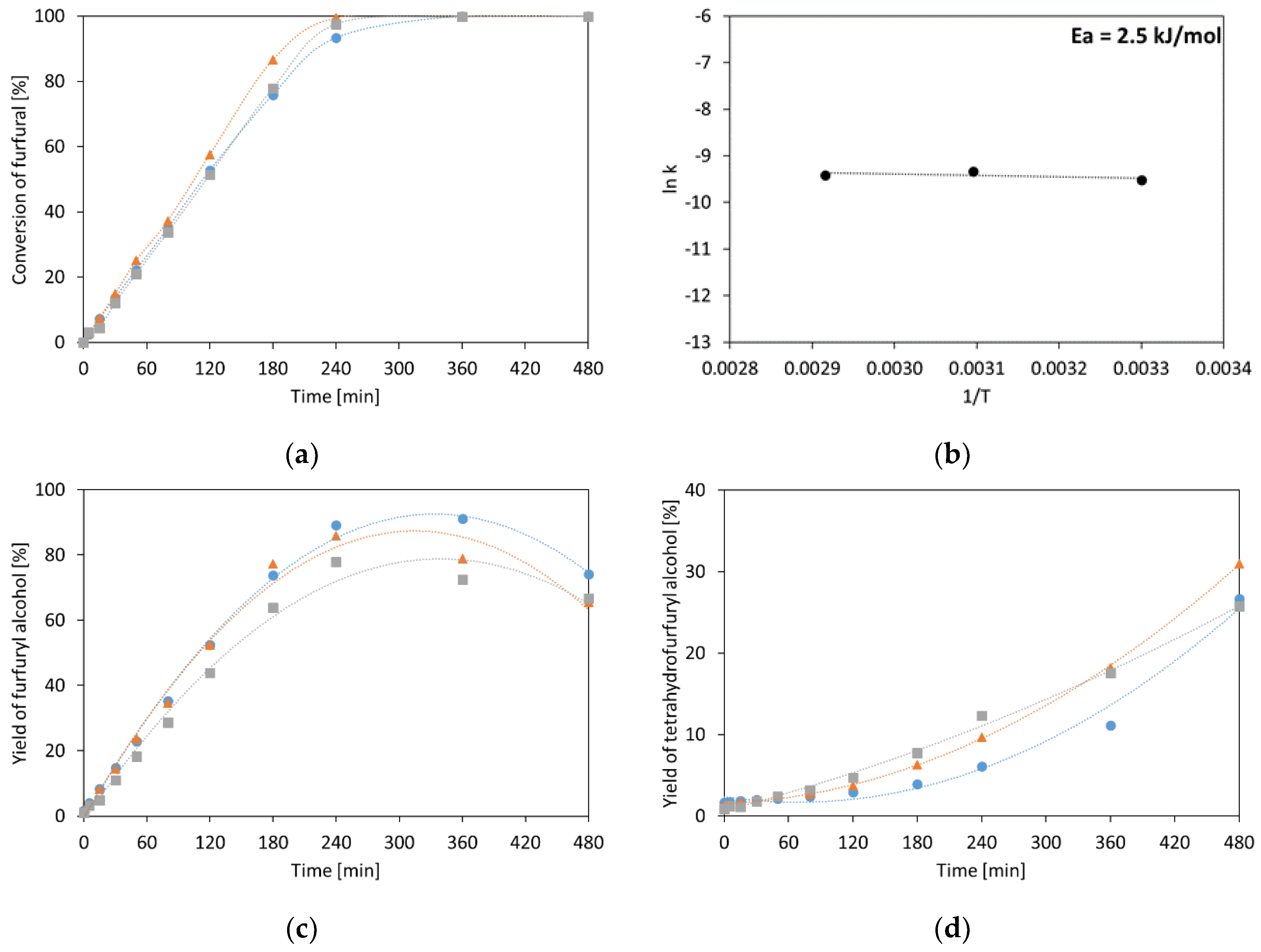
2.1.2. Furfural
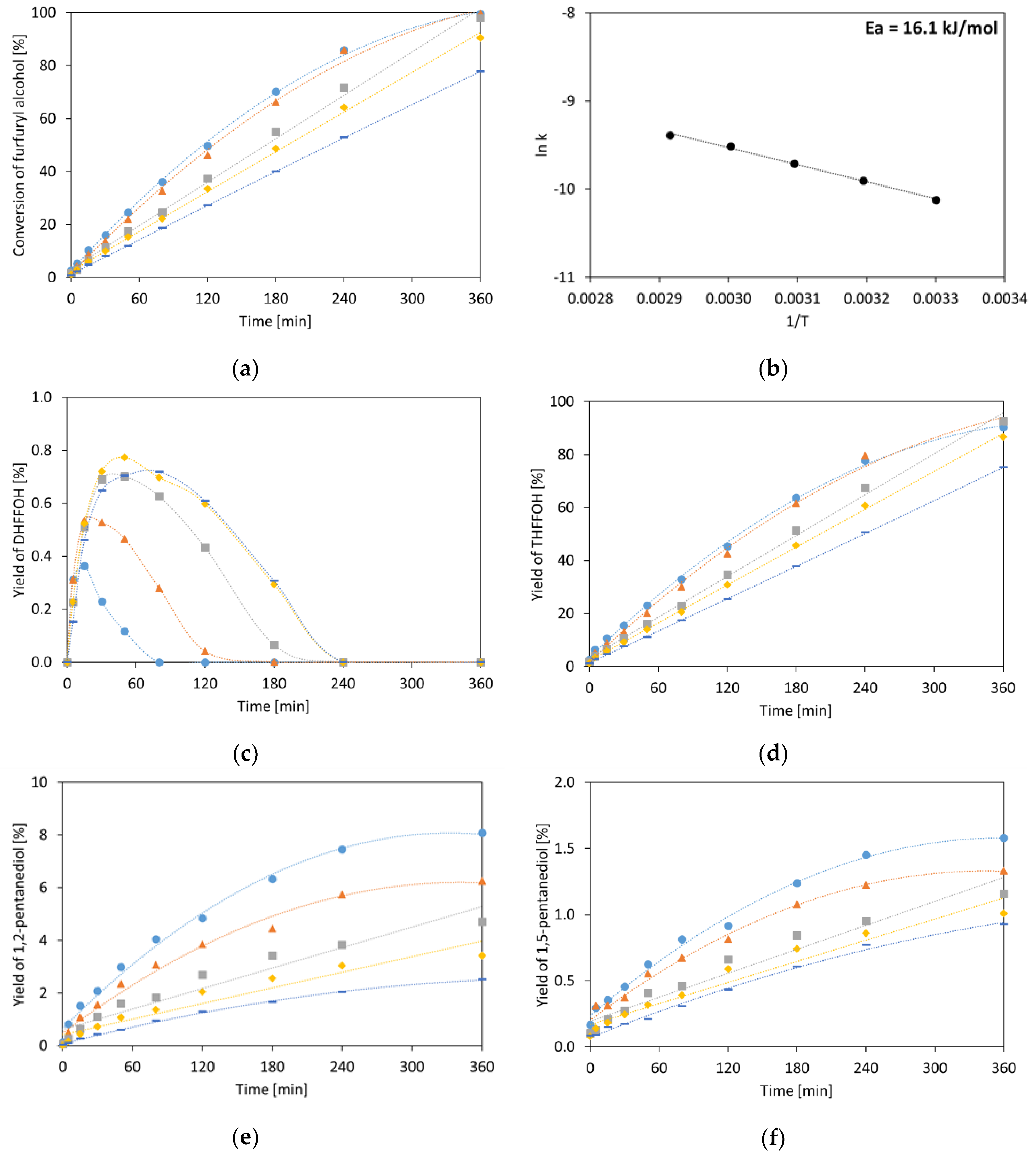
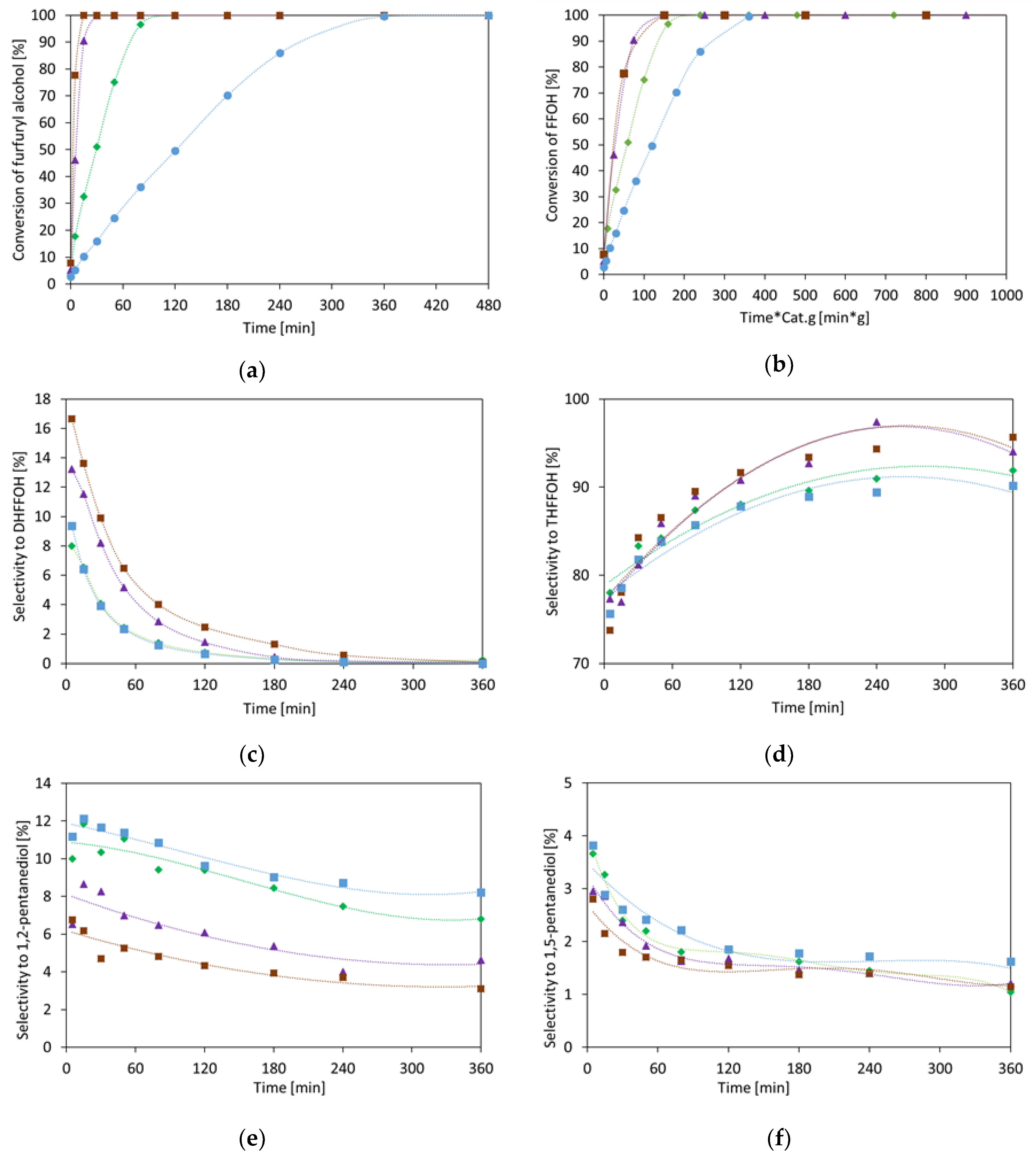
2.1.3. Furfuryl Alcohol
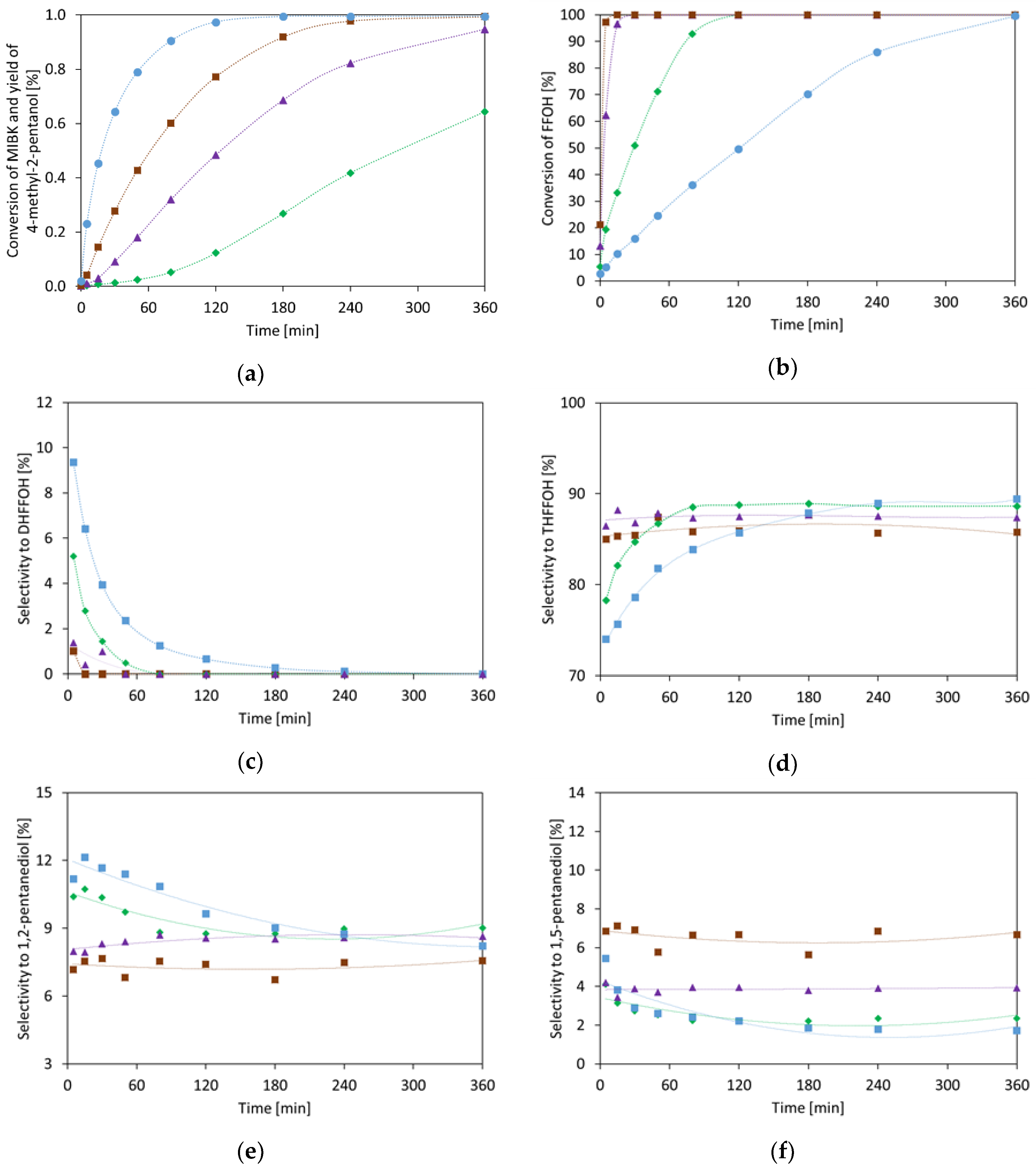
2.1.4. Methyl Isobutyl Ketone
2.2. Hydrogenation of Binary Mixtures
2.2.1. Guaiacol with Furfuryl Alcohol
2.2.2. Methyl Isobutyl Ketone with Furfuryl Alcohol
2.2.3. Guaiacol with Methyl Isobutyl Ketone
3. Materials and Methods
3.1. Chemicals
3.2. Experimental Setup
3.3. Qualitative and Quantitative Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Neste Renewable Diesel. Available online: http://gtsummitexpo.socialenterprises.net/assets/docs/past-events/GTSE-tacoma-2016/april-6/gtse-tacoma-2016-GTSE-Session-6B-Dayne-Delahoussaye-Neste-April-6th.pdf (accessed on 2 February 2019).
- Ecofining™ Process. Available online: https://www.uop.com/processing-solutions/renewables/green-diesel/ecofining/ (accessed on 2 February 2019).
- Venderbosch, R.; Ardiyant, A.; Wildschut, J.; Oasmaa, A.; Heeres, H. Stabilization of biomass-derived pyrolysis oils. J. Chem. Technol. Biotechnol. 2010, 85, 674–686. [Google Scholar] [CrossRef]
- Peacocke, G.; Bridgwater, A. Ablative plate pyrolysis of biomass for liquids. Biomass Bioenergy 1994, 7, 147–154. [Google Scholar] [CrossRef]
- Baldauf, W.; Balfanz, U.; Rupp, M. Upgrading of flash pyrolysis oil and utilization in refineries. Biomass Bioenergy 1994, 7, 237–244. [Google Scholar] [CrossRef]
- Ly, H.; Im, K.; Go, Y.; Galiwano, E.; Kim, S.; Kim, J.; Choi, J.; Woo, H. Spray pyrolysis synthesis of γ-Al2O3 supported metal and metal phosphide catalysts and their activity in the hydrodeoxygenation of a bio-oil model compound. Energ. Convers. Manage. 2016, 127, 545–553. [Google Scholar] [CrossRef]
- Rodríguez-Aguado, E.; Infantes-Molina, A.; Ballesteros-Plata, D.; Cecila, J.; Baroso-Martin, I.; Rodríguez-Castellón, E. Ni and Fe mixed phosphides catalysts for O-removal of a bio-oil model molecule from lignocellulosic biomass. Mol. Catal. 2017, 437, 130–139. [Google Scholar] [CrossRef]
- French, R.; Stunkel, J.; Black, S.; Myers, M.; Yung, M.; Iisa, K. Evaluate impact of catalyst type on oil yield and hydrogen consumption from mild hydrotreating. Energ. Fuels 2014, 28, 3086–3095. [Google Scholar] [CrossRef]
- Ardiyanti, A.; Khromova, S.; Venderbosch, H.; Yakovlev, V.; Melián-Cabrera, I.; Heeres, H. Catalytic hydrotreatment of fast pyrolysis oil using bimetallic Ni–Cu catalysts on various supports. Appl. Catal. A 2012, 449, 121–130. [Google Scholar] [CrossRef]
- Lei, H.; Ren, S.J.; Julson, J. The effects of reaction temperature and time and particle size of corn stover on microwave pyrolysis. Energ. Fuels 2009, 23, 3254–3261. [Google Scholar] [CrossRef]
- Pandey, J.; Kumar, R.; Wate, S.; Chakrabarti, T. Methane Emissions from Wastewater, Wetlands, Mangroves and Hydroelectric Dams: Developing Appropriate Emission Factors for Region-Specific GHGS (ERG). Asia Pac J. Manag. Res. Innov. 2010, 6, 29–41. [Google Scholar] [CrossRef]
- Hubert, G.; Iborra, S.; Corma, A. Synthesis of Transportation Fuels from Biomass: Chemistry, Catalysts, and Engineering. Chem. Rev. 2006, 106, 4044–4098. [Google Scholar] [CrossRef] [PubMed]
- Holladay, J.; White, J.; Bozell, J.; Johnson, D. Top Value-Added Chemicals from Biomass - Volume II—Results of Screening for Potential Candidates from Biorefinery Lignin. PNNL 2007, 106, 4044–4098. [Google Scholar]
- Effendi, A.; Gerhauser, H.; Bridgwater, A. Production of renewable phenolic resins by thermochemical conversion of biomass: A review. Renew. Sustain. Energy Rev. 2008, 8, 2092–2116. [Google Scholar] [CrossRef]
- Auersvald, M.; Shumeiko, B.; Staš, M.; Kubička, D.; Chudoba, J.; Šimáček, P. Quantitative Study of Straw Bio-oil Hydrodeoxygenation over a Sulfided NiMo Catalyst. ACS Sustain. Chem. Eng. 2019. submitted. [Google Scholar] [CrossRef]
- Lup, A.; Abnisa, F.; Daud, W.; Aroua, M. A review on reaction mechanisms of metal-catalyzed deoxygenation process in bio-oil model compounds. Appl. Catal. A 2019, 541, 87–106. [Google Scholar] [CrossRef]
- Liu, L.; Liu, Y.; Zhang, R.; Zhai, Y. Hydrodeoxygenation of bio-oil model compounds over amorphous NiB/SiO2-Al2O3 catalyst in oil-water biphasic system. J. Fuel Chem. Technol. 2017, 45, 932–938. [Google Scholar] [CrossRef]
- Ozagac, M.; Bertino-Ghera, C.; Uzio, D.; Rivallan, M.; Laurenti, D.; Geantet, C. Understanding macromolecules formation from the catalytic hydroconversion of pyrolysis bio-oil model compounds. Biomass Bioenergy 2016, 95, 182–193. [Google Scholar] [CrossRef]
- Lee, C.; Yoon, Y.; Suh, Y.; Choi, J.; Ha, J.; Suh, D.; Park, Y. Catalytic roles of metals and supports on hydrodeoxygenation of lignin monomer guaiacol. Catal. Commun. 2012, 17, 54–58. [Google Scholar] [CrossRef]
- Kao, G.N.; Tilak, B.D.; Venkataraman, K. Raney nickel reductions. Math. Sci. 1953, 38, 244–256. [Google Scholar]
- Ahmed, M.; Khadom, A.; Kadhum, A. Optimization hydrogenation process of D-glucose to D-sorbitol over Raney nickel catalyst. Eur. J. Sci. Res. 2009, 30, 294–304. [Google Scholar]
- Yu, Y.; Xu, Y.; Wang, T.; Ma, L.; Zhang, Q.; Zhang, X.; Zhang, X. In-situ hydrogenation of lignin depolymerization model compounds to cyclohexanol. J. Fuel Chem. Technol. 2013, 41, 443–447. [Google Scholar] [CrossRef]
- Xu, Y.; Long, J.; Liu, Q.; Li, Y.; Wang, C.; Zhang, Q.; Lv, W. In situ hydrogenation of model compounds and raw bio-oil over Raney Ni catalyst. Energy Convers. Manage. 2015, 89, 188–196. [Google Scholar] [CrossRef]
- Wang, L.; Ye, P.; Fang, Y.; Li, S.; Ye, Z. Liquid phase in-situ hydrodeoxygenation of bio-derived phenol over Raney Ni and Nafion/SiO2. Int. J. hydrogen energy 2015, 40, 14790–14797. [Google Scholar] [CrossRef]
- Feng, J.; Yang, Z.; Hse, C.; Su, Q.; Wang, K.; Jiang, J.; Xu, J. In situ catalytic hydrogenation of model compounds and biomassderived phenolic compounds for bio-oil upgrading. Renew. Energy 2017, 105, 140–148. [Google Scholar] [CrossRef]
- Bai, Y.; Lu, C.; Ma, L.; Chen, P.; Zheng, Y.; Li, X. Hydrogen production by aqueous-phase reforming of ethylene glycol over Pt catalysts supported on gamma-Al2O3 modified with Ce and Mg. Chin. J. Catal. 2006, 27, 275–280. [Google Scholar]
- Lup, A.; Abnisa, F.; Daud, W.; Aroua, M. A review on reactivity and stability of heterogeneous metal catalysts for deoxygenation of bio-oil model compounds. J. Ind. Eng. Chem. 2017, 56, 1–34. [Google Scholar] [CrossRef]
- Morais, A.; Costa Lopes, A.; Costa, P.; Fonseca, I.; Nogueira, I.; Oliveira, A.; Bogel-Lukasik, R. Cattle fat valorisation through biofuel production by hydrogenation in supercritical carbon dioxide. RSC adv. 2014, 4, 32081–32091. [Google Scholar] [CrossRef]
- Wang, X.; Rinaldi, R. Exploiting H-transfer reactions with Raney-Ni for upgrade of phenolic and aromatic biorefinery feeds under unusual, low-severity conditions. Energy Environ. Sci. 2012, 5, 8244. [Google Scholar] [CrossRef]
- Mebane, R.; Holte, K.; Gross, B. Transfer Hydrogenation of Ketones with 2-Propanol and Raney® Nickel. Synth. Commun. 2007, 37, 2787–2791. [Google Scholar] [CrossRef]
- Rahman, A.; Jonnalagadda, S. Rapid and selective reduction of adehydes, ketones, phenol, and alkenes with Ni–boride–silica catalysts system at low temperature. J. Mol. Catal. A: Chem. 2009, 299, 98–101. [Google Scholar] [CrossRef]
- Rinaldi, R.; Wang, X.; Bridgwater, A. Solvent effects on the hydrogenolysis of diphenyl ether with Raney nickel and their implications for the conversion of lignin. Chemsuschem 2012, 5, 1455–1466. [Google Scholar]
- Song, Y.; Li, W.; Zhang, M.; Tao, K. Hydrogenation of furfuryl alcohol to tetrahydrofurfuryl alcohol on NiB/SiO2 amorphous alloy catalyst. Front. Chem. Eng. China 2007, 1, 151–154. [Google Scholar] [CrossRef]
- Alotaibi, M.; Kozhevnikova, E.; Kozhevnikov, I. Hydrogenation of methyl isobutyl ketone over bifunctional Pt–zeolite catalyst. J. Catal. 2012, 293, 141–144. [Google Scholar] [CrossRef]

© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shumeiko, B.; Schlackl, K.; Kubička, D. Hydrogenation of Bio-Oil Model Compounds over Raney-Ni at Ambient Pressure. Catalysts 2019, 9, 268. https://doi.org/10.3390/catal9030268
Shumeiko B, Schlackl K, Kubička D. Hydrogenation of Bio-Oil Model Compounds over Raney-Ni at Ambient Pressure. Catalysts. 2019; 9(3):268. https://doi.org/10.3390/catal9030268
Chicago/Turabian StyleShumeiko, Bogdan, Klaus Schlackl, and David Kubička. 2019. "Hydrogenation of Bio-Oil Model Compounds over Raney-Ni at Ambient Pressure" Catalysts 9, no. 3: 268. https://doi.org/10.3390/catal9030268
APA StyleShumeiko, B., Schlackl, K., & Kubička, D. (2019). Hydrogenation of Bio-Oil Model Compounds over Raney-Ni at Ambient Pressure. Catalysts, 9(3), 268. https://doi.org/10.3390/catal9030268