1. Introduction
To enable a shift away from fossil fuels to clean-energy alternatives with reduced environmental impact, it is important to search for safe, efficient and economical energy conversion technologies that can be commercialized. Fuel cells are electrochemical devices that directly convert stored chemical energy into electricity with high efficiency. They have received a significant amount of attention for many applications, including transportation, where the proton exchange membrane fuel cell (PEMFC) has dominated the market. However, the broad commercialization of PEMFCs has been hindered by high cost, which is partially caused by the use of platinum group metal (PGM) catalysts at both the anode and cathode [
1]. In recent years, anion exchange membrane fuel cells (AEMFCs) have been proposed as a potentially lower-cost alternative to PEMFCs. It is believed that the alkaline electrolyte would make it possible to use non-PGM electrocatalysts from a broader selection of materials that are unstable in acid as well as enable lower cost hardware and membranes. This combination of non-PGM catalysts with potentially low-cost membranes and other cell components may allow AEMFCs to be much lower cost than PEMFCs at scale.
Although discovering non-PGM catalysts for both fuel cell reactions is important, a majority of the work to date has focused on the oxygen reduction reaction (ORR), which is the cathode reaction not only for AEMFCs, but other technologies as well, such as metal air batteries. This suggests that advancements made at the AEMFC cathode could have a wide-reaching impact on a number of electrochemical technologies [
2]. Researchers have developed non-PGM ORR catalysts with a wide array of chemistries and structures [
3,
4,
5]. Several groups have reported Fe- and Co-doped catalysts derived from the high temperature annealing of a polymeric precursor [
6,
7], resulting in highly porous or hierarchical structures, but with many active sites buried within the bulk material or pores that are unreachable by the ionomer. Such catalysts have often shown very high ex-situ ORR activity [
6,
7], but one limitation of these materials has been how they are implemented into electrodes. Hence, a majority of non-PGM electrocatalysts have not been successfully transitioned to operating AEMFCs due to the inability of researchers to create properly structured electrodes with sufficient mass transport, porosity, electronic conductivity and catalyst layer (CL) thickness [
8], with a few notable exceptions [
8,
9,
10]. Another potential issue with these materials has been their low durability, and their likely high production cost is also potentially problematic.
Another class of catalysts that has been explored is carbon-supported metal oxide nanoparticles [
11]. Recently, success has been reported using cobalt and manganese oxides grown on a N-doped graphene support [
12], creating covalent coupling between spinel oxide nanoparticles and N-doped graphene oxide sheets, which yielded higher alkaline ORR activity than commercial Pt/C. However, one of the limitations of the graphene approach is that graphene layers are prone to stacking during electrode fabrication. Hence, an expensive, hard-to-scale freeze-drying step was needed to form a 3D porous electrode [
12]. A recently successful approach that showed very high catalyst activity, AEMFC performance and durability was to create platelet-structured catalysts where the N-doped carbon was grown between templated metal oxide particles [
9]. Not only did the N-C-CoO
x catalyst in that study show excellent intrinsic activity, the electrode-level mass transport was also enhanced by integrating a powder-based ionomer during electrode fabrication, which was a previously successful approach with another non-PGM cathode [
10]. Although the creation of the N-C-CoO
x catalyst did not require any exotic reagents, like nearly all other high-activity non-PGM catalysts it does require high-temperature processing that may be undesirable in some manufacturing environments.
In this study, we have created a highly active non-PGM catalyst at very low temperature through the solvothermal synthesis of cobalt ferrite nanoparticles supported by Vulcan carbon (CF-VC), which is an easily scalable approach. CF particles were targeted because it is believed that their magnetic nature would facilitate the magneto hydrodynamic movement of the reactants, resulting in high ORR activity [
13]. Vulcan carbon was chosen as the catalyst support as it provides a unique combination of high surface area, high electrical conductivity and mesoporous structure, which can be beneficial for reducing electron transfer and mass transport resistances when integrated into fuel cell catalyst layers. The CF-VC catalyst was first characterized through a combination of imaging and X-ray diffraction (XRD). The catalyst was then deposited onto a rotating disk electrode (RDE) where its activity was probed ex-situ. Next, the catalyst was integrated with a powder ionomer [
14] to create gas-diffusion electrodes and tested for its ORR performance in operating AEMFCs.
2. Results and Discussion
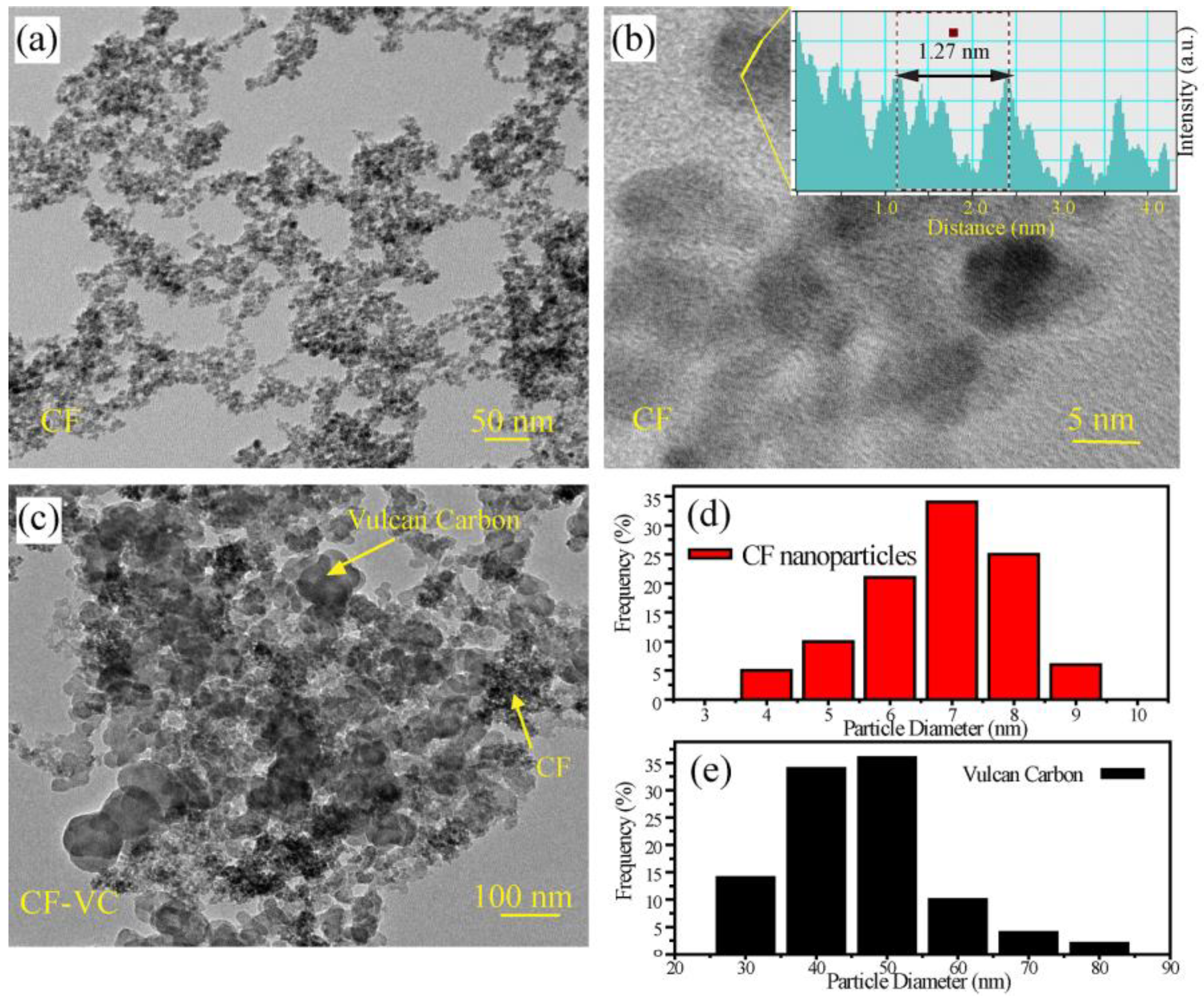
The morphology, structure and size distribution of the CF nanoparticles were analyzed by transmission electron microscopy (TEM) and the resulting images are presented in
Figure 1. The ferrite nanoparticles were spherical in structure and well distributed. Because they are magnetic in nature, they are generally restricted to individual particles. Hence, despite their close association, very little agglomeration was observed. A magnified image of the nanoparticles is presented in
Figure 1b and a corresponding line profile analysis is shown in the inset. Careful analysis of the CF particles showed a d-spacing of 0.254 nm, corresponding to the (311) plane of the cobalt ferrite (0.252 nm) [
15]. The small variation in the d-spacing can be explained by the presence of oxygen vacancies and crystal relaxation due to the nanoparticle nature of the CF.
In order to create the final catalyst, the CF nanoparticles were physically mixed with Vulcan XC-72R carbon (50:50 wt%) and annealed at 150 °C for 12 h. Transmission electron microscopy (TEM) analysis of the final CF-VC catalyst clearly showed the distribution of the CF nanoparticles and the Vulcan carbon (
Figure 1c). The particle size distribution of the CF and Vulcan carbon were determined through analysis of multiple images (
Figure S1 in the supporting information) using the Digital Micrograph software, which are presented in
Figure 1d,e. The CF nanoparticles and Vulcan carbon had a primary size distribution of 6–8 nm and 40–50 nm, respectively. The annealing of the cobalt ferrite nanoparticles with Vulcan carbon provides an electrically conducting network, which allows for the active material to be better utilized and the apparent ORR activity to be improved. Also, the spherical shape of the Vulcan carbon does not allow for the extensive catalyst stacking and densification that can occur with graphene-based catalysts.
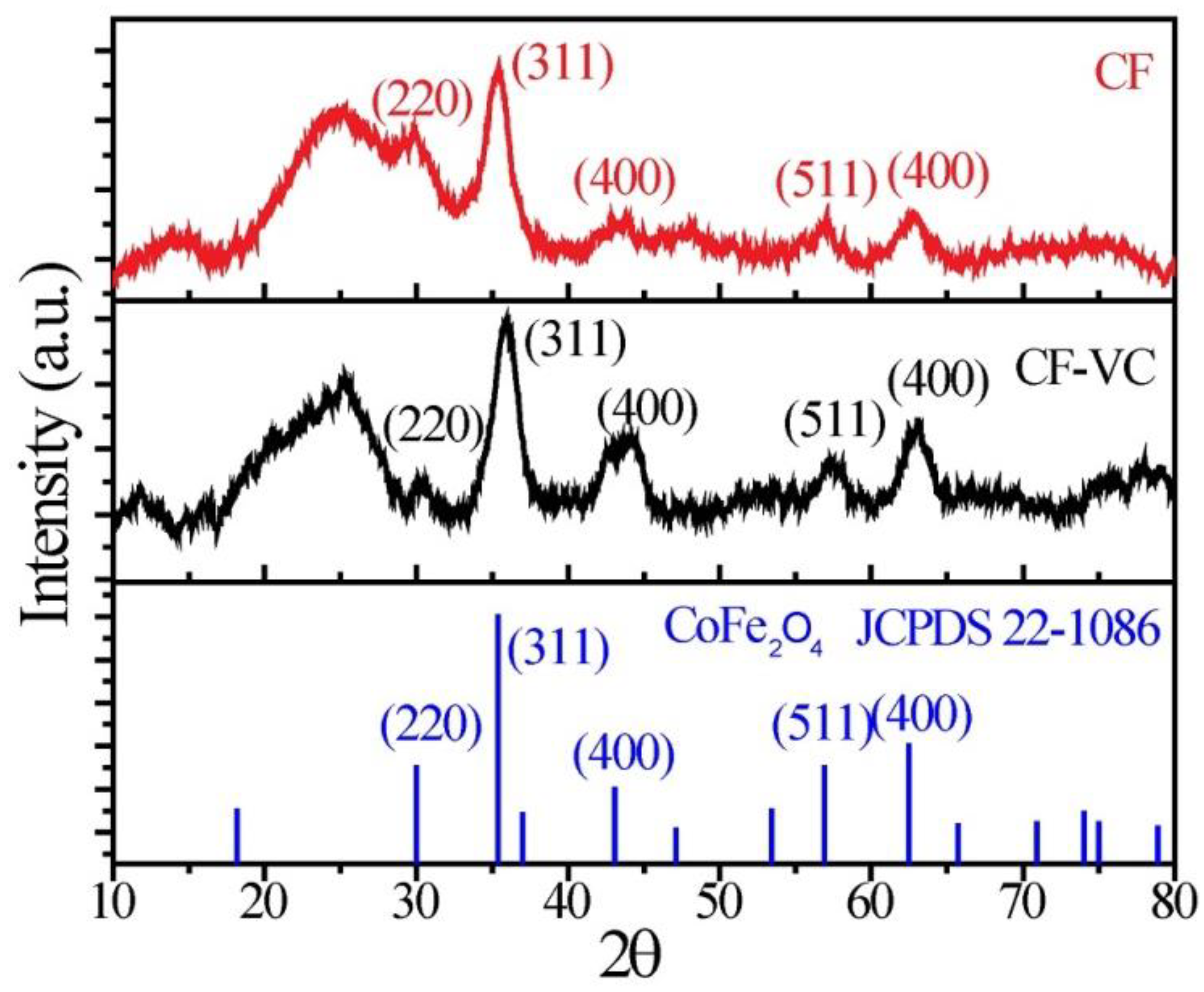
Powder X-ray diffraction (XRD) patterns for raw CF and CF-VC are presented in
Figure 2. Being nanocrystalline, as observed in the TEM images, the XRD peaks were generally broad, although the characteristic peak for spinel cobalt ferrite (JCPDS 22-1086) at 35.3 2θ for the (311) plane was well resolved. Peaks corresponding to the (400), (511) and (440) reflections were also clearly observed at 43.8, 57.2 and 62.8 2θ, respectively. Comparing the XRD pattern of the raw CF and the CF-VC, the intensity of each of the individual peaks was increased in the CF-VC catalyst, most likely due to the additional heat treatment, which increased the CF crystallinity.
Information regarding the catalyst surface was obtained using X-ray photoelectron spectroscopy (XPS), with a summary of the results presented in
Figure 3. Broad survey spectra for both CF and CF-VC,
Figure 3a, revealed peaks corresponding to C 1s, O 1s, Fe 2p and Co 2p at 282.7, 531, 711.2 and 785 eV, respectively [
16]. From the survey spectra, it was clear that the O 1s intensity was higher for CF and the C 1s intensity was higher for the CF-VC catalyst. Significant changes were observed to the oxygen-binding environment for the CF by annealing with Vulcan carbon, which can be shown in the high resolution O1s spectra (
Figure 3b). In the deconvolution, O
L represents the lattice oxygen, O
V denotes oxygen vacancies and O
MC signifies metal carbonates (formed during the solvothermal process) [
17]. It appears that the annealing of the CF nanoparticles decomposed the metal carbonates and, consequently, the relative O
MC intensity was decreased in the CF-VC compared to CF. The deconvoluted Co 2p and Fe 2p spectra are presented in
Figure 3c and
Figure 3d, respectively. The peaks corresponding to the Co and Fe atoms were less intense in the CF-VC catalyst, as half of the catalyst mass was Vulcan carbon. The binding energy for Co
3+ and Co
2+ in CF were observed at 780.2 and 782.3 eV [
18]. In the CF-VC catalyst, Co
3+ and Co
2+ appeared at very similar binding energies, 780.2 and 781.8 eV, respectively. The annealing of CF with Vulcan carbon changed the Co
3+/Co
2+ratio, which is clearly shown in the area occupied by the two states in the deconvoluted spectra (
Figure 3c). This change in the oxidation state was not limited to cobalt; a significant change in the iron oxidation state was also observed. Peaks for Fe
2+ and Fe
3+ were observed at 709.7 and 710.8 eV in the CF nanocrystals. However, in the CF-VC, the peak positions were slightly shifted to 709.8 and 711.2 eV. The peak areas and peak widths for each of the metal oxidation states are given in
Table S1. Moreover, the change in the oxidation state of the cobalt and iron—from the XPS peak intensity and area—show structural changes in the nanocrystals. The Fe
+3/Fe
+2 ratio also changed from CF to CF-VC, also as a result of annealing. The change in oxidation states after annealing makes perfect sense. Because the annealing decomposed the surface carbonates, oxygen vacancies were formed that—because of the need for crystal neutrality—led to the change in the oxidation state of the metal atoms.
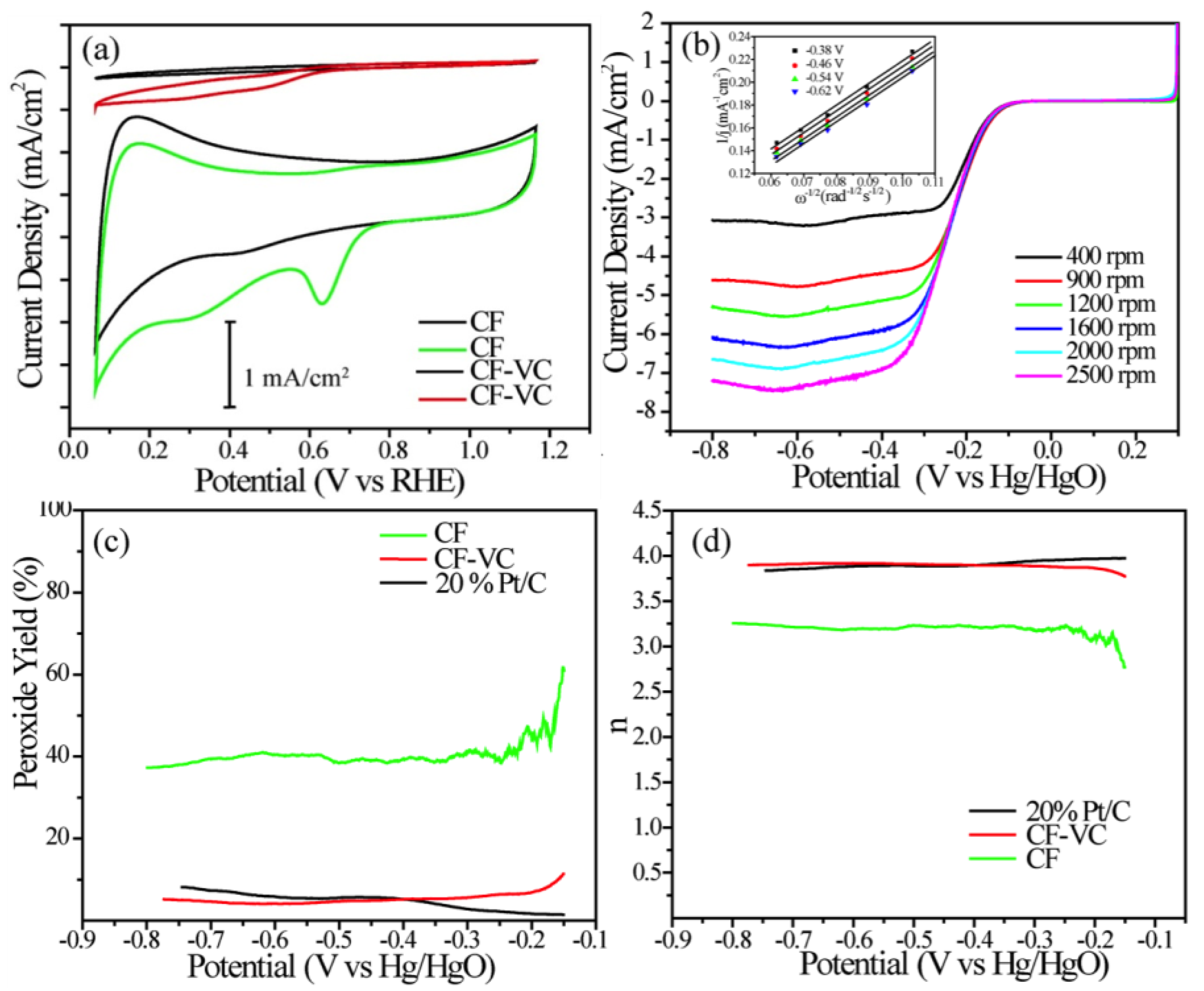
The intrinsic activity of the catalysts towards the oxygen reduction reaction was investigated electrochemically using a combination of cyclic voltammetry (CV) and linear sweep voltammetry (LSV) on RDEs and rotating ring disk electrodes (RRDEs). The electrolyte used in this work was 0.1 M KOH, and the experiments were done in nitrogen- and oxygen-saturated environments sequentially (
Figure 4). The CVs for CF and CF-VC clearly showed two characteristic processes at high potentials: pseudocapacitance from the high surface area carbon and oxygen reduction. The CF catalyst showed very little overall activity for the oxygen reduction reaction, which is evidenced by both the low current densities in the CVs (
Figure 4a) and the high peroxide yield (
Figure 4c). On the other hand, the CF-VC catalyst showed a significantly improved voltammetric response. First, the electrode capacitance was greatly enhanced due to the presence of the Vulcan carbon. Second, a clear ORR peak emerged at 0.62 V (vs. the reversible hydrogen electrode, RHE), which is likely enabled by both higher electronic conductivity (increasing catalyst utilization) and surface modification from the annealing step.
Because of the enhanced voltammetric response and a clear ORR peak, the behavior of the CF-VC catalyst was also probed by LSV under rotation, and the results are shown in
Figure 4b. Electrode rotation is an important part of the ex situ kinetic evaluation of ORR catalysts because it enables homogenous oxygen accessibility over the catalyst surface, removing any uncertainties in the experimental data related to mass transport, allowing pure kinetic information to be extracted from the RDE and RRDE data. The first kinetic parameters that were extracted were the onset and half-wave potentials—0.78 and 0.71 V, respectively. It should be noted that these values are ca. 100 mV negative of current state-of-the-art PGM-free ORR catalysts [
19,
20,
21,
22], although they are still very good relative to most PGM-free catalysts reported in the literature. RDE data was collected at multiple rotation rates and a Kouteckey–Levich (K–L) analysis was performed to probe the reaction mechanism. A K–L plot, which relates the inverse of the current density at various potentials to the square root of the rotation rate of the working electrode [
23,
24], is shown as an inset of
Figure 4b. The K–L plot clearly showed both linearity and parallelism over a wide potential range, showing that the ORR on these materials is first order with respect to oxygen and have a near constant number of electrons transferred per oxygen atom. The dominant pathway for the ORR on the CF-VC catalyst was further probed using a RRDE (
Figure 4c,d) [
10]. One advantage of RRDE over an RDE is that it is possible to quantify the amount of unwanted peroxide that forms from the two electron reduction of oxygen. High peroxide yields can degrade both the carbon support and membrane in the operating fuel cell; hence, it is desirable to reduce the amount of peroxide produced for device durability and to have catalysts that facilitate the complete four electron reduction of oxygen (n = 4). Positively, the CF-VC catalyst showed nearly a four electron reduction of oxygen over the entire potential window, with peroxide yields as low as commercial Pt/C.
Next, the CF-VC catalysts were incorporated into gas diffusion electrodes (GDEs). To do this, the catalyst was mixed with a poly(ethylenetetrafluoroethylene) (ETFE) solid powder ionomer [
14] (20 wt% ionomer), dispersed in solvent and sprayed onto Toray 60 gas diffusion layers as described in our previous publications [
9,
25,
26,
27]. The ETFE powder was used as the ionomer to support (along with the Vulcan carbon support) the formation of a porous electrode with facile product and reactant mass transport, which is important for AEMFC performance [
26]. Scanning electron microscope (SEM) images of the CF-VC GDEs (
Figure 5a,b) showed a uniform distribution of catalyst and ionomer particles as well as a very porous architecture. The ETFE ionomer appeared to be very well integrated with the CF-VC catalyst (
Figure 5c), suggesting that these electrodes are likely to have a well-formed triple-phase boundary in operating AEMFCs. An energy-dispersive x-ray spectroscopy (EDS) fluorine elemental map for a typical catalyst-covered ionomer particle is shown in
Figure 5d, which clearly shows the size, structure and distribution of the solid anionomer in the electrode.
The CF-VC GDEs were used as the cathode electrode in laboratory scale 5 cm
2, single-cell AEMFCs. The CF-VC catalyst loading on the GDE was 2.4 mg
CF/cm
2. The anode electrode consisted of a PtRu/C catalyst and ETFE ionomer (20 wt% ionomer). The PtRu loading on the anode was very low, only 0.07 mg
PtRu/cm
2. The membrane used in this work was a radiation-grafted polyethylene-based film with a covalently-bound benzyltrimethylammonium (BTMA) cationic head-groups (25 μm; ion exchange capacity, IEC = 2.44 ± 0.04 mmol g
−1). The development of this class of AEM was first reported in 2017 [
28]. The operating AEMFCs were fed with pure H
2 and O
2 reacting gases, and the resulting CF-VC containing GDE was able to achieve a peak power density of 1350 mW cm
−2 with a peak current density over 4 A cm
−2 (
Figure 6a), which is the best performance for an AEMFC with a non-PGM cathode reported in the literature to date [
8,
9,
10,
20,
29,
30]. Using H
2 and air (CO
2-free) reacting gases, the CF-VC non-PGM cathode GDE was able to achieve a peak power density of 670 mW cm
−2 (
Figure 6b). Compared to state-of-the-art non-PGM AEMFCs in the literature (
Figure S2) [
8,
9,
10,
29], the CF-VC cell in this work showed greatly enhanced single-cell performance—despite lower intrinsic activity. The most impressive metrics for the CF-VC based AEMFC was its peak power density and mass transport limited current density. Again, both were the highest values reported in the literature to date, which was most likely due to the formation of a porous electrode architecture created by the combination of Vulcan carbon [
9] and the solid powder anionomer, allowing for facile mass transport.
3. Materials and Methods
Chemicals: iron(II) acetate and cobalt(II) acetate tetrahydrate were purchased from Sigma-Aldrich (Delhi, India). Absolute ethanol and isopropyl alcohol were purchased from Thomas Baker (Delhi, India) and were used without any further purification. Deionized (DI) water was produced by a Millipore Milli-Q unit.
Synthesis of the cobalt ferrite (CF) nanoparticles: for the synthesis of the cobalt ferrite nanoparticles, cobalt acetate and iron acetate salts were dissolved in a 1:2 ratio in a solution of 1:1 (by volume) water-ethanol. 50 mL of the resulting solution was placed in a 100 mL autoclave, which was heated to 130 °C and maintained at that temperature for 12 h. A solid precipitate was obtained and separated from the solution supernatant by centrifugation. The solid was washed with a copious amount of ethanol. The sample was dried at 60 °C and the resulting cobalt ferrite (CF) nanoparticles [
12] were used without any further treatment.
Synthesis of the cobalt ferrite/Vulcan carbon (CF-VC) catalyst: CF nanoparticles were first dispersed in a 3:2 water–isopropyl alcohol (IPA) solution. Then, Vulcan XC-72R carbon was added to the dispersion (1:1 ratio of CF and Vulcan carbon by weight), followed by sonication for 1 h. After this ensured that the CF and Vulcan carbon were well mixed, the solvent was evaporated and the solids were annealed at 150 °C for 12 h in air.
Glassy carbon RDE ink preparation and deposition: 10 mg of the catalyst was dispersed in 1 mL of a water–IPA solution (3:2) followed by the addition of 40 μL of 5% Nafion dispersion. 10 μL of the resulting ink was placed onto a glassy carbon RDE (0.196 cm2) and dried under an infrared (IR) lamp.
Material characterization: transmission electron microscopy (TEM) was performed using a FEI TECHNI (Delhi, India) G2 F20 instrument. The instrument was operated at 200 kV and has a resolution of 1.7 Å (Cs = 0.6 mm). Before TEM analysis, samples were drop-coated over a Cu grid and dried under an IR lamp. Powder X-ray diffraction analysis was performed with a PAN (Delhi, India) analytical X’pert Pro instrument equipped with a Cu Kα (λ = 1.54 Å) X-ray source. The samples were scanned for 30 min between 10 and 80 degrees (2θ). X-ray photoelectron spectroscopy (XPS) was performed with a Thermo Fisher Scientific (Delhi, India) instrument (Model: K ALPHA+) equipped with Mg Kα X-ray source (hν = 1.2356 keV). All of the RDE electrochemical studies were electrochemically controlled using a Biologic (Delhi, India) VMp3 instrument.
Koutecky–Levich analysis: the average number of electrons transferred during the ORR (
n) was calculated from the Koutecky–Levich equations:
where
j is the measured current density,
jK is the kinetic current density,
jL is the mass transport limited current density at a given rotation rate,
is the angular velocity of the rotating disk,
F is Faraday’s constant (96485.3 C/mol),
Co is the bulk concentration of O
2 in 0.1 M KOH at room temperature (1.2 × 10
−6 mol/cm
3),
Do is the diffusion coefficient of O
2 in 0.1 M KOH at room temperature (1.9 × 10
−5 cm
2/s), and
is the kinematic viscosity for 0.1 M KOH (0.01 cm
2/s).
Rotating ring-disk electrode (RRDE) measurements were made in the same RDE setup as mentioned above. The disk electrode was rotated at 1600 rpm with scan rate of 10 mV/s. The ring electrode potential was set to 1.1 V vs. RHE. The hydrogen peroxide yield (%H
2O
2) and average number of electrons transferred (
n) were calculated by the following equations:
where
id and
ir are the disk and ring current densities, N is the ring H
2O
2 collection efficiency, which was 37%.
GDE fabrication and AEMFC testing: First, the ETFE-g-poly(VBTMAC) powder anionomer (IEC = 1.24 ± 0.06 mmol/g) [
14] was ground with a well-cleaned mortar and pestle for 10 min to reduce the number of aggregated particles. Next, the catalyst and 1 mL of DI water was added to the ground anionomer and ground for an additional 10 min until a visually and texturally homogeneous slurry was formed. The ETFE powder mass comprised 20% of the total solid mass of all of the catalyst layers (CLs) in this study. After the slurry was homogenized, 1.5 mL of IPA was added into the mortar followed by another 5 min grinding. A final 5 mL of IPA was added to the mortar and the final ink mixture was transferred to a PTFE-lined vial and sonicated for 1 h in an ice bath. The prepared ink was then sprayed onto the gas diffusion layer (GDL, Toray 60, 5% PTFE wetproofing) using an air-assisted sprayer (Iwata Eclipse HP CS dual action airbrush gun; ANEST IWATA, Yokohama, Japan) to fabricate the GDEs. A Pt-Ru catalyst (Alfa Aesar, Haverhill, MA, USA, HiSPEC 10000, Pt nominally 40 wt%, and Ru, nominally 20 wt%, supported on Vulcan XC-72R carbon) was used at the anode. The anode GDEs, cathode GDEs and membrane were hydrated in DI water for 20 min and then soaked three times in aqueous 1.0 M KOH for a total of 1 h to remove impurities and ion exchange the quaternary ammonium hydroxide groups before cell assembly.
AEMFCs with 5 cm
2 active area were assembled in single-cell hardware with a single channel, serpentine flow field. The anion exchange membranes used in this work were radiation-grafted polyethylene-based films with a covalently-bound benzyltrimethylammonium (BTMA) cationic head-groups (25 μm, IEC = 2.44 ± 0.04 mmol g
−1). The average thickness of the anode and cathode were measured to be 216 and 270 μm, respectively. Thus, 152 and 203 μm Teflon gaskets were used on the anode and cathode, respectively, to keep the cell pinch at around 25% of the total GDE thickness. The AEMFCs were tested using a Scribner 850e fuel cell test station at a cell temperature of 65 °C under H
2/O
2 or H
2/Air flow at 1.0 L/min. The cell was broken in at a voltage of 0.5 V and the relative humidity (RH) of both the cathode and anode were adjusted to allow the cell to be operated at optimal conditions [
26].

 ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}





