Fe-Doping in Double Perovskite PrBaCo2(1-x)Fe2xO6-δ: Insights into Structural and Electronic Effects to Enhance Oxygen Evolution Catalyst Stability

 , ,
, ,  , ,
, ,
Abstract
1. Introduction
2. Results and Discussion
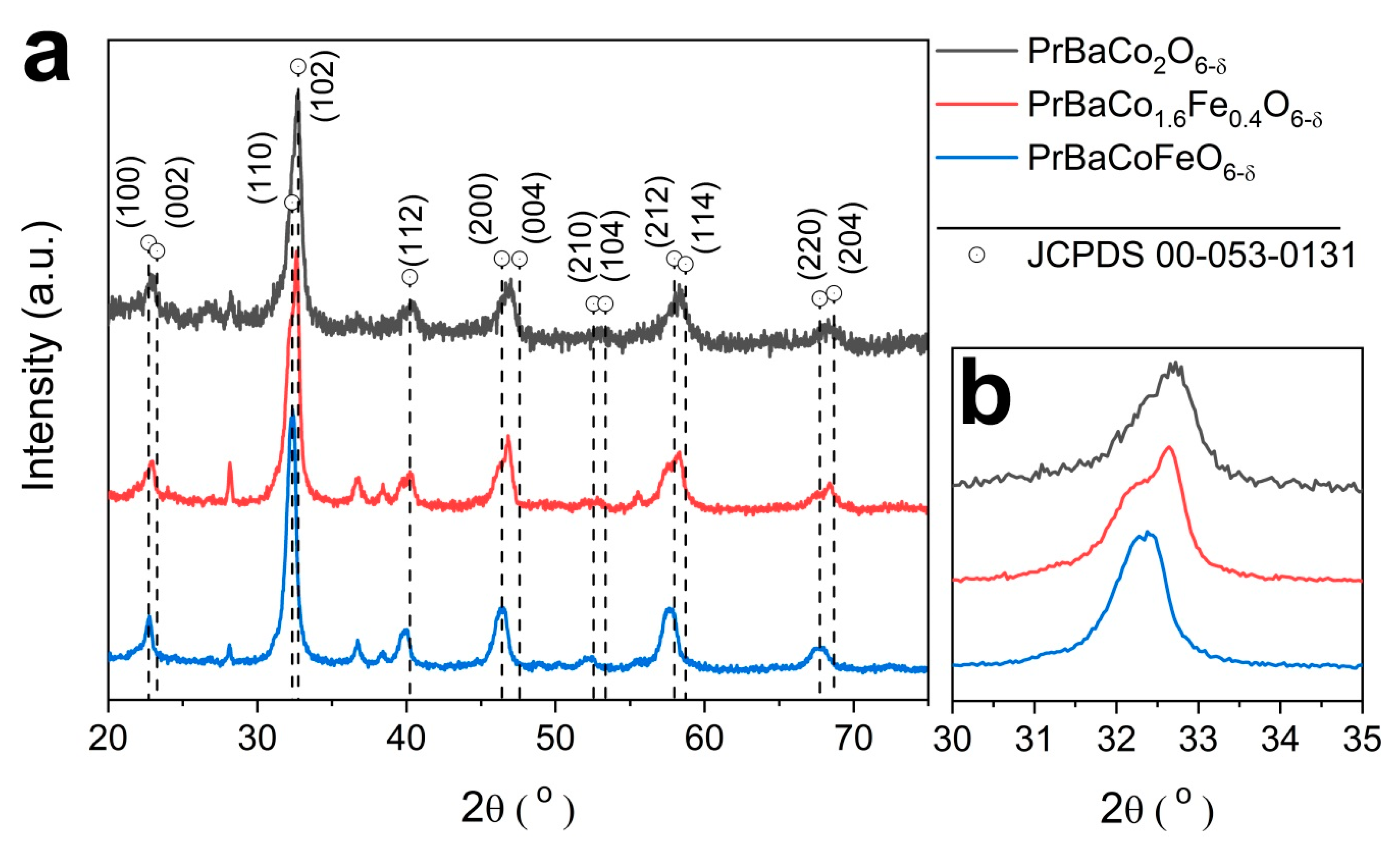
2.1. Physcial Characterization
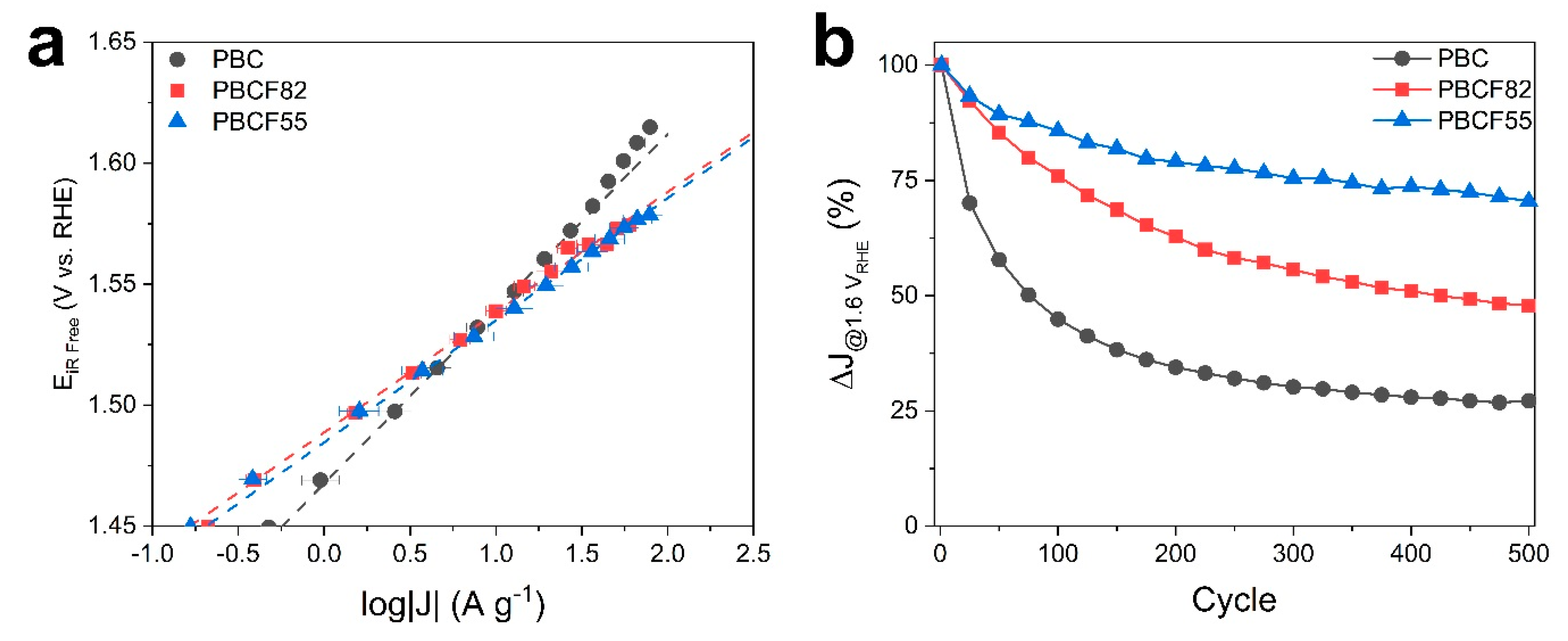
2.2. Electrochemical Study
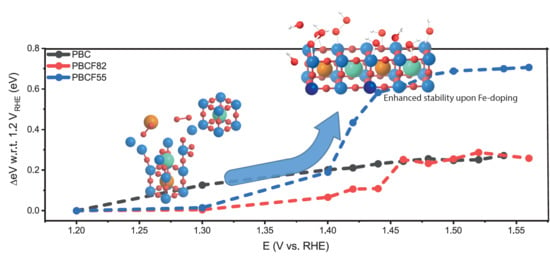
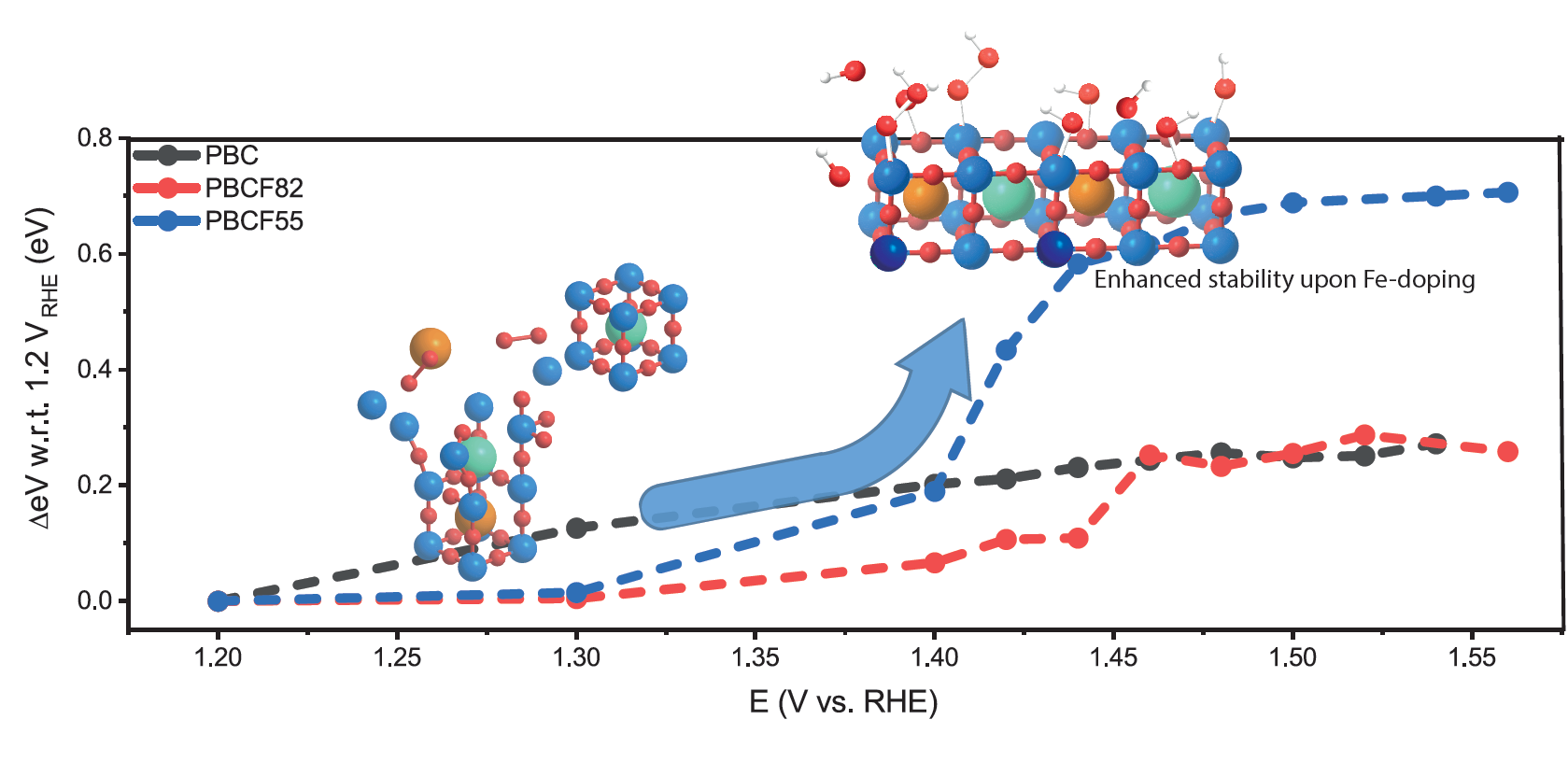
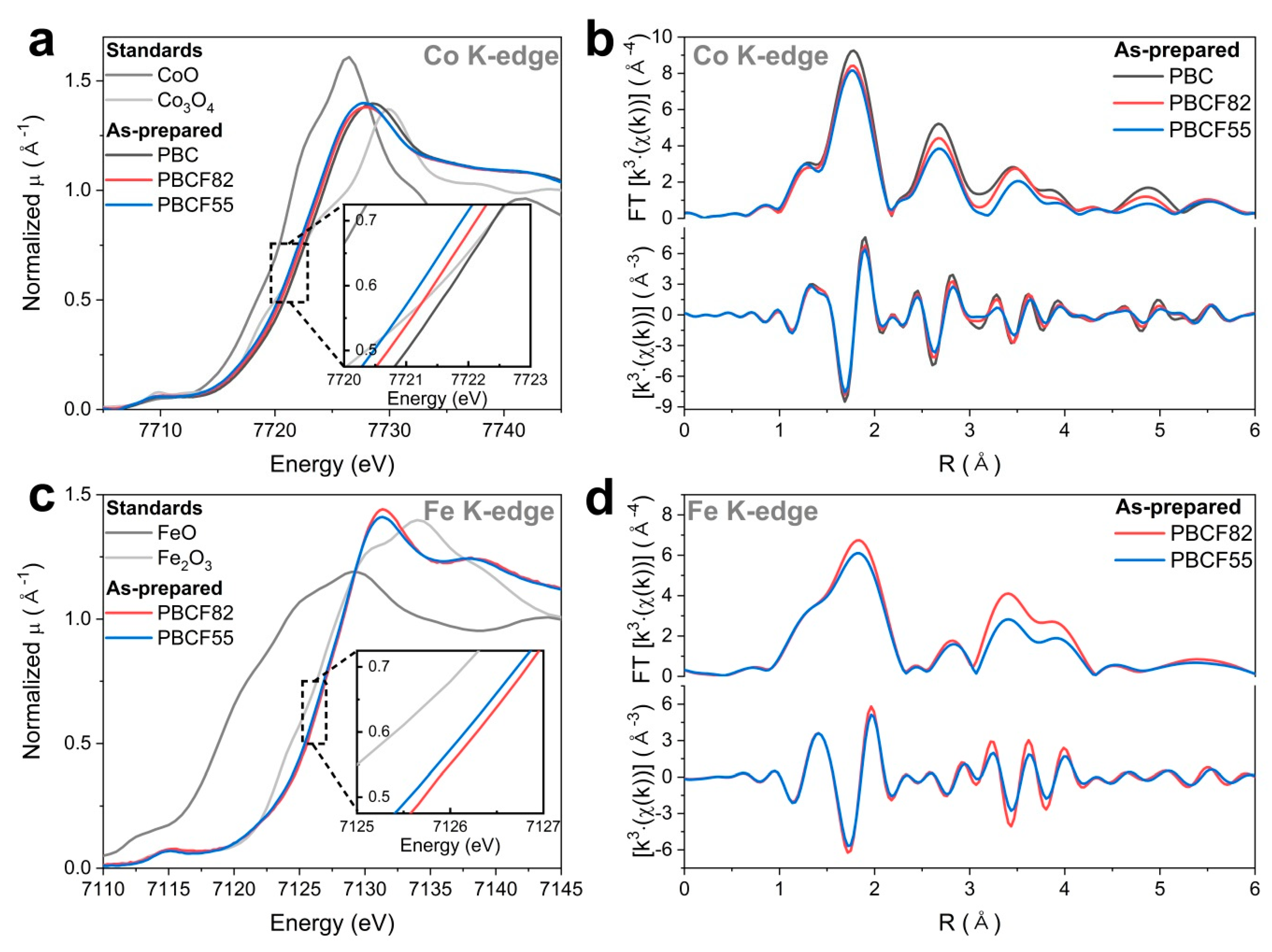
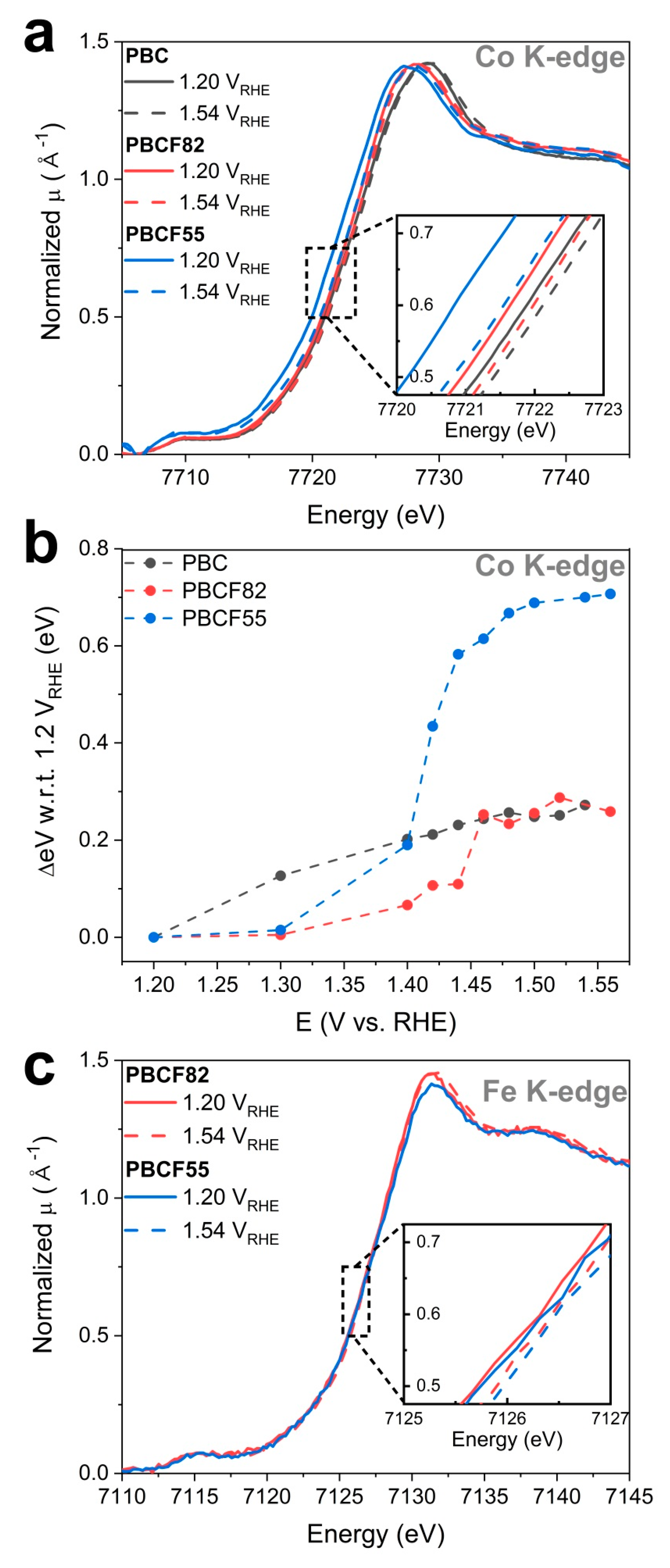
2.3. Operando X-ray Absorption Spectroscopy Study
3. Materials and Methods
3.1. Material Synthesis
3.2. Material Characterizations
3.3. Electrochemical Characterization
3.4. Operando Flow Cell Study
3.5. Density-Functional Theory—Pourbaix Diagrams
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Babic, U.; Suermann, M.; Buehi, F.N.; Gubler, L.; Schmidt, T.J. Review-identifying critical gaps for polymer electrolyte water electrolysis development. J. Electrochem. Soc. 2017, 164, F387–F399. [Google Scholar] [CrossRef]
- Kim, B.-J.; Cheng, X.; Abbott, D.F.; Fabbri, E.; Bozza, F.; Graule, T.; Castelli, I.E.; Wiles, L.; Danilovic, N.; Ayers, K.E.; et al. Highly active nanoperovskite catalysts for oxygen evolution reaction: Insights into activity and stability of ba0.5sr0.5co0.8fe0.2o2+δ and prbaco2o5+δ. Adv. Funct. Mater. 2018, 28, 1804355. [Google Scholar] [CrossRef]
- Cheng, X.; Fabbri, E.; Yamashita, Y.; Castelli, I.E.; Kim, B.; Uchida, M.; Haumont, R.; Puente-Orench, I.; Schmidt, T.J. Oxygen evolution reaction on perovskites: A multieffect descriptor study combining experimental and theoretical methods. ACS Catal. 2018, 8, 9567–9578. [Google Scholar] [CrossRef]
- Suntivich, J.; Gasteiger, H.A.; Yabuuchi, N.; Shao-Horn, Y. Electrocatalytic measurement methodology of oxide catalysts using a thin-film rotating disk electrode. J. Electrochem. Soc. 2010, 157, B1263–B1268. [Google Scholar] [CrossRef]
- Grimaud, A.; May, K.J.; Carlton, C.E.; Lee, Y.L.; Risch, M.; Hong, W.T.; Zhou, J.G.; Shao-Horn, Y. Double perovskites as a family of highly active catalysts for oxygen evolution in alkaline solution. Nat. Commun. 2013, 4, 2439. [Google Scholar] [CrossRef] [PubMed]
- Huang, X.B.; Zhao, G.X.; Wang, G.; Irvine, J.T.S. Synthesis and applications of nanoporous perovskite metal oxides. Chem. Sci. 2018, 9, 3623–3637. [Google Scholar] [CrossRef] [PubMed]
- Hwang, J.; Rao, R.R.; Giordano, L.; Katayama, Y.; Yu, Y.; Shao-Horn, Y. Perovskites in catalysis and electrocatalysis. Science 2017, 358, 751–756. [Google Scholar] [CrossRef] [PubMed]
- Vasala, S.; Karppinen, M. A2b′b″o6 perovskites: A review. Prog. Solid State Chem. 2015, 43, 1–36. [Google Scholar] [CrossRef]
- Fabbri, E.; Habereder, A.; Waltar, K.; Kotz, R.; Schmidt, T.J. Developments and perspectives of oxide-based catalysts for the oxygen evolution reaction. Catal. Sci. Technol. 2014, 4, 3800–3821. [Google Scholar] [CrossRef]
- King, G.; Woodward, P.M. Cation ordering in perovskites. J. Mater. Chem. 2010, 20, 5785–5796. [Google Scholar] [CrossRef]
- Davies, P.K.; Wu, H.; Borisevich, A.Y.; Molodetsky, I.E.; Farber, L. Crystal chemistry of complex perovskites: New cation-ordered dielectric oxides. Annu. Rev. Mater. Res. 2008, 38, 369–401. [Google Scholar] [CrossRef]
- Fabbri, E.; Schmidt, T.J. Oxygen evolution reaction—the enigma in water electrolysis. ACS Catal. 2018, 8, 9765–9774. [Google Scholar] [CrossRef]
- Grimaud, A.; Diaz-Morales, O.; Han, B.H.; Hong, W.T.; Lee, Y.L.; Giordano, L.; Stoerzinger, K.A.; Koper, M.T.M.; Shao-Horn, Y. Activating lattice oxygen redox reactions in metal oxides to catalyse oxygen evolution. Nat. Chem. 2017, 9, 457–465. [Google Scholar] [CrossRef] [PubMed]
- Taskin, A.A.; Lavrov, A.N.; Ando, Y. Achieving fast oxygen diffusion in perovskites by cation ordering. Appl. Phys. Lett. 2005, 86, 091910. [Google Scholar] [CrossRef]
- Kim, G.; Wang, S.; Jacobson, A.J.; Yuan, Z.; Donner, W.; Chen, C.L.; Reimus, L.; Brodersen, P.; Mims, C.A. Oxygen exchange kinetics of epitaxial prbaco(2)o(5+delta) thin films. Appl. Phys. Lett. 2006, 88. [Google Scholar] [CrossRef]
- Man, I.C.; Su, H.Y.; Calle-Vallejo, F.; Hansen, H.A.; Martinez, J.I.; Inoglu, N.G.; Kitchin, J.; Jaramillo, T.F.; Norskov, J.K.; Rossmeisl, J. Universality in oxygen evolution electrocatalysis on oxide surfaces. ChemCatChem 2011, 3, 1159–1165. [Google Scholar] [CrossRef]
- Rossmeisl, J.; Qu, Z.W.; Zhu, H.; Kroes, G.J.; Norskov, J.K. Electrolysis of water on oxide surfaces. J. Electroanal. Chem. 2007, 607, 83–89. [Google Scholar] [CrossRef]
- Damjanovic, A.; Jovanovic, B. Anodic oxide-films as barriers to charge-transfer in o2 evolution at pt in acid solutions. J. Electrochem. Soc. 1976, 123, 374–381. [Google Scholar] [CrossRef]
- Han, B.; Shao-Horn, Y. (invited) in-situ study of the activated lattice oxygen redox reactions in metal oxides during oxygen evolution catalysis. Meeting Abstracts 2018, MA2018-01, 1935. [Google Scholar]
- Yoo, J.S.; Rong, X.; Liu, Y.; Kolpak, A.M. Role of lattice oxygen participation in understanding trends in the oxygen evolution reaction on perovskites. ACS Catal. 2018, 8, 4628. [Google Scholar] [CrossRef]
- Hong, W.T.; Stoerzinger, K.A.; Lee, Y.L.; Giordano, L.; Grimaud, A.; Johnson, A.M.; Hwang, J.; Crumlin, E.J.; Yang, W.L.; Shao-Horn, Y. Charge-transfer-energy-dependent oxygen evolution reaction mechanisms for perovskite oxides. Energ. Environ. Sci. 2017, 10, 2190–2200. [Google Scholar] [CrossRef]
- Suntivich, J.; May, K.J.; Gasteiger, H.A.; Goodenough, J.B.; Shao-Horn, Y. A perovskite oxide optimized for oxygen evolution catalysis from molecular orbital principles. Science 2011, 334, 1383–1385. [Google Scholar] [CrossRef] [PubMed]
- Han, B.H.; Risch, M.; Lee, Y.L.; Ling, C.; Jia, H.F.; Shao-Horn, Y. Activity and stability trends of perovskite oxides for oxygen evolution catalysis at neutral ph. Phys. Chem. Chem. Phys. 2015, 17, 22576–22580. [Google Scholar] [CrossRef] [PubMed]
- Kim, B.; Fabbri, E.; Abbott, D.F.; Cheng, X.; Clark, A.H.; Nachtegaal, M.; Borlaf, M.; Castelli, I.E.; Graule, T.; Schmidt, T.J. Functional role of fe-doping in co-based perovskite oxide cata-lysts for oxygen evolution reaction. J. Am. Chem. Soc. 2019. accepted. [Google Scholar]
- May, K.J.; Carlton, C.E.; Stoerzinger, K.A.; Risch, M.; Suntivich, J.; Lee, Y.L.; Grimaud, A.; Shao-Horn, Y. Influence of oxygen evolution during water oxidation on the surface of perovskite oxide catalysts. J. Phys. Chem. Lett. 2012, 3, 3264–3270. [Google Scholar] [CrossRef]
- Risch, M.; Grimaud, A.; May, K.J.; Stoerzinger, K.A.; Chen, T.J.; Mansour, A.N.; Shao-Horn, Y. Structural changes of cobalt-based perovskites upon water oxidation investigated by exafs. J. Phys. Chem. C 2013, 117, 8628–8635. [Google Scholar] [CrossRef]
- Fabbri, E.; Nachtegaal, M.; Binninger, T.; Cheng, X.; Kim, B.; Durst, J.; Bozza, F.; Graule, T.; Schäublin, R.; Wiles, L.; et al. Dynamic surface self-reconstruction is the key of highly active perovskite nano-electrocatalysts for water splitting. Nat. Mater. 2017, 16, 925–932. [Google Scholar] [CrossRef] [PubMed]
- Suntivich, J.; Gasteiger, H.A.; Yabuuchi, N.; Nakanishi, H.; Goodenough, J.B.; Shao-Horn, Y. Design principles for oxygen-reduction activity on perovskite oxide catalysts for fuel cells and metal-air batteries. Nat. Chem. 2011, 3, 546–550. [Google Scholar] [CrossRef] [PubMed]
- Burke, M.S.; Kast, M.G.; Trotochaud, L.; Smith, A.M.; Boettcher, S.W. Cobalt-iron (oxy)hydroxide oxygen evolution electrocatalysts: The role of structure and composition on activity, stability, and mechanism. J. Am. Chem. Soc. 2015, 137, 3638–3648. [Google Scholar] [CrossRef] [PubMed]
- Gong, M.; Li, Y.G.; Wang, H.L.; Liang, Y.Y.; Wu, J.Z.; Zhou, J.G.; Wang, J.; Regier, T.; Wei, F.; Dai, H.J. An advanced ni-fe layered double hydroxide electrocatalyst for water oxidation. J. Am. Chem. Soc. 2013, 135, 8452–8455. [Google Scholar] [CrossRef] [PubMed]
- Zou, S.H.; Burke, M.S.; Kast, M.G.; Fan, J.; Danilovic, N.; Boettcher, S.W. Fe (oxy)hydroxide oxygen evolution reaction electrocatalysis: Intrinsic activity and the roles of electrical conductivity, substrate, and dissolution. Chem. Mater. 2015, 27, 8011–8020. [Google Scholar] [CrossRef]
- Trotochaud, L.; Young, S.L.; Ranney, J.K.; Boettcher, S.W. Nickel-iron oxyhydroxide oxygen-evolution electrocatalysts: The role of intentional and incidental iron incorporation. J. Am. Chem. Soc. 2014, 136, 6744–6753. [Google Scholar] [CrossRef] [PubMed]
- Dionigi, F.; Strasser, P. Nife-based (oxy)hydroxide catalysts for oxygen evolution reaction in non-acidic electrolytes. Adv. Energy Mater. 2016, 6, 1600621. [Google Scholar] [CrossRef]
- Chen, J.Y.C.; Dang, L.N.; Liang, H.F.; Bi, W.L.; Gerken, J.B.; Jin, S.; Alp, E.E.; Stahl, S.S. Operando analysis of nife and fe oxyhydroxide electrocatalysts for water oxidation: Detection of fe4+ by mossbauer spectroscopy. J. Am. Chem. Soc. 2015, 137, 15090–15093. [Google Scholar] [CrossRef] [PubMed]
- Gorlin, M.; Chernev, P.; de Araujo, J.F.; Reier, T.; Dresp, S.; Paul, B.; Krahnert, R.; Dau, H.; Strasser, P. Oxygen evolution reaction dynamics, faradaic charge efficiency, and the active metal redox states of ni-fe oxide water splitting electrocatalysts. J. Am. Chem. Soc. 2016, 138, 5603–5614. [Google Scholar] [CrossRef] [PubMed]
- Gorlin, M.; de Araujo, J.F.; Schmies, H.; Bernsmeier, D.; Dresp, S.; Gliech, M.; Jusys, Z.; Chernev, P.; Kraehnert, R.; Dau, H.; et al. Tracking catalyst redox states and reaction dynamics in ni-fe oxyhydroxide oxygen evolution reaction electrocatalysts: The role of catalyst support and electrolyte ph. J. Am. Chem. Soc. 2017, 139, 2070–2082. [Google Scholar] [CrossRef] [PubMed]
- Abbott, D.F.; Fabbri, E.; Borlaf, M.; Bozza, F.; Schäublin, R.; Nachtegaal, M.; Graule, T.; Schmidt, T.J. Operando x-ray absorption investigations into the role of fe in the electrochemical stability and oxygen evolution activity of ni1−xfexoy nanoparticles. J. Mater. Chem. A 2018, 6, 24534–24549. [Google Scholar] [CrossRef]
- Abbott, D.F.; Meier, M.; Meseck, G.R.; Fabbri, E.; Seeger, S.; Schmidt, T.J. Silicone nanofilament-supported mixed nickel-metal oxides for alkaline water electrolysis. J. Electrochem. Soc. 2017, 164, F203–F208. [Google Scholar] [CrossRef]
- Heel, A.; Vital, A.; Holtappels, P.; Graule, T. Flame spray synthesis and characterisation of stabilised zro2 and ceo2 electrolyte nanopowders for sofc applications at intermediate temperatures. J. Electroceram. 2009, 22, 40–46. [Google Scholar] [CrossRef]
- Han, B.H.; Grimaud, A.; Giordano, L.; Hong, W.T.; Diaz-Morales, O.; Yueh-Lin, L.; Hwang, J.; Charles, N.; Stoerzinger, K.A.; Yang, W.L.; et al. Iron-based perovskites for catalyzing oxygen evolution reaction. J. Phys. Chem. C 2018, 122, 8445–8454. [Google Scholar] [CrossRef]
- Binninger, T.; Mohamed, R.; Waltar, K.; Fabbri, E.; Levecque, P.; Kotz, R.; Schmidt, T.J. Thermodynamic explanation of the universal correlation between oxygen evolution activity and corrosion of oxide catalysts. Sci. Rep. 2015, 5, 12167. [Google Scholar] [CrossRef] [PubMed]
- Kim, B.J.; Abbott, D.F.; Cheng, X.; Fabbri, E.; Nachtegaal, M.; Bozza, F.; Castelli, I.E.; Lebedev, D.; Schaublin, R.; Coperet, C.; et al. Unraveling thermodynamics, stability, and oxygen evolution activity of strontium ruthenium perovskite oxide. ACS Catal. 2017, 7, 3245–3256. [Google Scholar] [CrossRef]
- Bick, D.S.; Kindsmuller, A.; Staikov, G.; Gunkel, F.; Muller, D.; Schneller, T.; Waser, R.; Valov, I. Stability and degradation of perovskite electrocatalysts for oxygen evolution reaction. Electrochim. Acta. 2016, 218, 156–162. [Google Scholar] [CrossRef]
- Battle, P.D.; Gibb, T.C.; Strange, R. A study of a new incommensurate phase in the system srmn1-xcoxo3-y. J. Solid State Chem. 1989, 81, 217–229. [Google Scholar] [CrossRef]
- Gibb, T.C. Evidence for an unusual phase in the perovskite-related system bacoxmn1-xo3-y from exafs spectroscopy. J. Mater. Chem. 1992, 2, 387–393. [Google Scholar] [CrossRef]
- Gangopadhayay, S.; Inerbaev, T.; Masunov, A.E.; Altilio, D.; Orlovskaya, N. Structural characterization combined with the first principles simulations of barium/strontium cobaltite/ferrite as promising material for solid oxide fuel cells cathodes and high-temperature oxygen permeation membranes. ACS Appl. Mater. Interfaces 2009, 1, 1512–1519. [Google Scholar] [CrossRef] [PubMed]
- Ganopadhyay, S.; Masunov, A.E.; Inerbaev, T.; Mesit, J.; Guha, R.K.; Sleiti, A.K.; Kapat, J.S. Understanding oxygen vacancy migration and clustering in barium strontium cobalt iron oxide. Solid State Ionics 2010, 181, 1067–1073. [Google Scholar] [CrossRef]
- Mueller, D.N.; De Souza, R.A.; Brendt, J.; Samuelis, D.; Martin, M. Oxidation states of the transition metal cations in the highly nonstoichiometric perovskite-type oxide ba0.1sr0.9co0.8fe0.2o3-delta. J. Mater. Chem. 2009, 19, 1960–1963. [Google Scholar] [CrossRef]
- Arnold, M.; Xu, Q.; Tichelaar, F.D.; Feldhoff, A. Local charge disproportion in a high-performance perovskite. Chem. Mater. 2009, 21, 635–640. [Google Scholar] [CrossRef]
- Jun, A.; Kim, J.; Shin, J.; Kim, G. Perovskite as a cathode material: A review of its role in solid-oxide fuel cell technology. ChemElectroChem 2016, 3, 511–530. [Google Scholar] [CrossRef]
- Kuklja, M.M.; Kotomin, E.A.; Merkle, R.; Mastrikov, Y.A.; Maier, J. Combined theoretical and experimental analysis of processes determining cathode performance in solid oxide fuel cells. Phys. Chem. Chem. Phys. 2013, 15, 5443–5471. [Google Scholar] [CrossRef] [PubMed]
- Glazer, A.M. Simple ways of determining perovskite structures. Acta Cryst. Sect. A 1975, 31, 756–762. [Google Scholar] [CrossRef]
- Risch, M. Perovskite electrocatalysts for the oxygen reduction reaction in alkaline media. Catalysts 2017, 7, 154. [Google Scholar] [CrossRef]
- Heel, A.; Holtappels, P.; Hug, P.; Graule, T. Flame spray synthesis of nanoscale la0.65sr0.4co0.2fe0.8o3-delta and ba0.5sr0.5co0.8fe0.2o3-delta as cathode materials for intermediate temperature solid oxide fuel cells. Fuel Cells 2010, 10, 419–432. [Google Scholar] [CrossRef]
- Muller, O.; Nachtegaal, M.; Just, J.; Lutzenkirchen-Hecht, D.; Frahm, R. Quick-exafs setup at the superxas beamline for in situ x-ray absorption spectroscopy with 10 ms time resolution. J. Synchrotron Radiat. 2016, 23, 260–266. [Google Scholar] [CrossRef] [PubMed]
- Ravel, B.; Newville, M. Athena, artemis, hephaestus: Data analysis for x-ray absorption spectroscopy using ifeffit. J. Synchrotron Radiat. 2005, 12, 537–541. [Google Scholar] [CrossRef] [PubMed]
- Delaplane, R.G.; Ibers, J.A.; Ferraro, J.R.; Rush, J.J. Diffraction and spectroscopic studies of cobaltic acid system hcoo2-dcoo2. J. Chem. Phys. 1969, 50, 1920–1927. [Google Scholar] [CrossRef]
- Schmidt, T.J.; Gasteiger, H.A.; Stab, G.D.; Urban, P.M.; Kolb, D.M.; Behm, R.J. Characterization of high-surface area electrocatalysts using a rotating disk electrode configuration. J. Electrochem. Soc. 1998, 145, 2354–2358. [Google Scholar] [CrossRef]
- Binninger, T.; Fabbri, E.; Patru, A.; Garganourakis, M.; Han, J.; Abbott, D.F.; Sereda, O.; Kotz, R.; Menzel, A.; Nachtegaal, M.; et al. Electrochemical flow-cell setup for in situ x-ray investigations i. Cell for saxs and xas at synchrotron facilities. J. Electrochem. Soc. 2016, 163, H906–H912. [Google Scholar] [CrossRef]
- Castelli, I.E.; Huser, F.; Pandey, M.; Li, H.; Thygesen, K.S.; Seger, B.; Jain, A.; Persson, K.A.; Ceder, G.; Jacobsen, K.W. New light-harvesting materials using accurate and efficient bandgap calculations. Adv. Energy Mater. 2015, 5, 1400915. [Google Scholar] [CrossRef]
- The Materials Project. Available online: https://materialsproject.org/ (accessed on 17 November 2017).
- Johnson, J.W.; Oelkers, E.H.; Helgeson, H.C. Supcrt92 - a software package for calculating the standard molal thermodynamic properties of minerals, gases, aqueous species, and reactions from 1-bar to 5000-bar and 0-degrees-c to 1000-degrees-c. Comput. Geosci. 1992, 18, 899–947. [Google Scholar] [CrossRef]
- Pourbaix, M. Atlas of Electrochemical Equilibria in Aqueous Solutions. Pergamon Press: Oxford, UK, 1966. [Google Scholar]
- Larsen, A.H.; Mortensen, J.J.; Blomqvist, J.; Castelli, I.E.; Christensen, R.; Dulak, M.; Friis, J.; Groves, M.N.; Hammer, B.; Hargus, C.; et al. The atomic simulation environment-a python library for working with atoms. J. Phys. Condens. Mat. 2017, 29, 273002. [Google Scholar]
- Persson, K.A.; Waldwick, B.; Lazic, P.; Ceder, G. Prediction of solid-aqueous equilibria: Scheme to combine first-principles calculations of solids with experimental aqueous states. Phys. Rev. B 2012, 85. [Google Scholar] [CrossRef]
- Castelli, I.E.; Thygesen, K.S.; Jacobsen, K.W. Calculated optical absorption of different perovskite phases. J. Mater. Chem. A 2015, 3, 12343–12349. [Google Scholar] [CrossRef]
- Giannozzi, P.; Baroni, S.; Bonini, N.; Calandra, M.; Car, R.; Cavazzoni, C.; Ceresoli, D.; Chiarotti, G.L.; Cococcioni, M.; Dabo, I.; et al. Quantum espresso: A modular and open-source software project for quantum simulations of materials. J. Phys. Condens. Mat. 2009, 21, 395502. [Google Scholar] [CrossRef] [PubMed]
- Perdew, J.P.; Ruzsinszky, A.; Csonka, G.I.; Vydrov, O.A.; Scuseria, G.E.; Constantin, L.A.; Zhou, X.L.; Burke, K. Restoring the density-gradient expansion for exchange in solids and surfaces. Phys. Rev. Lett. 2008, 100. [Google Scholar] [CrossRef] [PubMed]
- Standard Solid State Pseudopotential Library. Available online: http://materialscloud.org/sssp (accessed on 17 November 2017).






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Catalysts | OER | Stability | ||
|---|---|---|---|---|
| j0 | Tafel slope | j @ 1.55 VRHE | △ J | |
| (A g−1) | (mV dec−1) | (A g−1) | (%) | |
| PBC | 1.47 | 72 | 13.8 | −73% |
| PBCF82 | 1.49 | 50 | 17.1 | −52% |
| PBCF55 | 1.48 | 50 | 19.7 | −32% |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kim, B.-J.; Fabbri, E.; Castelli, I.E.; Borlaf, M.; Graule, T.; Nachtegaal, M.; Schmidt, T.J. Fe-Doping in Double Perovskite PrBaCo2(1-x)Fe2xO6-δ: Insights into Structural and Electronic Effects to Enhance Oxygen Evolution Catalyst Stability. Catalysts 2019, 9, 263. https://doi.org/10.3390/catal9030263
Kim B-J, Fabbri E, Castelli IE, Borlaf M, Graule T, Nachtegaal M, Schmidt TJ. Fe-Doping in Double Perovskite PrBaCo2(1-x)Fe2xO6-δ: Insights into Structural and Electronic Effects to Enhance Oxygen Evolution Catalyst Stability. Catalysts. 2019; 9(3):263. https://doi.org/10.3390/catal9030263
Chicago/Turabian StyleKim, Bae-Jung, Emiliana Fabbri, Ivano E. Castelli, Mario Borlaf, Thomas Graule, Maarten Nachtegaal, and Thomas J. Schmidt. 2019. "Fe-Doping in Double Perovskite PrBaCo2(1-x)Fe2xO6-δ: Insights into Structural and Electronic Effects to Enhance Oxygen Evolution Catalyst Stability" Catalysts 9, no. 3: 263. https://doi.org/10.3390/catal9030263
APA StyleKim, B.-J., Fabbri, E., Castelli, I. E., Borlaf, M., Graule, T., Nachtegaal, M., & Schmidt, T. J. (2019). Fe-Doping in Double Perovskite PrBaCo2(1-x)Fe2xO6-δ: Insights into Structural and Electronic Effects to Enhance Oxygen Evolution Catalyst Stability. Catalysts, 9(3), 263. https://doi.org/10.3390/catal9030263