Synthetic Biomimetic Coenzymes and Alcohol Dehydrogenases for Asymmetric Catalysis

 ,
,  , ,
, ,  and
and
Abstract
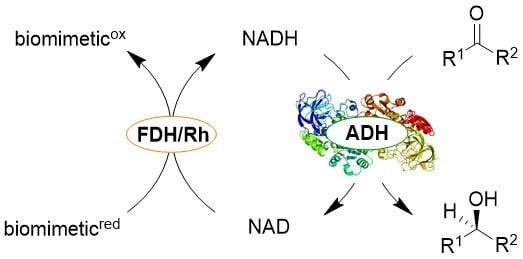
{kind=link}
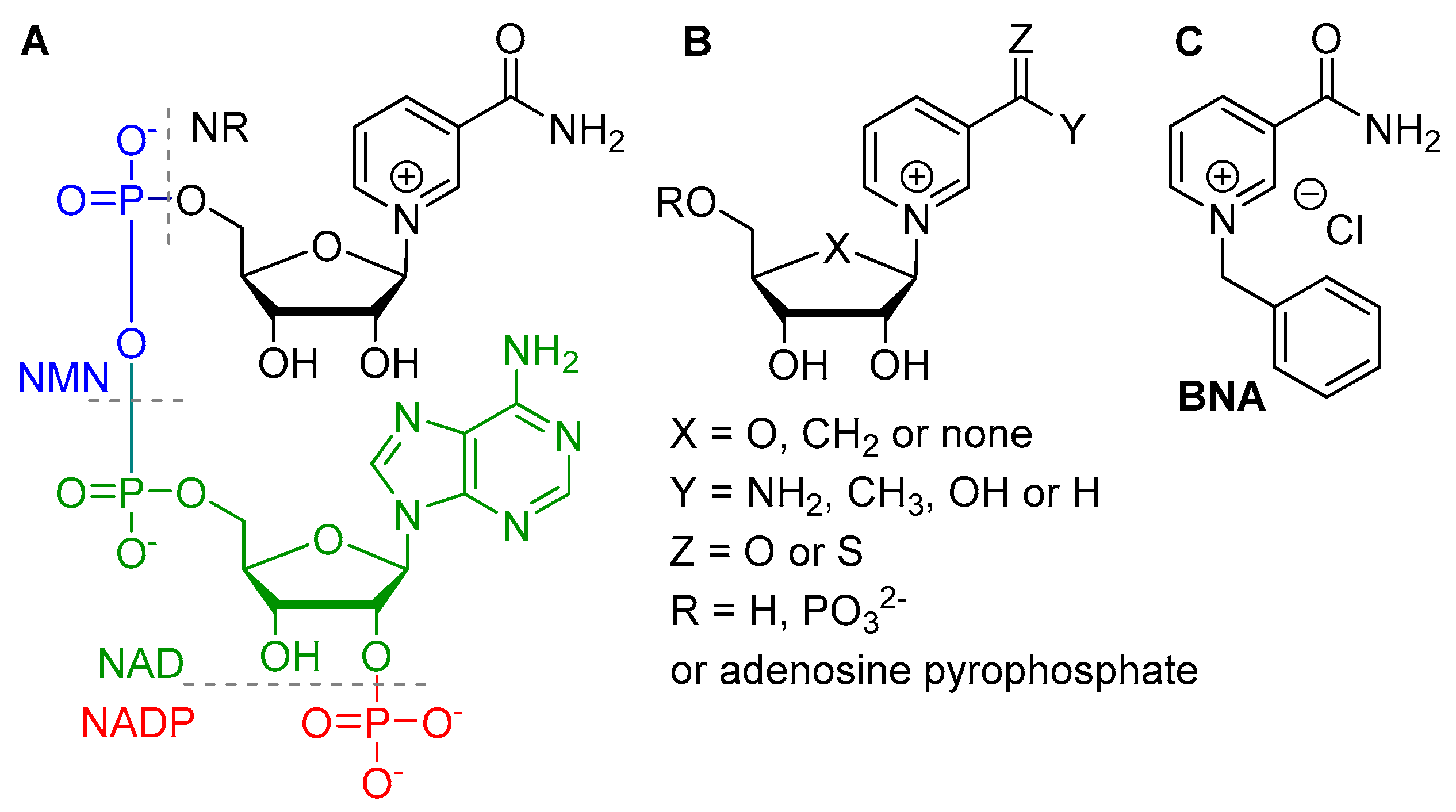{kind=link}
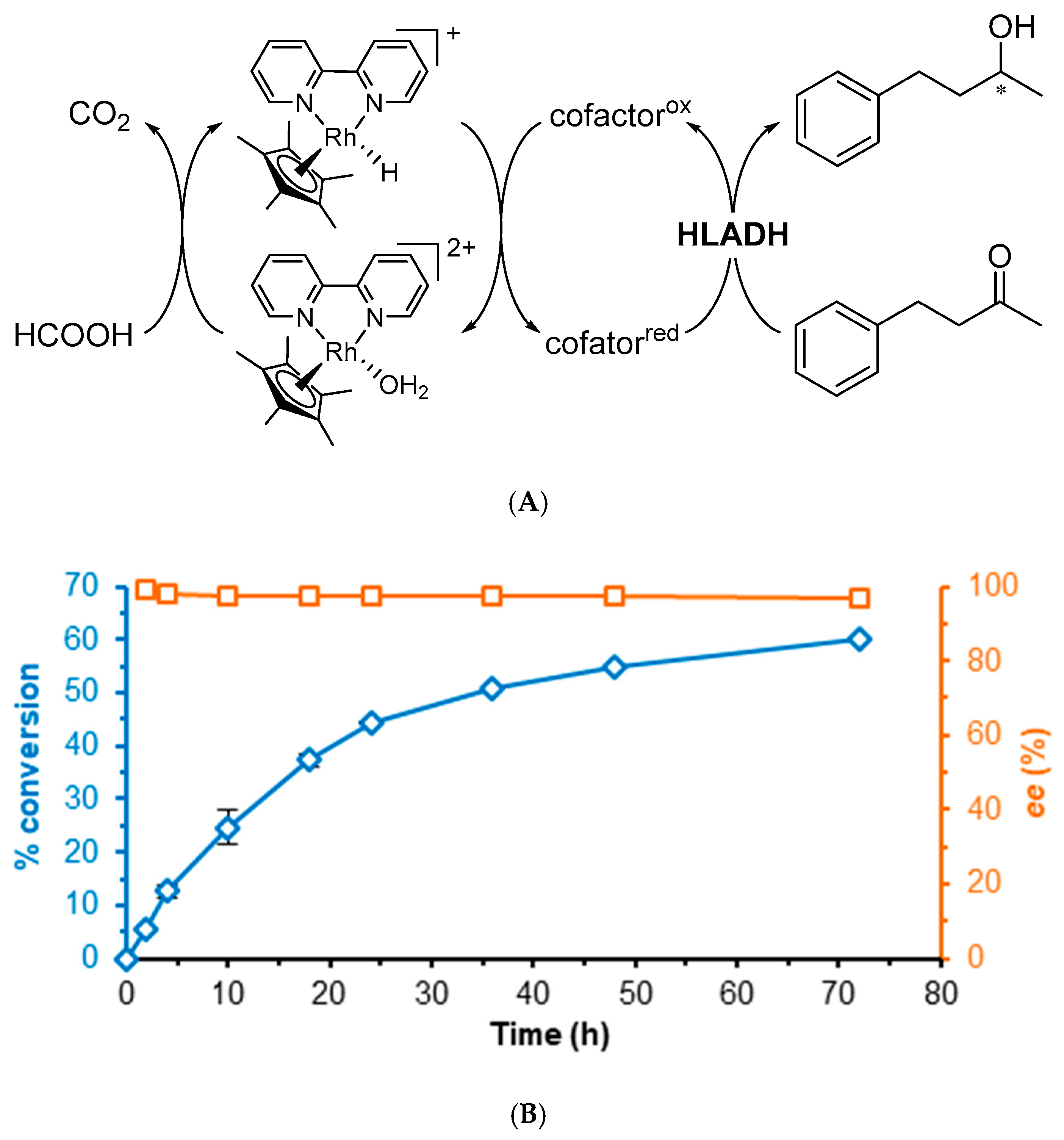{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
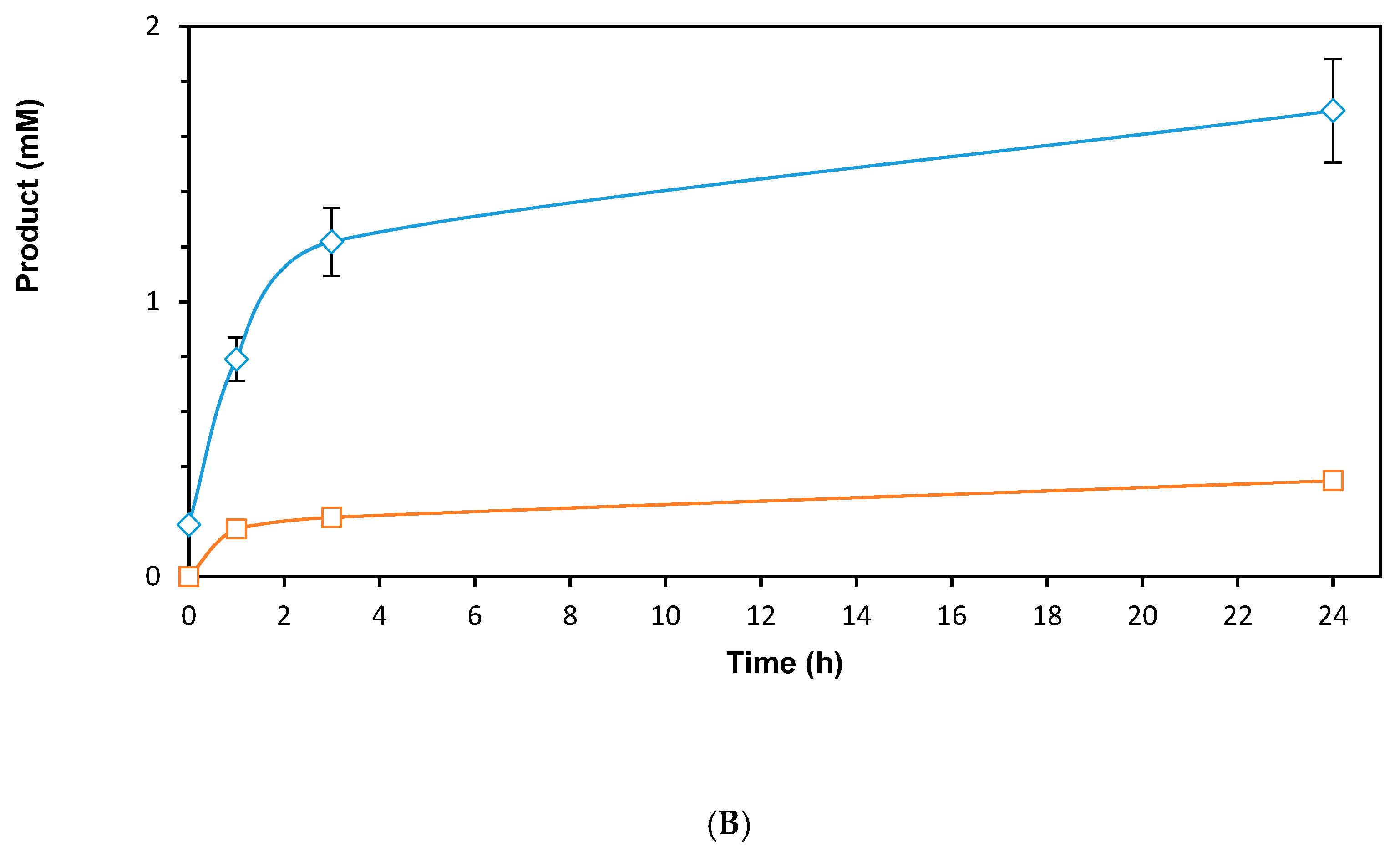
2. Results and Discussion
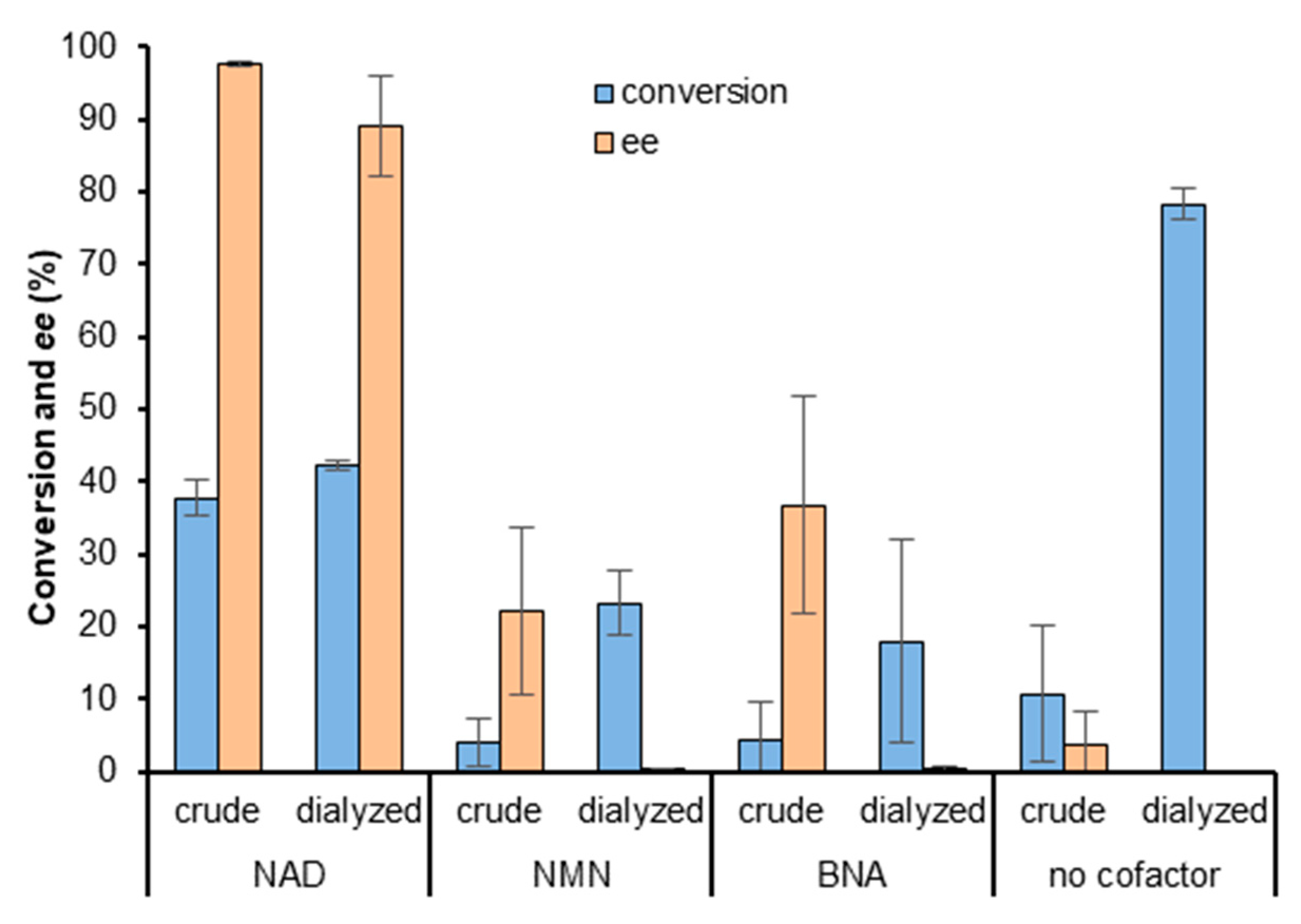
2.1. Rh-Catalyzed Cofactor Recycling
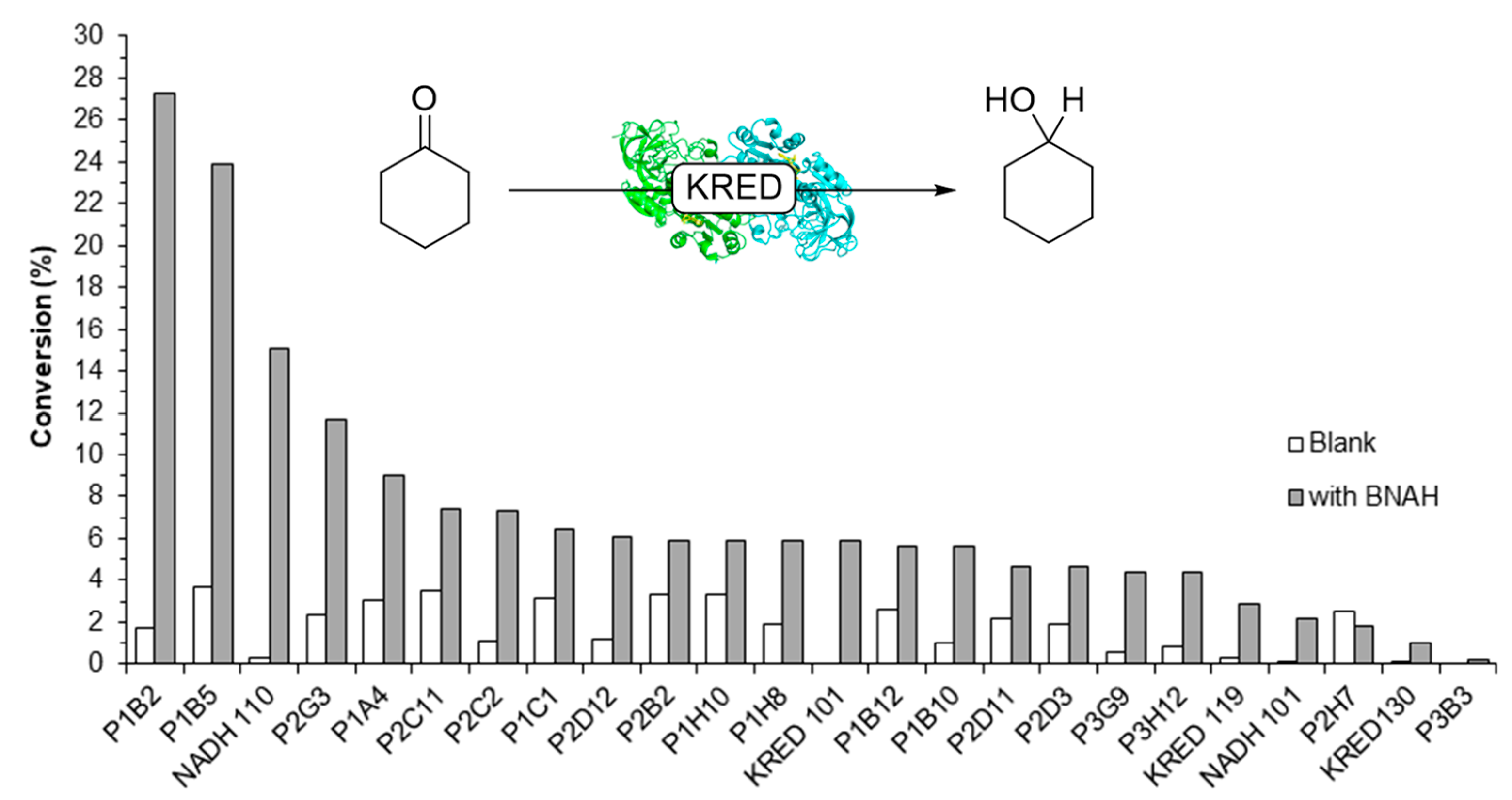
2.2. Screening of ADHs and Ketoreductases
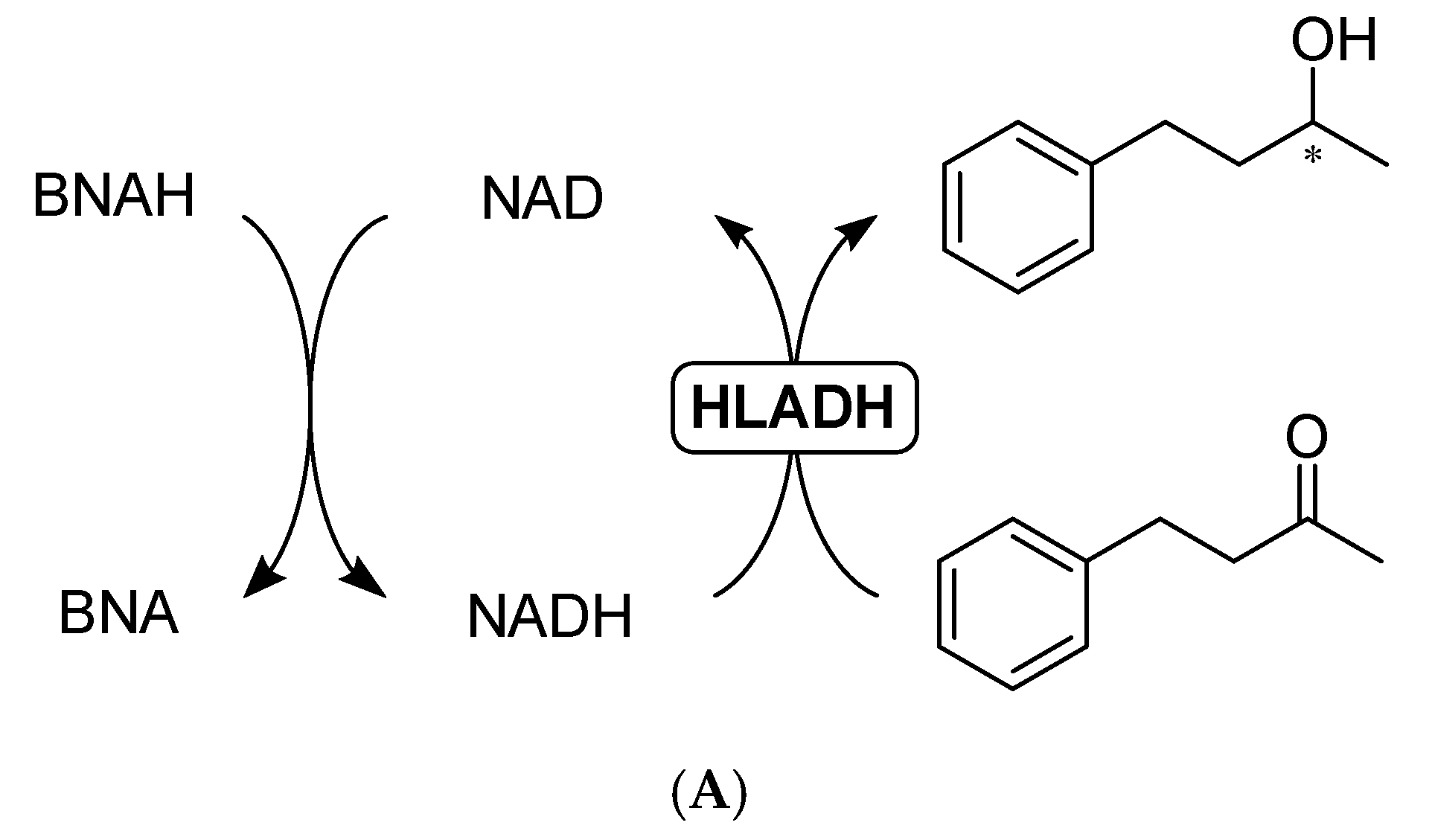
2.3. BNAH as a Hydride Donor
2.3.1. Direct Hydride Transfer to NAD
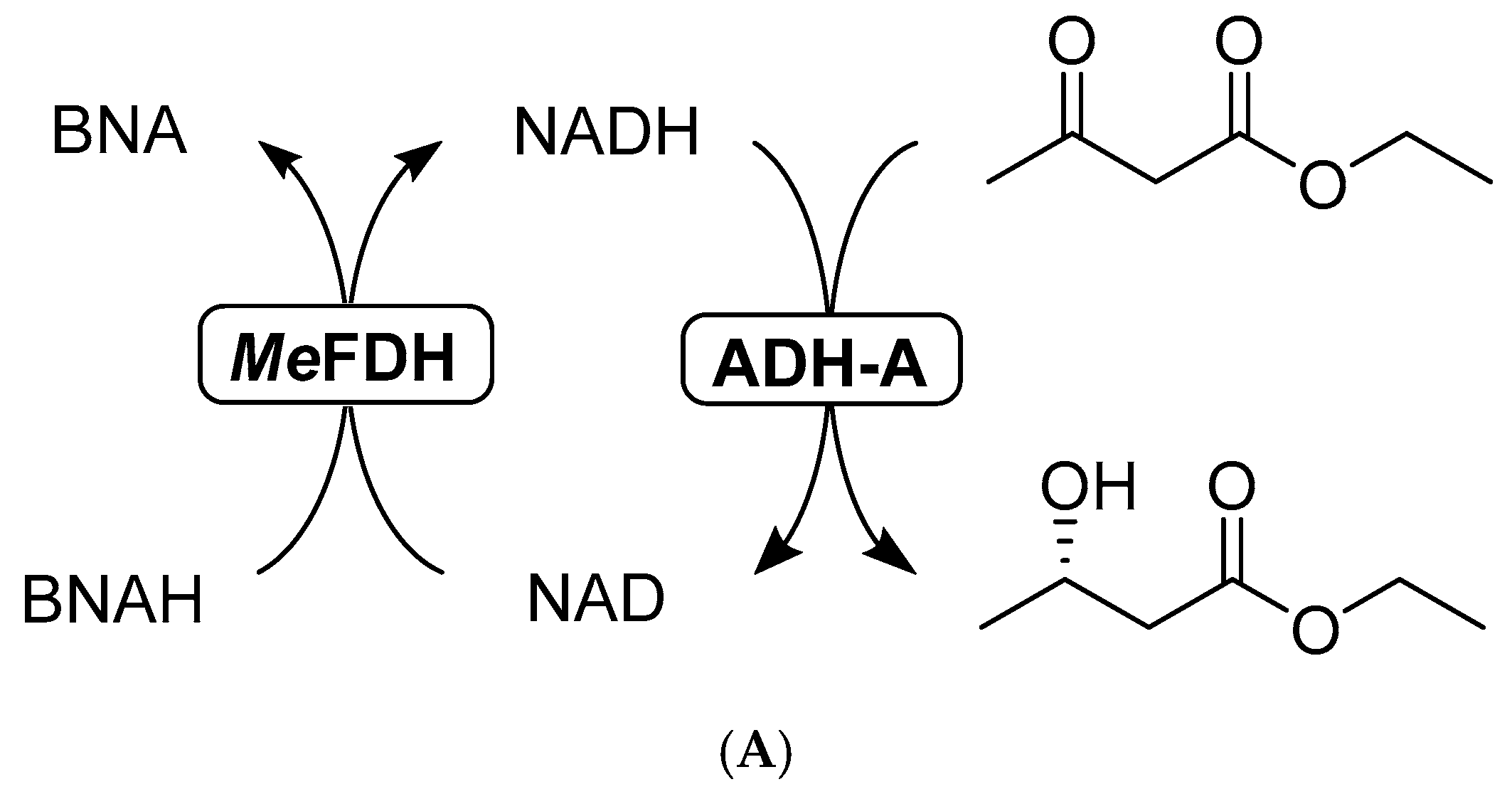
2.3.2. Formate Dehydrogenase with BNAH
3. Materials and Methods
3.1. Chemicals and Enzymes
3.2. Instrumentation
3.3. Experimental Setup
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Faber, K. Biotransformations in Organic Chemistry; Springer: Berlin, Germany, 2011; ISBN 978-3-642-17393-6. [Google Scholar]
- Gröger, H.; Hummel, W.; Borchert, S.; Kraußer, M. Reduction of ketones and aldehydes to alcohols. In Enzyme Catalysis in Organic Synthesis; Drauz, K., Gröger, H., May, O., Eds.; Wiley-VCH: Weinheim, Germany, 2012; pp. 1035–1110. [Google Scholar]
- Hollmann, F.; Bühler, K.; Bühler, B. Oxidation of alcohols, aldehydes, and acids. In Enzyme Catalysis in Organic Synthesis; Drauz, K., Gröger, H., May, O., Eds.; Wiley-VCH: Weinheim, Germany, 2012; pp. 1325–1437. [Google Scholar]
- Oppenheimer, N.J. NAD hydrolysis: Chemical and enzymatic mechanisms. Mol. Cell. Biochem. 1994, 138, 245–251. [Google Scholar] [CrossRef] [PubMed]
- Knox, R.J.; Friedlos, F.; Jarman, M.; Davies, L.C.; Goddard, P.; Anlezark, G.M.; Melton, R.G.; Sherwood, R.F. Virtual cofactors for an Escherichia coli nitroreductase enzyme—Relevance to reductively activated prodrugs in antibody directed enzyme prodrug therapy (ADEPT). Biochem. Pharmacol. 1995, 49, 1641–1647. [Google Scholar] [CrossRef]
- Friedlos, F.; Jarman, M.; Davies, L.C.; Boland, M.P.; Knox, R.J. Identification of novel reduced pyridinium derivatives as synthetic cofactors for the enzyme DT diaphorase (NAD(P)H dehydrogenase (quinone), EC 1.6.99.2). Biochem. Pharmacol. 1992, 44, 25–31. [Google Scholar] [CrossRef]
- Knox, R.J.; Jenkins, T.C.; Hobbs, S.M.; Chen, S.A.; Melton, R.G.; Burke, P.J. Bioactivation of 5-(aziridin-1-yl)-2,4-dinitrobenzamide (CB 1954) by human NAD(P)H quinone oxidoreductase 2: A novel co-substrate-mediated antitumor prodrug therapy. Cancer Res. 2000, 60, 4179–4186. [Google Scholar] [PubMed]
- Ryan, J.D.; Fish, R.H.; Clark, D.S. Engineering cytochrome P450 enzymes for improved activity towards biomimetic 1,4-NADH cofactors. ChemBioChem 2008, 9, 2579–2582. [Google Scholar] [CrossRef] [PubMed]
- Paul, C.E.; Gargiulo, S.; Opperman, D.J.; Lavandera, I.; Gotor-Fernández, V.; Gotor, V.; Taglieber, A.; Arends, I.W.C.E.; Hollmann, F. Mimicking nature: Synthetic nicotinamide cofactors for C=C bioreduction using enoate reductases. Org. Lett. 2013, 15, 180–183. [Google Scholar] [CrossRef] [PubMed]
- Knaus, T.; Paul, C.E.; Levy, C.W.; de Vries, S.; Mutti, F.G.; Hollmann, F.; Scrutton, N.S. Better than nature: Nicotinamide biomimetics that outperform natural coenzymes. J. Am. Chem. Soc. 2016, 138, 1033–1039. [Google Scholar] [CrossRef] [PubMed]
- Geddes, A.; Paul, C.E.; Hay, S.; Hollmann, F.; Scrutton, N.S. Donor-acceptor distance sampling enhances the performance of “better than Nature” nicotinamide coenzyme biomimetics. J. Am. Chem. Soc. 2016, 138, 11089–11092. [Google Scholar] [CrossRef] [PubMed]
- Löw, S.A.; Löw, I.M.; Weissenborn, M.J.; Hauer, B. Enhanced ene-reductase activity through alteration of artificial nicotinamide cofactor substituents. ChemCatChem 2016, 8, 911–915. [Google Scholar] [CrossRef]
- Scholtissek, A.; Tischler, D.; Westphal, A.H.; van Berkel, W.J.H.; Paul, C.E. Old yellow enzyme-catalysed asymmetric hydrogenation: Linking family roots with improved catalysis. Catalysts 2017, 7, 130. [Google Scholar] [CrossRef]
- Scholtissek, A.; Gadke, E.; Paul, C.E.; Westphal, A.H.; van Berkel, W.J.H.; Tischler, D. Catalytic performance of a class III old yellow enzyme and its cysteine variants. Front. Microbiol. 2018, 9, 2410. [Google Scholar] [CrossRef] [PubMed]
- Scholtissek, A.; Ullrich, S.R.; Mühling, M.; Schlömann, M.; Paul, C.E.; Tischler, D. A thermophilic-like ene-reductase originating from an acidophilic iron oxidizer. Appl. Microbiol. Biotechnol. 2017, 101, 609–619. [Google Scholar] [CrossRef] [PubMed]
- Falcone, N.; She, Z.; Syed, J.; Lough, A.; Kraatz, H.-B. Synthesis and biochemical evaluation of nicotinamide derivatives as NADH analogues in ene reductase. ChemBioChem 2019, 20. [Google Scholar] [CrossRef] [PubMed]
- Paul, C.E.; Tischler, D.; Riedel, A.; Heine, T.; Itoh, N.; Hollmann, F. Nonenzymatic regeneration of styrene monooxygenase for catalysis. ACS Catal. 2015, 5, 2961–2965. [Google Scholar] [CrossRef]
- Paul, C.E.; Hollmann, F. A survey of synthetic nicotinamide cofactors in enzymatic processes. Appl. Microbiol. Biotechnol. 2016, 100, 4773–4778. [Google Scholar] [CrossRef] [PubMed]
- Paul, C.E.; Arends, I.W.C.E.; Hollmann, F. Is simpler better? Synthetic nicotinamide cofactor analogues for redox chemistry. ACS Catal. 2014, 4, 788–797. [Google Scholar] [CrossRef]
- Hollmann, F.; Paul, C.E. Synthetische nikotinamide in der biokatalyse. BIOspektrum 2015, 21, 376–378. [Google Scholar] [CrossRef]
- Qi, J.X.; Paul, C.E.; Hollmann, F.; Tischler, D. Changing the electron donor improves azoreductase dye degrading activity at neutral pH. Enzyme Microb. Technol. 2017, 100, 17–19. [Google Scholar] [CrossRef] [PubMed]
- Lutz, J.; Hollmann, F.; Ho, T.V.; Schnyder, A.; Fish, R.H.; Schmid, A. Bioorganometallic chemistry: Biocatalytic oxidation reactions with biomimetic NAD+/NADH co-factors and [Cp*Rh (bpy) H]+ for selective organic synthesis. J. Organomet. Chem. 2004, 689, 4783–4790. [Google Scholar] [CrossRef]
- Taylor, K.E.; Jones, J.B. Nicotinamide coenzyme regeneration by dihydropyridine and pyridinium compounds. J. Am. Chem. Soc. 1976, 98, 5689–5694. [Google Scholar] [CrossRef] [PubMed]
- Jones, J.B.; Taylor, K.E. Nicotinamide coenzyme regeneration-rates of some 1,4-dihydropyridine, pyridinium salt, and flavin mononucleotide hydrogen-transfer reactions. Can. J. Chem. 1976, 54, 2974–2980. [Google Scholar] [CrossRef]
- Jones, J.B.; Taylor, K.E. Nicotinamide coenzyme regeneration-flavin mononucleotide (riboflavin phosphate) as an efficient, economical, and enzyme-compatible recycling agent. Can. J. Chem. 1976, 54, 2969–2973. [Google Scholar] [CrossRef]
- Jones, J.B.; Taylor, K.E. Use of pyridinium and flavin derivatives for recycling of catalytic amounts of NAD+ during preparative-scale horse liver alcohol dehydrogenase-catalyzed oxidations of alcohols. J. Chem. Soc. Chem. Commun. 1973, 6, 205–206. [Google Scholar] [CrossRef]
- Lo, H.C.; Fish, R.H. Biomimetic NAD+ models for tandem cofactor regeneration, horse liver alcohol dehydrogenase recognition of 1,4-NADH derivatives, and chiral synthesis. Angew. Chem. Int. Ed. 2002, 41, 478–481. [Google Scholar] [CrossRef]
- Ansell, R.J.; Lowe, C.R. Artificial redox coenzymes: Biomimetic analogues of NAD+. Appl. Microbiol. Biotechnol. 1999, 51, 703–710. [Google Scholar] [CrossRef]
- Sunderland, J.R.; Tao, X.; Butrick, E.E.; Keilich, L.C.; Villa, C.E.; Miecznikowski, J.R.; Jain, S.S. Investigation of liver alcohol dehydrogenase catalysis using an NADH biomimetic and comparison with a synthetic zinc model complex. Polyhedron 2016, 114, 145–151. [Google Scholar] [CrossRef]
- Campbell, E.; Meredith, M.; Minteer, S.D.; Banta, S. Enzymatic biofuel cells utilizing a biomimetic cofactor. Chem. Commun. 2012, 48, 1898–1900. [Google Scholar] [CrossRef] [PubMed]
- Campbell, E.; Wheeldon, I.R.; Banta, S. Broadening the cofactor specificity of a thermostable alcohol dehydrogenase using rational protein design introduces novel kinetic transient behavior. Biotechnol. Bioeng. 2010, 107, 763–774. [Google Scholar] [CrossRef] [PubMed]
- Malver, O.; Sebastian, M.J.; Oppenheimer, N.J. Alteration in substrate specificity of horse liver alcohol dehydrogenase by an acyclic nicotinamide analog of NAD. DNA Repair 2014, 23, 95–100. [Google Scholar] [CrossRef] [PubMed]
- Nowak, C.; Beer, B.; Pick, A.; Roth, T.; Lommes, P.; Sieber, V. A water-forming NADH oxidase from Lactobacillus pentosus suitable for the regeneration of synthetic biomimetic cofactors. Front. Microbiol. 2015, 6, 957. [Google Scholar] [CrossRef] [PubMed]
- Nowak, C.; Pick, A.; Csepei, L.I.; Sieber, V. Characterization of biomimetic cofactors according to stability, redox potentials, and enzymatic conversion by NADH oxidase from Lactobacillus pentosus. ChemBioChem 2017, 18, 1944–1949. [Google Scholar] [CrossRef] [PubMed]
- Nowak, C.; Pick, A.; Lommes, P.; Sieber, V. Enzymatic reduction of nicotinamide biomimetic cofactors using an engineered glucose dehydrogenase: Providing a regeneration system for artificial cofactors. ACS Catal. 2017, 7, 5202–5208. [Google Scholar] [CrossRef]
- Zachos, I.; Nowak, C.; Sieber, V. Biomimetic cofactors and methods for their recycling. Curr. Opin. Chem. Biol. 2018, 49, 59–66. [Google Scholar] [CrossRef] [PubMed]
- Sicsic, S.; Durand, P.; Langrene, S.; Legoffic, F. A new approach for using cofactor dependent enzymes—Example of alcohol-dehydrogenase. FEBS Lett. 1984, 176, 321–324. [Google Scholar] [CrossRef]
- Sicsic, S.; Durand, P.; Langrene, S.; Legoffic, F. Activity of NMN+, nicotinamide ribose and analogs in alcohol oxidation promoted by horse-liver alcohol-dehydrogenase—Improvement of this activity and structural requirements of the pyridine-nucleotide part of the NAD+ coenzyme. Eur. J. Biochem. 1986, 155, 403–407. [Google Scholar] [CrossRef] [PubMed]
- Agarwal, P.K.; Webb, S.P.; Hammes-Schiffer, S. Computational studies of the mechanism for proton and hydride transfer in liver alcohol dehydrogenase. J. Am. Chem. Soc. 2000, 122, 4803–4812. [Google Scholar] [CrossRef]
- Poizat, M.; Arends, I.W.C.E.; Hollmann, F. On the nature of mutual inactivation between [Cp*Rh (bpy) (H2O)]2+ and enzymes-analysis and potential remedies. J. Mol. Catal. B Enzym. 2010, 63, 149–156. [Google Scholar] [CrossRef]
- Srinivasan, R.; Fisher, H.F. Configurational, conformational, and solvent effects on the reduction of a Schiff-base by reduced pyridine-nucleotide analogs. Arch. Biochem. Biophys. 1983, 223, 453–457. [Google Scholar] [CrossRef]
- Eklund, H.; Nordström, B.; Zeppezauer, E.; Söderlund, G.; Ohlsson, I.; Boiwe, T.; Söderberg, B.O.; Tapia, O.; Brändén, C.-I.; Akeson, A. 3-Dimensional structure of horse liver alcohol-dehydrogenase at 2.4 Å resolution. J. Mol. Biol. 1976, 102, 27–59. [Google Scholar] [CrossRef]
- Eklund, H.; Samama, J.-P.; Wallen, L.; Brändén, C.-I.; Akeson, A.; Jones, T.A. Structure of a triclinic ternary complex of horse liver alcohol-dehydrogenase at 2.9 Å resolution. J. Mol. Biol. 1981, 146, 561–587. [Google Scholar] [CrossRef]
- Abdallah, M.A.; Biellmann, J.-F.; Nördstrom, B.; Brändén, C.-I. Conformation of adenosine diphosphoribose and 8-bromoadenosine diphosphoribose when bound to liver alcohol-dehydrogenase. Eur. J. Biochem. 1975, 50, 475–481. [Google Scholar] [CrossRef] [PubMed]
- Samama, J.-P.; Zeppezauer, E.; Biellmann, J.-F.; Brändén, C.-I. Crystal-structure of complexes between horse liver alcohol-dehydrogenase and coenzyme analogs 3-iodopyridine-adenine dinucleotide and pyridine-adenine dinucleotide. Eur. J. Biochem. 1977, 81, 403–409. [Google Scholar] [CrossRef] [PubMed]
- Plapp, B.V. Conformational changes and catalysis by alcohol dehydrogenase. Arch. Biochem. Biophys. 2010, 493, 3–12. [Google Scholar] [CrossRef] [PubMed]
- Hayward, S.; Kitao, A. Molecular dynamics simulations of NAD+-induced domain closure in horse liver alcohol dehydrogenase. Biophys. J. 2006, 91, 1823–1831. [Google Scholar] [CrossRef] [PubMed]
- Timpson, L.M.; Liliensiek, A.K.; Alsafadi, D.; Cassidy, J.; Sharkey, M.A.; Liddell, S.; Allers, T.; Paradisi, F. A comparison of two novel alcohol dehydrogenase enzymes (ADH1 and ADH2) from the extreme halophile Haloferax volcanii. Appl. Microbiol. Biotechnol. 2013, 97, 195–203. [Google Scholar] [CrossRef] [PubMed]
- Quaglia, D.; Irwin, J.A.; Paradisi, F. Horse liver alcohol dehydrogenase: New perspectives for an old enzyme. Mol. Biotechnol. 2012, 52, 244–250. [Google Scholar] [CrossRef] [PubMed]








© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Josa-Culleré, L.; Lahdenperä, A.S.K.; Ribaucourt, A.; Höfler, G.T.; Gargiulo, S.; Liu, Y.-Y.; Xu, J.-H.; Cassidy, J.; Paradisi, F.; Opperman, D.J.; et al. Synthetic Biomimetic Coenzymes and Alcohol Dehydrogenases for Asymmetric Catalysis. Catalysts 2019, 9, 207. https://doi.org/10.3390/catal9030207
Josa-Culleré L, Lahdenperä ASK, Ribaucourt A, Höfler GT, Gargiulo S, Liu Y-Y, Xu J-H, Cassidy J, Paradisi F, Opperman DJ, et al. Synthetic Biomimetic Coenzymes and Alcohol Dehydrogenases for Asymmetric Catalysis. Catalysts. 2019; 9(3):207. https://doi.org/10.3390/catal9030207
Chicago/Turabian StyleJosa-Culleré, Laia, Antti S. K. Lahdenperä, Aubert Ribaucourt, Georg T. Höfler, Serena Gargiulo, Yuan-Yang Liu, Jian-He Xu, Jennifer Cassidy, Francesca Paradisi, Diederik J. Opperman, and et al. 2019. "Synthetic Biomimetic Coenzymes and Alcohol Dehydrogenases for Asymmetric Catalysis" Catalysts 9, no. 3: 207. https://doi.org/10.3390/catal9030207
APA StyleJosa-Culleré, L., Lahdenperä, A. S. K., Ribaucourt, A., Höfler, G. T., Gargiulo, S., Liu, Y.-Y., Xu, J.-H., Cassidy, J., Paradisi, F., Opperman, D. J., Hollmann, F., & Paul, C. E. (2019). Synthetic Biomimetic Coenzymes and Alcohol Dehydrogenases for Asymmetric Catalysis. Catalysts, 9(3), 207. https://doi.org/10.3390/catal9030207