Effect of Pt Promotion on the Ni-Catalyzed Deoxygenation of Tristearin to Fuel-Like Hydrocarbons

 , and
, and 
Abstract
1. Introduction
2. Results and Discussion
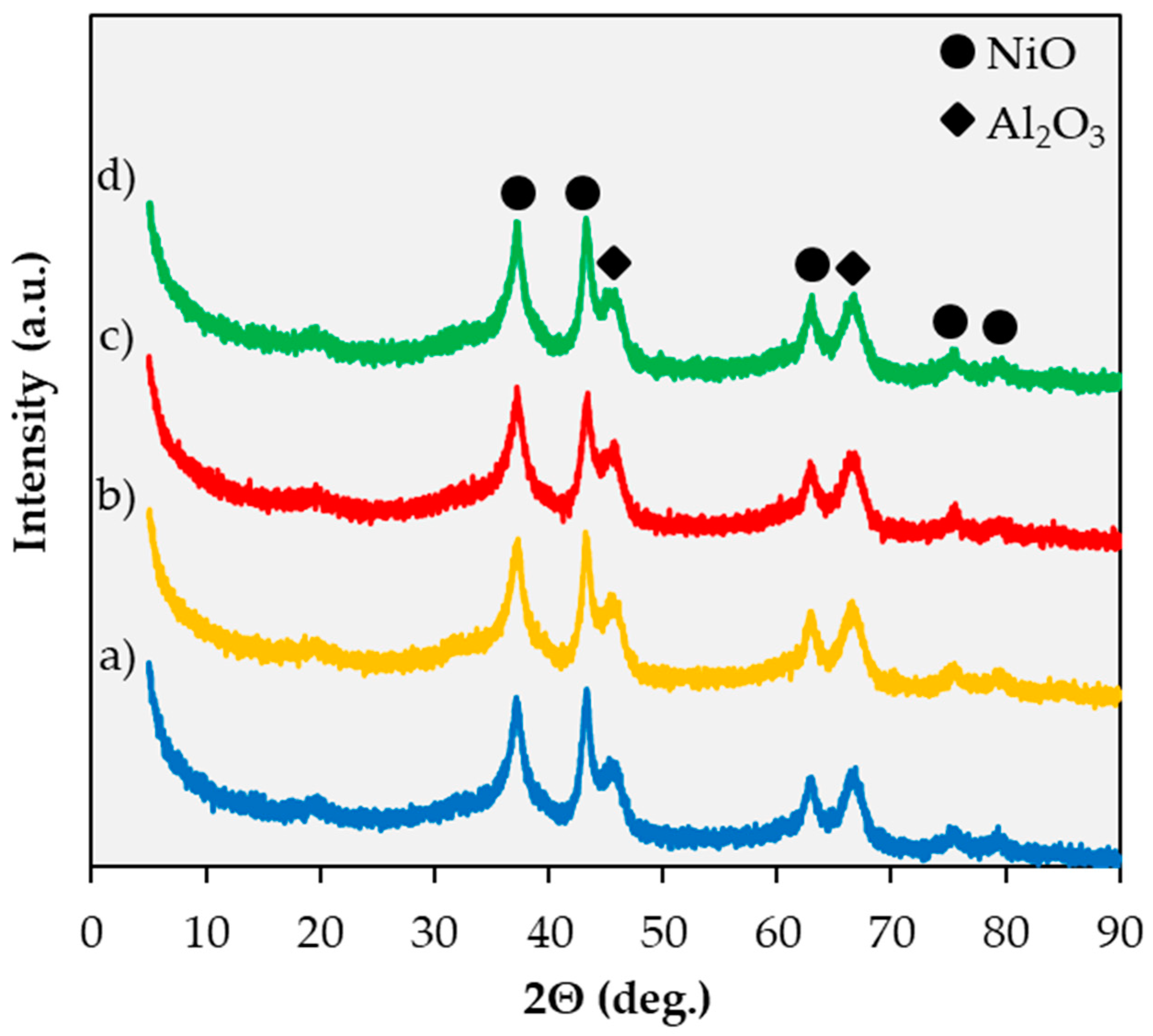
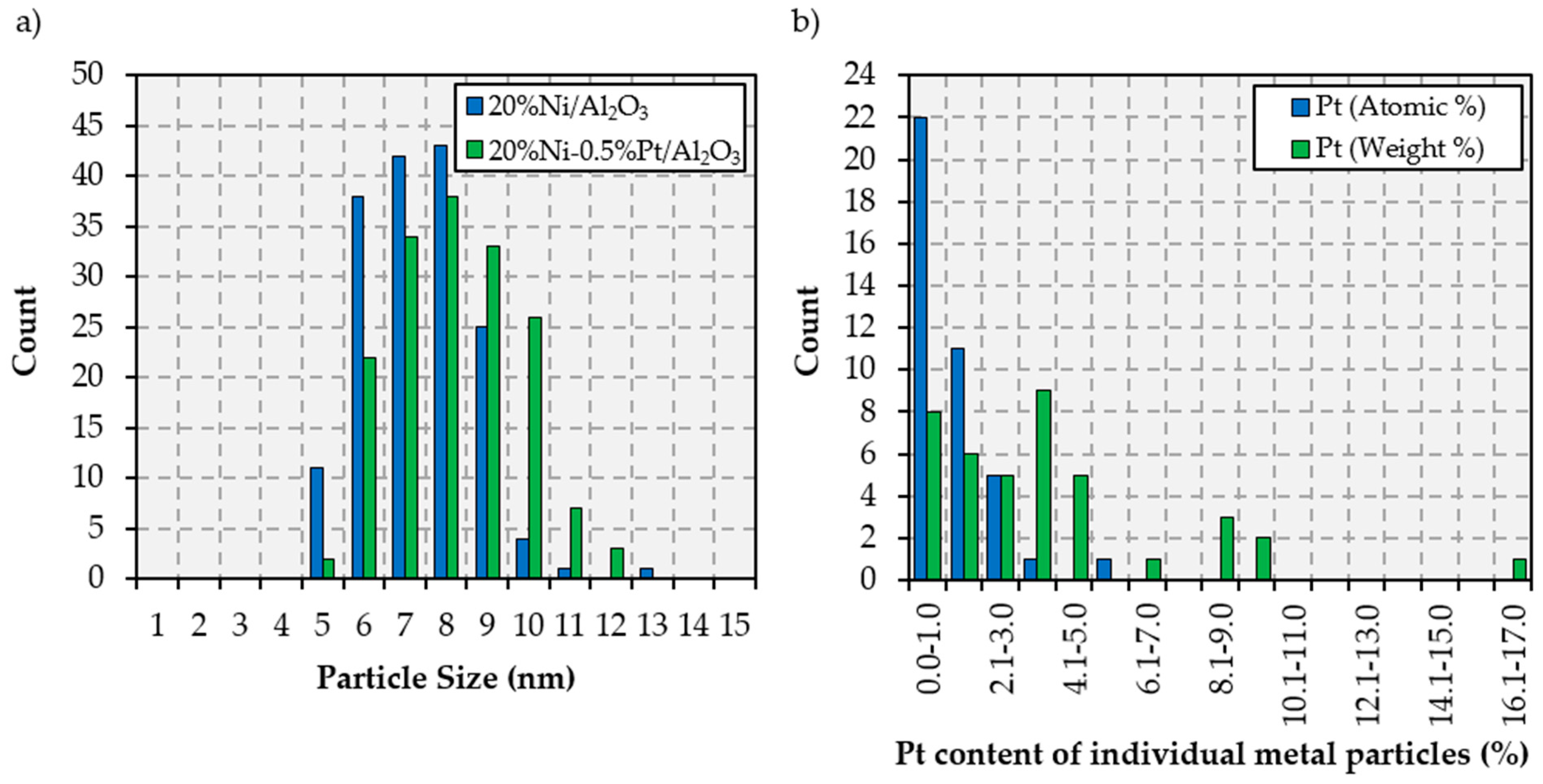
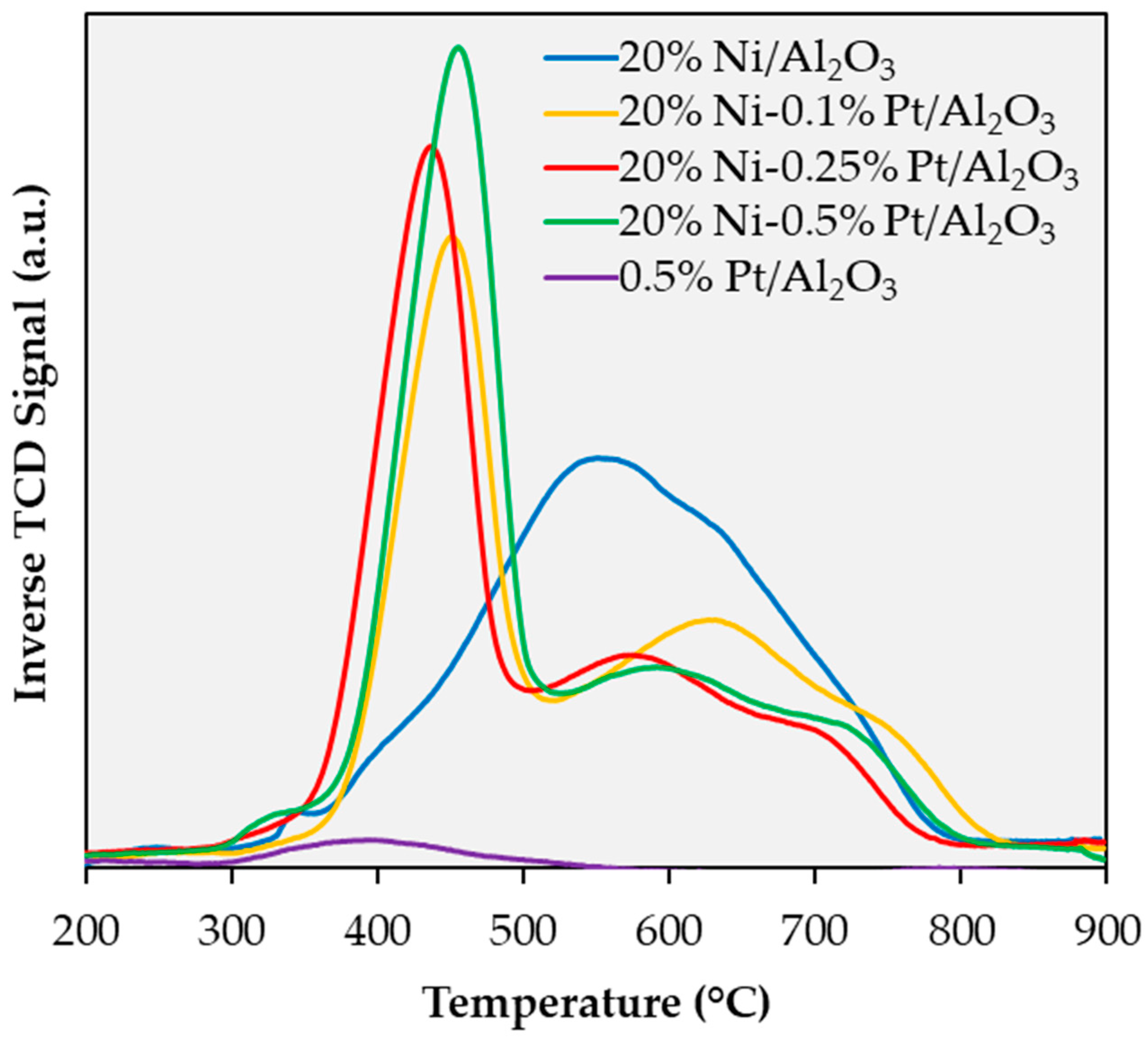
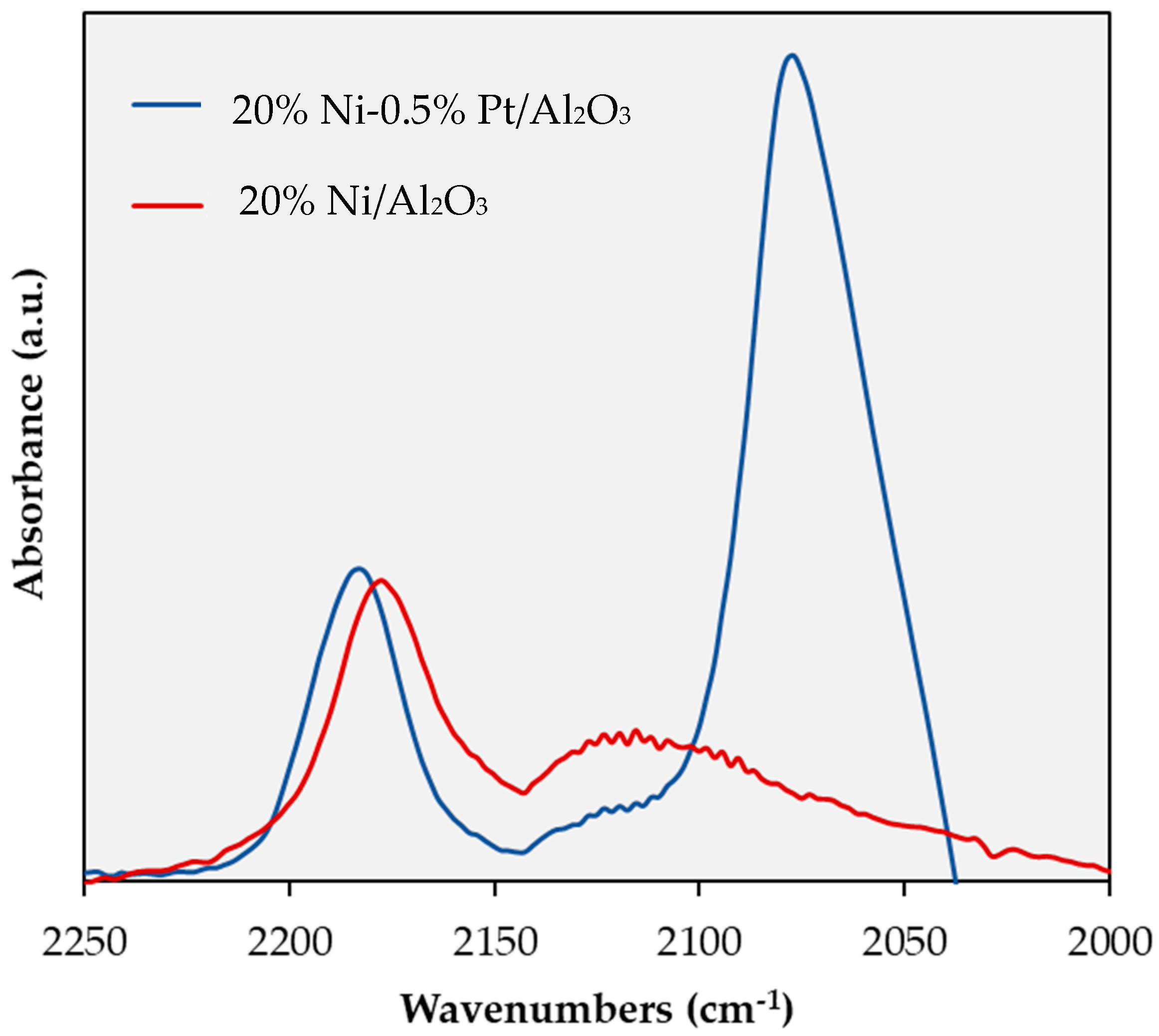
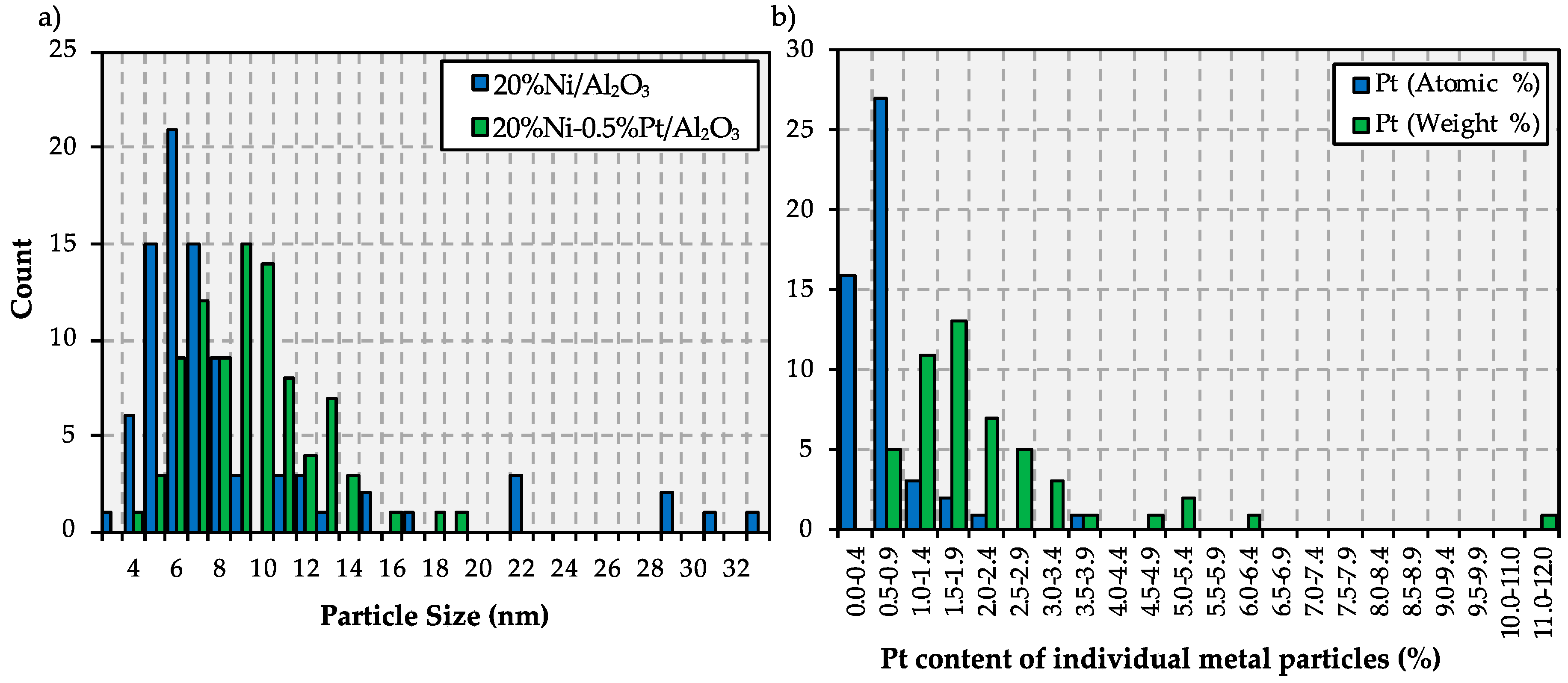
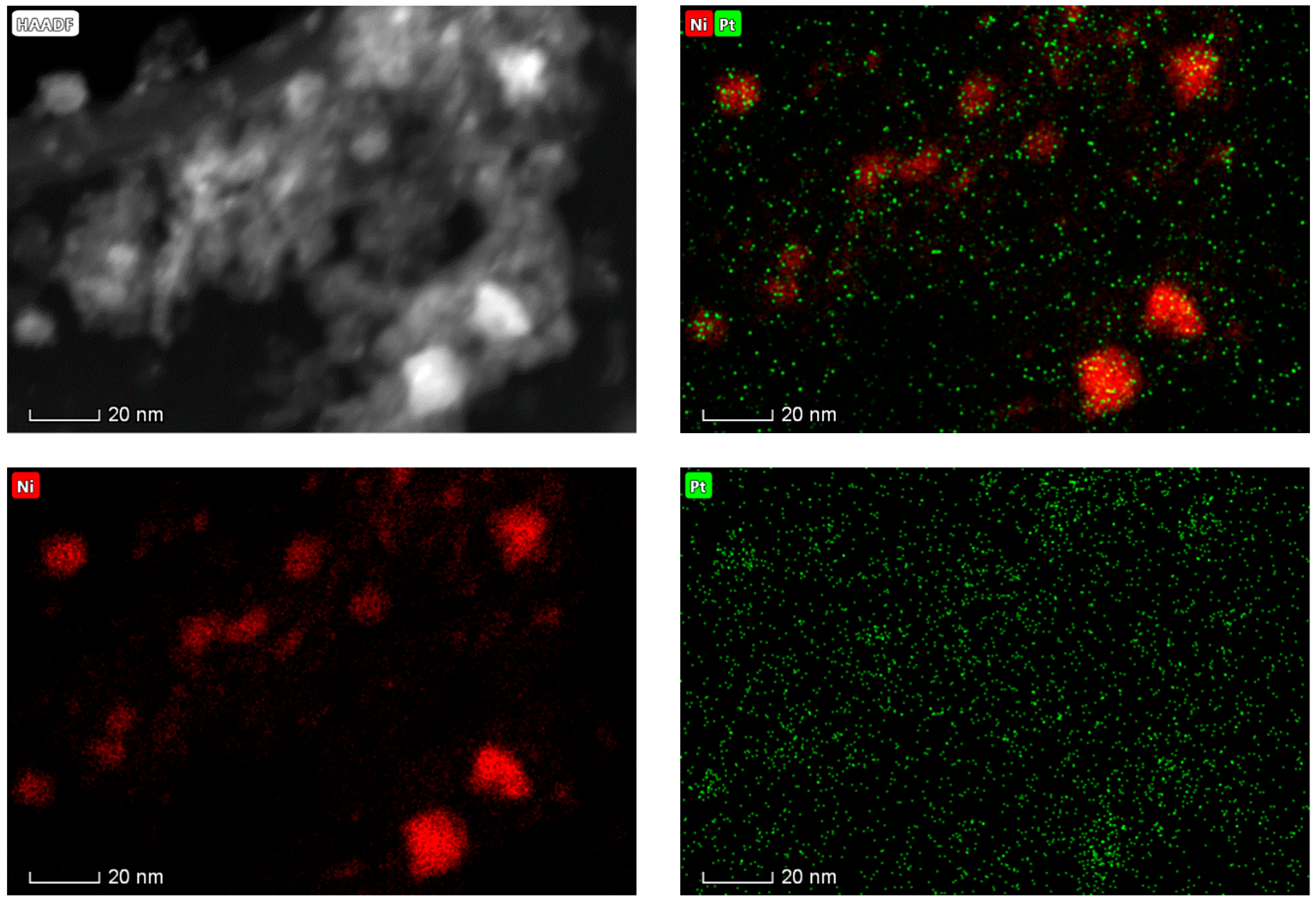
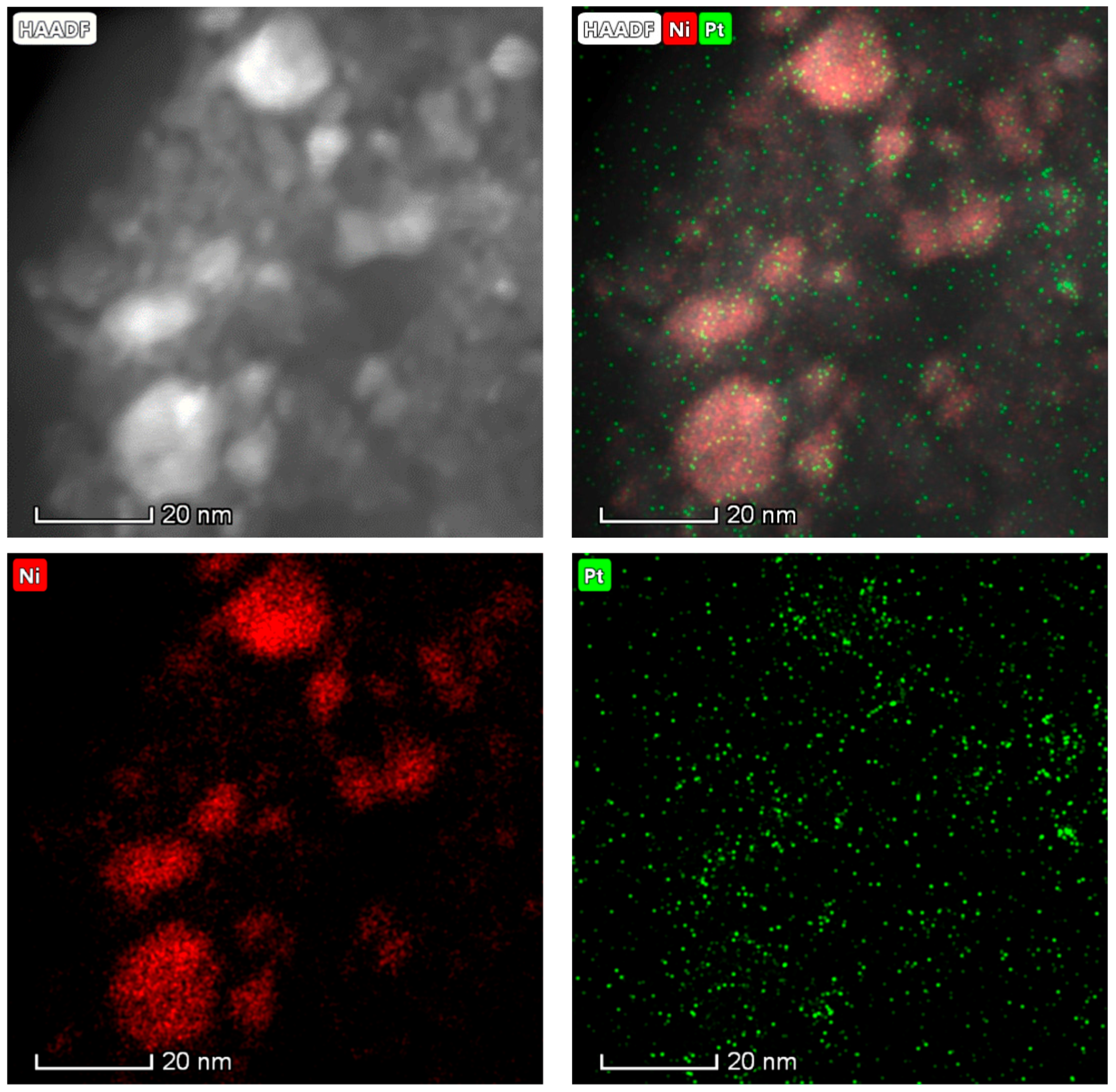
2.1. Catalyst Characterization
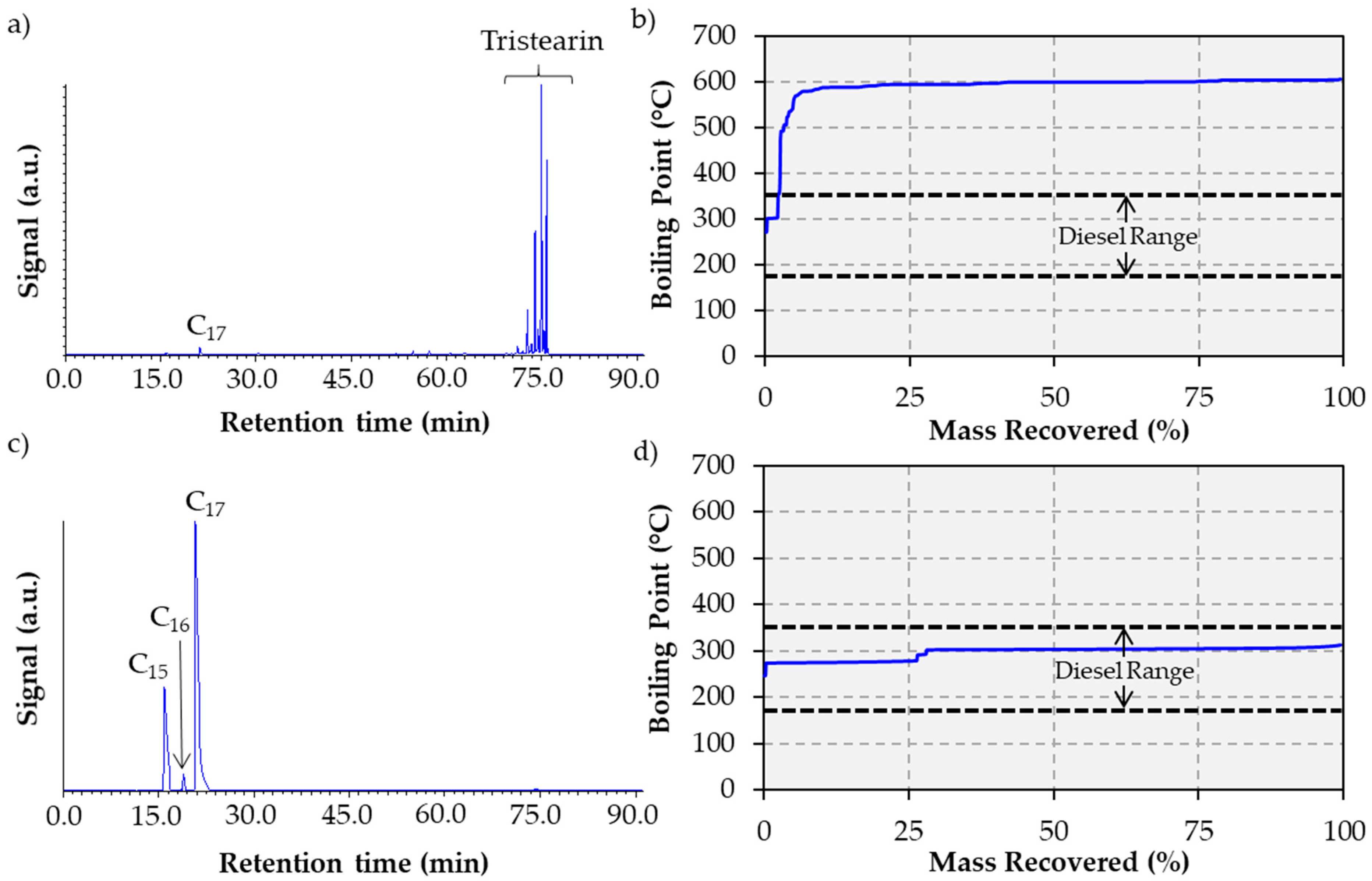
2.2. Tristearin Deoxygenation in Semi-batch Mode
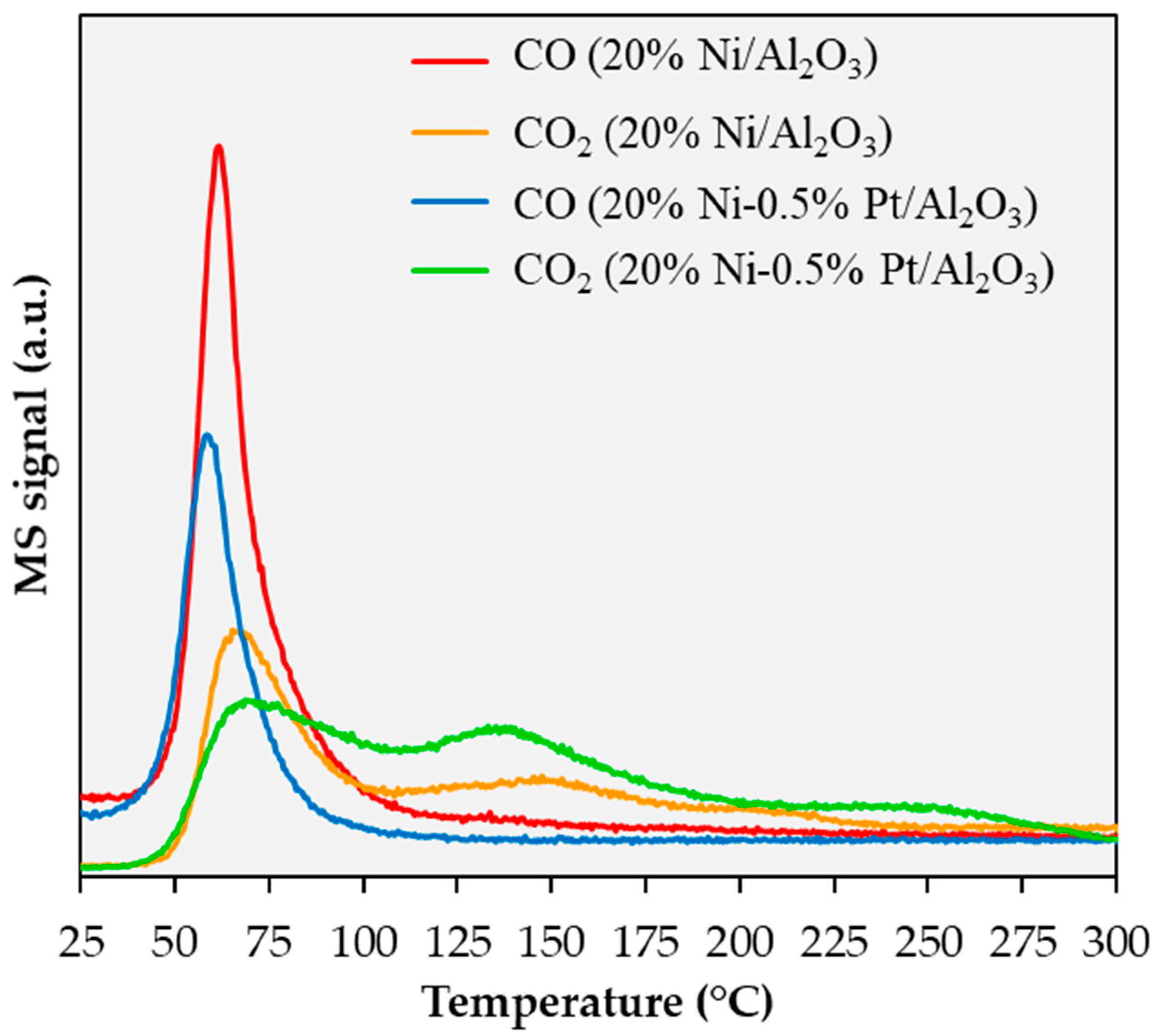
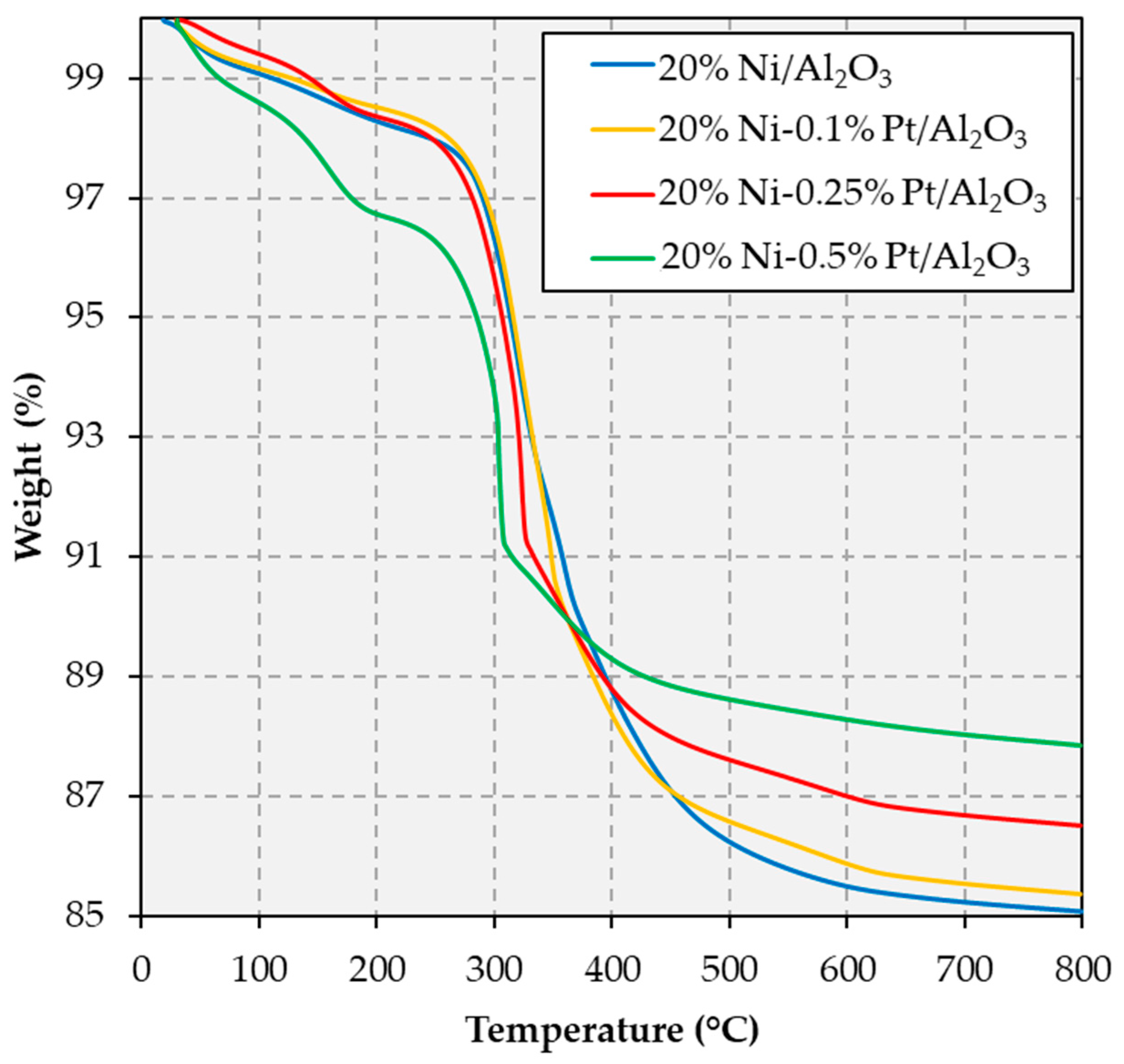
2.3. Spent Catalyst Characterization and Catalyst Stability
3. Materials and Methods
3.1. Catalyst Preparation
3.2. Catalyst Characterization
3.3. Deoxygenation Experiments
3.4. Liquid Product Analysis
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
Appendix A




References
- Fargione, J.; Hill, J.; Tilman, D.; Polasky, S.; Hawthorne, P. Land clearing and the biofuel carbon debt. Science 2008, 319, 1235–1238. [Google Scholar] [CrossRef] [PubMed]
- Tilman, D.; Hill, J.; Lehman, C. Carbon-negative biofuels from low-input high-diversity grassland biomass. Science 2006, 314, 1598–1600. [Google Scholar] [CrossRef] [PubMed]
- Berenblyum, A.S.; Podoplelova, T.A.; Shamsiev, R.S.; Katsman, E.A.; Danyushevsky, V.Y.; Flid, V.R. Catalytic chemistry of preparation of hydrocarbon fuels from vegetable oils and fats. Catal. Ind. 2012, 4, 209–214. [Google Scholar] [CrossRef]
- Benazzi, E.; Cameron, C. Boutique diesel in the on-road market. Hydrocarbon Process. 2006, 85, DD17–DD18. [Google Scholar]
- Kubičková, I.; Snåre, M.; Eränen, K.; Mäki-Arvela, P.; Murzin, D.Y. Hydrocarbons for diesel fuel via decarboxylation of vegetable oils. Catal. Today 2005, 106, 197–200. [Google Scholar] [CrossRef]
- Kordulis, C.; Bourikas, K.; Gousi, M.; Kordouli, E.; Lycourghiotis, A. Development of nickel based catalysts for the transformation of natural triglycerides and related compounds into green diesel: A critical review. Appl. Catal. B 2016, 181, 156–196. [Google Scholar] [CrossRef]
- Santillan-Jimenez, E.; Crocker, M. Catalytic deoxygenation of fatty acids and their derivatives to hydrocarbon fuels via decarboxylation/decarbonylation. J. Chem. Technol. Biotechnol. 2012, 87, 1041–1050. [Google Scholar] [CrossRef]
- Snåre, M.; Kubičková, I.; Mäki-Arvela, P.; Eränen, K.; Murzin, D.Y. Heterogeneous catalytic deoxygenation of stearic acid for production of biodiesel. Ind. Eng. Chem. Res. 2006, 45, 5708–5715. [Google Scholar] [CrossRef]
- Mäki-Arvela, P.; Kubickova, I.; Snåre, M.; Eränen, K.; Murzin, D.Y. Catalytic deoxygenation of fatty acids and their derivatives. Energy Fuels 2007, 21, 30–41. [Google Scholar] [CrossRef]
- Snåre, M.; Kubicková, I.; Mäki-Arvela, P.; Chichova, D.; Eränen, K.; Murzin, D.Y. Catalytic deoxygenation of unsaturated renewable feedstocks for production of diesel fuel hydrocarbons. Fuel 2008, 87, 933–945. [Google Scholar] [CrossRef]
- Simakova, I.; Simakova, O.; Mäki-Arvela, P.; Murzin, D.Y. Decarboxylation of fatty acids over Pd supported on mesoporous carbon. Catal. Today 2010, 150, 28–31. [Google Scholar] [CrossRef]
- Simakova, I.; Rozmysłowicz, B.; Simakova, O.; Mäki-Arvela, P.; Simakov, A.; Murzin, D.Y. Catalytic Deoxygenation of C18 Fatty Acids Over Mesoporous Pd/C Catalyst for Synthesis of Biofuels. Top. Catal. 2011, 54, 460–466. [Google Scholar] [CrossRef]
- Immer, J.G.; Lamb, H.H. Fed-batch catalytic deoxygenation of free fatty acids. Energy Fuels 2010, 24, 5291–5299. [Google Scholar] [CrossRef]
- Immer, J.G.; Kelly, M.J.; Lamb, H.H. Catalytic reaction pathways in liquid-phase deoxygenation of C18 free fatty acids. Appl. Catal. A 2010, 375, 134–139. [Google Scholar] [CrossRef]
- Hengst, K.; Arend, M.; Pfützenreuter, R.; Hoelderich, W.F. Deoxygenation and cracking of free fatty acids over acidic catalysts by single step conversion for the production of diesel fuel and fuel blends. Appl. Catal. B 2015, 174–175, 383–394. [Google Scholar] [CrossRef]
- Sari, E.; DiMaggio, C.; Kim, M.; Salley, S.O.; Ng, K.Y.S. Catalytic conversion of brown grease to green diesel via decarboxylation over activated carbon supported palladium catalyst. Ind. Eng. Chem. Res. 2013, 52, 11527–11536. [Google Scholar] [CrossRef]
- de Sousa, F.P.; Cardoso, C.C.; Pasa, V.M. Producing hydrocarbons for green diesel and jet fuel formulation from palm kernel fat over Pd/C. Fuel Process. Technol. 2016, 143, 35–42. [Google Scholar] [CrossRef]
- Shao, Y.; Xia, Q.; Liu, X.; Lu, G.; Wang, Y. Pd/Nb2O5/SiO2 Catalyst for the Direct Hydrodeoxygenation of Biomass-Related Compounds to Liquid Alkanes under Mild Conditions. ChemSusChem 2015, 8, 1761–1767. [Google Scholar] [CrossRef] [PubMed]
- Do, P.T.; Chiappero, M.; Lobban, L.L.; Resasco, D.E. Catalytic deoxygenation of methyl-octanoate and methyl-stearate on Pt/Al2O3. Catal. Lett. 2009, 130, 9–18. [Google Scholar] [CrossRef]
- Chiappero, M.; Do, P.T.M.; Crossley, S.; Lobban, L.L.; Resasco, D.E. Direct conversion of triglycerides to olefins and paraffins over noble metal supported catalysts. Fuel 2011, 90, 1155–1165. [Google Scholar] [CrossRef]
- Na, J.-G.; Yi, B.E.; Han, J.K.; Oh, Y.-K.; Park, J.-H.; Jung, T.S.; Han, S.S.; Yoon, H.C.; Kim, J.-N.; Lee, H.; et al. Deoxygenation of microalgal oil into hydrocarbon with precious metal catalysts: Optimization of reaction conditions and supports. Energy 2012, 47, 25–30. [Google Scholar] [CrossRef]
- Fu, J.; Lu, X.; Savage, P.E. Hydrothermal decarboxylation and hydrogenation of fatty acids over Pt/C. ChemSusChem 2011, 4, 481–486. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Carreon, M.A. Deoxygenation of palmitic and lauric acids over Pt/ZIF-67 membrane/zeolite 5A bead catalysts. ACS Appl. Mater. Int. 2017, 9, 31993–32000. [Google Scholar] [CrossRef] [PubMed]
- Ahmadi, M.; Nambo, A.; Jasinski, J.B.; Ratnasamy, P.; Carreon, M.A. Decarboxylation of oleic acid over Pt catalysts supported on small-pore zeolites and hydrotalcite. Catal. Sci. Technol. 2015, 5, 380–388. [Google Scholar] [CrossRef]
- Ahmadi, M.; Macias, E.E.; Jasinski, J.B.; Ratnasamy, P.; Carreon, M.A. Decarboxylation and further transformation of oleic acid over bifunctional, Pt/SAPO-11 catalyst and Pt/chloride Al2O3 catalysts. J. Mol. Catal. A Chem. 2014, 386, 14–19. [Google Scholar] [CrossRef]
- Tian, Q.; Qiao, K.; Zhou, F.; Chen, K.; Wang, T.; Fu, J.; Lu, X.; Ouyang, P. Direct production of aviation fuel range hydrocarbons and aromatics from oleic acid without an added hydrogen donor. Energy Fuels 2016, 30, 7291–7297. [Google Scholar] [CrossRef]
- Kon, K.; Onodera, W.; Takakusagi, S.; Shimizu, K.-i. Hydrodeoxygenation of fatty acids and triglycerides by Pt-loaded Nb2O5 catalysts. Catal. Sci. Technol. 2014, 4, 3705–3712. [Google Scholar] [CrossRef]
- Kon, K.; Toyao, T.; Onodera, W.; Siddiki, S.M.A.H.; Shimizu, K.-i. Hydrodeoxygenation of Fatty Acids, Triglycerides, and Ketones to Liquid Alkanes by a Pt–MoOx/TiO2 Catalyst. ChemCatChem 2017, 9, 2822–2827. [Google Scholar] [CrossRef]
- Janampelli, S.; Darbha, S. Metal Oxide-Promoted Hydrodeoxygenation Activity of Platinum in Pt-MOx/Al2O3 Catalysts for Green Diesel Production. Energy Fuels 2018, 32, 12630–12643. [Google Scholar] [CrossRef]
- Morgan, T.; Grubb, D.; Santillan-Jimenez, E.; Crocker, M. Conversion of triglycerides to hydrocarbons over supported metal catalysts. Top. Catal. 2010, 53, 820–829. [Google Scholar] [CrossRef]
- Santillan-Jimenez, E.; Morgan, T.; Lacny, J.; Mohapatra, S.; Crocker, M. Catalytic deoxygenation of triglycerides and fatty acids to hydrocarbons over carbon-supported nickel. Fuel 2013, 103, 1010–1017. [Google Scholar] [CrossRef]
- Loe, R.; Santillan-Jimenez, E.; Morgan, T.; Sewell, L.; Ji, Y.; Jones, S.; Isaacs, M.A.; Lee, A.F.; Crocker, M. Effect of Cu and Sn promotion on the catalytic deoxygenation of model and algal lipids to fuel-like hydrocarbons over supported Ni catalysts. Appl. Catal. B 2016, 191, 147–156. [Google Scholar] [CrossRef]
- Santillan-Jimenez, E.; Morgan, T.; Shoup, J.; Harman-Ware, A.E.; Crocker, M. Catalytic deoxygenation of triglycerides and fatty acids to hydrocarbons over Ni–Al layered double hydroxide. Catal. Today 2014, 237, 136–144. [Google Scholar] [CrossRef]
- Santillan-Jimenez, E.; Morgan, T.; Loe, R.; Crocker, M. Continuous catalytic deoxygenation of model and algal lipids to fuel-like hydrocarbons over Ni–Al layered double hydroxide. Catal. Today 2015, 258, 284–293. [Google Scholar] [CrossRef]
- Morgan, T.; Santillan-Jimenez, E.; Harman-Ware, A.E.; Ji, Y.; Grubb, D.; Crocker, M. Catalytic deoxygenation of triglycerides to hydrocarbons over supported nickel catalysts. Chem. Eng. J. 2012, 189–190, 346–355. [Google Scholar] [CrossRef]
- Peng, B.; Zhao, C.; Kasakov, S.; Foraita, S.; Lercher, J.A. Manipulating catalytic pathways: Deoxygenation of palmitic acid on multifunctional catalysts. Chem. Eur. J. 2013, 19, 4732–4741. [Google Scholar] [CrossRef] [PubMed]
- Peng, B.; Yuan, X.; Zhao, C.; Lercher, J.A. Stabilizing catalytic pathways via redundancy: Selective reduction of microalgae oil to alkanes. J. Am. Chem. Soc. 2012, 134, 9400–9405. [Google Scholar] [CrossRef]
- Peng, B.; Yao, Y.; Zhao, C.; Lercher, J.A. Towards quantitative conversion of microalgae oil to diesel-range alkanes with bifunctional catalysts. Angew. Chem. 2012, 124, 2114–2117. [Google Scholar] [CrossRef]
- Ford, J.P.; Immer, J.G.; Lamb, H.H. Palladium catalysts for fatty acid deoxygenation: Influence of the support and fatty acid chain length on decarboxylation kinetics. Top. Catal. 2012, 55, 175–184. [Google Scholar] [CrossRef]
- Hollak, S.A.W.; de Jong, K.P.; van Es, D.S. Catalytic deoxygenation of fatty acids: Elucidation of the inhibition process. ChemCatChem 2014, 6, 2648–2655. [Google Scholar] [CrossRef]
- Song, W.; Zhao, C.; Lercher, J.A. Importance of size and distribution of Ni nanoparticles for the hydrodeoxygenation of microalgae oil. Chem. Eur. J. 2013, 19, 9833–9842. [Google Scholar] [CrossRef] [PubMed]
- Santillan-Jimenez, E.; Loe, R.; Garrett, M.; Morgan, T.; Crocker, M. Effect of Cu promotion on cracking and methanation during the Ni-catalyzed deoxygenation of waste lipids and hemp seed oil to fuel-like hydrocarbons. Catal. Today 2018, 302, 261–271. [Google Scholar] [CrossRef]
- Guo, Q.; Wu, M.; Wang, K.; Zhang, L.; Xu, X. Catalytic Hydrodeoxygenation of Algae Bio-oil over Bimetallic Ni–Cu/ZrO2 Catalysts. Ind. Eng. Chem. Res. 2015, 54, 890–899. [Google Scholar] [CrossRef]
- Pan, Z.; Wang, R.; Nie, Z.; Chen, J. Effect of a second metal (Co, Fe, Mo and W) on performance of Ni2P/SiO2 for hydrodeoxygenation of methyl laurate. J. Energy Chem. 2016, 25, 418–426. [Google Scholar] [CrossRef]
- Shi, Y.; Xing, E.; Cao, Y.; Liu, M.; Wu, K.; Yang, M.; Wu, Y. Tailoring product distribution during upgrading of palmitic acid over bi-functional metal/zeolite catalysts. Chem. Eng. Sci. 2017, 166, 262–273. [Google Scholar] [CrossRef]
- Chen, H.; Zhang, X.; Zhang, J.; Wang, Q. Tuning Decarboxylation Selectivity for Deoxygenation of Vegetable Oil over Pt-Ni Bimetal Catalysts via Surface Engineering. Catal. Sci. Technol. 2018. [Google Scholar] [CrossRef]
- Li, X.-F.; Luo, X.-G. Preparation of mesoporous activated carbon supported Ni catalyst for deoxygenation of stearic acid into hydrocarbons. Environ. Prog. Sustain. Energy 2015, 34, 607–612. [Google Scholar] [CrossRef]
- Lu, S.; Lonergan, W.W.; Bosco, J.P.; Wang, S.; Zhu, Y.; Xie, Y.; Chen, J.G. Low temperature hydrogenation of benzene and cyclohexene: A comparative study between γ-Al2O3 supported PtCo and PtNi bimetallic catalysts. J. Catal. 2008, 259, 260–268. [Google Scholar] [CrossRef]
- Tanksale, A.; Beltramini, J.; Dumesic, J.; Lu, G. Effect of Pt and Pd promoter on Ni supported catalysts—A TPR/TPO/TPD and microcalorimetry study. J. Catal. 2008, 258, 366–377. [Google Scholar] [CrossRef]
- Vizcaíno, A.; Carrero, A.; Calles, J. Hydrogen production by ethanol steam reforming over Cu–Ni supported catalysts. Int. J. Hydrogen Energy 2007, 32, 1450–1461. [Google Scholar] [CrossRef]
- Rynkowski, J.M.; Paryjczak, T.; Lenik, M. On the nature of oxidic nickel phases in NiO/γ-Al2O3 catalysts. Appl. Catal. A 1993, 106, 73–82. [Google Scholar] [CrossRef]
- Ko, E.-Y.; Park, E.D.; Seo, K.W.; Lee, H.C.; Lee, D.; Kim, S. Pt–Ni/γ-Al2O3 catalyst for the preferential CO oxidation in the hydrogen stream. Catal. Lett. 2006, 110, 275–279. [Google Scholar] [CrossRef]
- Jackson, S.; Glanville, B.; Willis, J.; McLellan, G.; Webb, G.; Moyes, R.; Simpson, S.; Wells, P.; Whyman, R. Supported metal catalysts: Preparation, characterization, and function: II. Carbon monoxide and dioxygen adsorption on platinum catalysts. J. Catal. 1993, 139, 207–220. [Google Scholar] [CrossRef]
- Dias, J.A.; Assaf, J.M. Autothermal reforming of methane over Ni/γ-Al2O3 catalysts: The enhancement effect of small quantities of noble metals. J. Power Sources 2004, 130, 106–110. [Google Scholar] [CrossRef]
- Parizotto, N.; Zanchet, D.; Rocha, K.; Marques, C.; Bueno, J. The effects of Pt promotion on the oxi-reduction properties of alumina supported nickel catalysts for oxidative steam-reforming of methane: Temperature-resolved XAFS analysis. Appl. Catal. A 2009, 366, 122–129. [Google Scholar] [CrossRef]
- Riguetto, B.A.; Damyanova, S.; Gouliev, G.; Marques, C.M.; Petrov, L.; Bueno, J.M.C. Surface behavior of alumina-supported Pt catalysts modified with cerium as revealed by X-ray diffraction, X-ray photoelectron spectroscopy, and Fourier transform infrared spectroscopy of CO adsorption. J. Phys. Chem. B 2004, 108, 5349–5358. [Google Scholar] [CrossRef]
- Xu, J.; White, T.; Li, P.; He, C.; Yu, J.; Yuan, W.; Han, Y.-F. Biphasic Pd−Au alloy catalyst for low-temperature CO oxidation. J. Am. Chem. Soc. 2010, 132, 10398–10406. [Google Scholar] [CrossRef]
- Fisk, C.A.; Morgan, T.; Ji, Y.; Crocker, M.; Crofcheck, C.; Lewis, S.A. Bio-oil upgrading over platinum catalysts using in situ generated hydrogen. Appl. Catal. A 2009, 358, 150–156. [Google Scholar] [CrossRef]
- Sugami, Y.; Minami, E.; Saka, S. Renewable diesel production from rapeseed oil with hydrothermal hydrogenation and subsequent decarboxylation. Fuel 2016, 166, 376–381. [Google Scholar] [CrossRef]
- Huber, G.W.; O’Connor, P.; Corma, A. Processing biomass in conventional oil refineries: Production of high quality diesel by hydrotreating vegetable oils in heavy vacuum oil mixtures. Appl. Catal. A 2007, 329, 120–129. [Google Scholar] [CrossRef]
- Berenblyum, A.S.; Danyushevsky, V.Y.; Katsman, E.A.; Podoplelova, T.A.; Flid, V.R. Production of engine fuels from inedible vegetable oils and fats. Pet. Chem. 2010, 50, 305–311. [Google Scholar] [CrossRef]
- Kikhtyanin, O.V.; Rubanov, A.E.; Ayupov, A.B.; Echevsky, G.V. Hydroconversion of sunflower oil on Pd/SAPO-31 catalyst. Fuel 2010, 89, 3085–3092. [Google Scholar] [CrossRef]
- Zhang, H.; Shao, S.; Xiao, R.; Shen, D.; Zeng, J. Characterization of coke deposition in the catalytic fast pyrolysis of biomass derivates. Energy Fuels 2013, 28, 52–57. [Google Scholar] [CrossRef]
- Chen, L.; Li, H.; Fu, J.; Miao, C.; Lv, P.; Yuan, Z. Catalytic hydroprocessing of fatty acid methyl esters to renewable alkane fuels over Ni/HZSM-5 catalyst. Catal. Today 2016, 259, 266–276. [Google Scholar] [CrossRef]
- Loe, R.; Lavoignat, Y.; Maier, M.; Abdallah, M.; Morgan, T.; Qian, D.; Pace, R.; Santillan-Jimenez, E.; Crocker, M. Continuous Catalytic Deoxygenation of Waste Free Fatty Acid-Based Feeds to Fuel-Like Hydrocarbons Over a Supported Ni-Cu Catalyst. Catalysts 2019, 9, 123. [Google Scholar] [CrossRef]
- Grosvenor, A.P.; Biesinger, M.C.; Smart, R.S.C.; McIntyre, N.S. New interpretations of XPS spectra of nickel metal and oxides. Surf. Sci. 2006, 600, 1771–1779. [Google Scholar] [CrossRef]
- Morgan, T.; Santillan-Jimenez, E.; Huff, K.; Javed, K.R.; Crocker, M. Use of dual detection in the gas chromatographic analysis of oleaginous biomass feeds and biofuel products to enable accurate simulated distillation and lipid profiling. Energy Fuels 2017, 31, 9498–9506. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Catalyst | N2 Physisorption | Avg. NiO Particle Size (nm) | ||
|---|---|---|---|---|
| BET Surface Area (m2/g) | Pore Vol. (cm3/g) | Avg. Pore Diameter (nm) | ||
| 20% Ni/Al2O3 | 135 | 0.29 | 7.0 | 6.8 |
| 20% Ni-0.1% Pt/Al2O3 | 129 | 0.27 | 7.0 | 7.4 |
| 20% Ni-0.25% Pt/Al2O3 | 138 | 0.29 | 7.0 | 6.1 |
| 20% Ni-0.5% Pt/Al2O3 | 134 | 0.29 | 6.9 | 8.7 |
| 0.5% Pt/Al2O3 | 196 | 0.13 | 4.6 | - |
| Catalyst | H2 Ads. (mL/g) | H2 Ads. on Pt for 50% Pt Dispersion (mL/g) 1 | H2 Ads. on Pt for 100% Pt Dispersion (mL/g) 1 | H2 Ads. on Ni-Pt for 100% Pt Dispersion (mL/g) 2 |
|---|---|---|---|---|
| 20% Ni/Al2O3 | 0.095 | - | - | - |
| 20% Ni-0.1% Pt/Al2O3 | 0.461 | 0.028 | 0.057 | 0.152 |
| 20% Ni-0.25% Pt/Al2O3 | 0.560 | 0.071 | 0.144 | 0.239 |
| 20% Ni-0.5% Pt/Al2O3 | 0.639 | 0.144 | 0.287 | 0.382 |
| 0.5% Pt/Al2O3 | 0.371 | 0.144 | 0.287 | 0.287 |
| Catalyst | Conversion (%) 2 | Selectively to (Yield of) C10-C17 (%) 3,6 | Selectively to (Yield of) C17 (%) 4,6 | Selectively to (Yield of) C18 (%) 5,6 |
|---|---|---|---|---|
| 20% Ni/Al2O3 | 2 | 83 (2) | 83 (2) | 3 (<1) |
| 20% Ni-0.1% Pt/Al2O3 | 32 | 96 (23) | 72 (15) | 0 (0) |
| 20% Ni-0.25% Pt/Al2O3 | 66 | 99 (65) | 72 (48) | 0 (0) |
| 20% Ni-0.5% Pt/Al2O3 | 100 | 96 (96) | 65 (65) | 2 (2) |
| 0.5% Pt/Al2O3 | 5 | 70 (4) | 65 (3) | 3 (<1) |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Loe, R.; Huff, K.; Walli, M.; Morgan, T.; Qian, D.; Pace, R.; Song, Y.; Isaacs, M.; Santillan-Jimenez, E.; Crocker, M. Effect of Pt Promotion on the Ni-Catalyzed Deoxygenation of Tristearin to Fuel-Like Hydrocarbons. Catalysts 2019, 9, 200. https://doi.org/10.3390/catal9020200
Loe R, Huff K, Walli M, Morgan T, Qian D, Pace R, Song Y, Isaacs M, Santillan-Jimenez E, Crocker M. Effect of Pt Promotion on the Ni-Catalyzed Deoxygenation of Tristearin to Fuel-Like Hydrocarbons. Catalysts. 2019; 9(2):200. https://doi.org/10.3390/catal9020200
Chicago/Turabian StyleLoe, Ryan, Kelsey Huff, Morgan Walli, Tonya Morgan, Dali Qian, Robert Pace, Yang Song, Mark Isaacs, Eduardo Santillan-Jimenez, and Mark Crocker. 2019. "Effect of Pt Promotion on the Ni-Catalyzed Deoxygenation of Tristearin to Fuel-Like Hydrocarbons" Catalysts 9, no. 2: 200. https://doi.org/10.3390/catal9020200
APA StyleLoe, R., Huff, K., Walli, M., Morgan, T., Qian, D., Pace, R., Song, Y., Isaacs, M., Santillan-Jimenez, E., & Crocker, M. (2019). Effect of Pt Promotion on the Ni-Catalyzed Deoxygenation of Tristearin to Fuel-Like Hydrocarbons. Catalysts, 9(2), 200. https://doi.org/10.3390/catal9020200