The Challenge of Achieving a High Density of Fe-Based Active Sites in a Highly Graphitic Carbon Matrix



 ,
,
Abstract
1. Introduction
2. Results
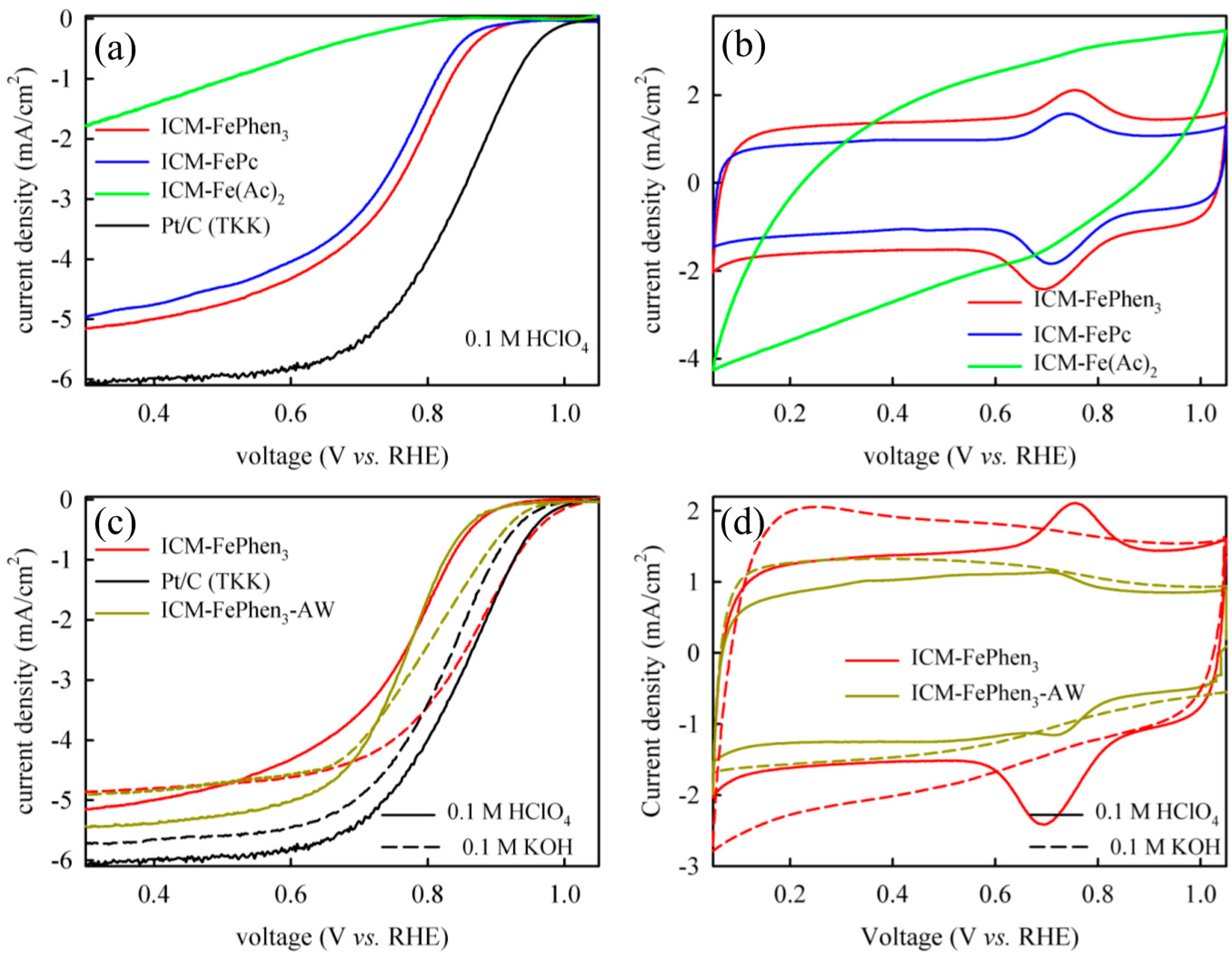
2.1. ORR Activity
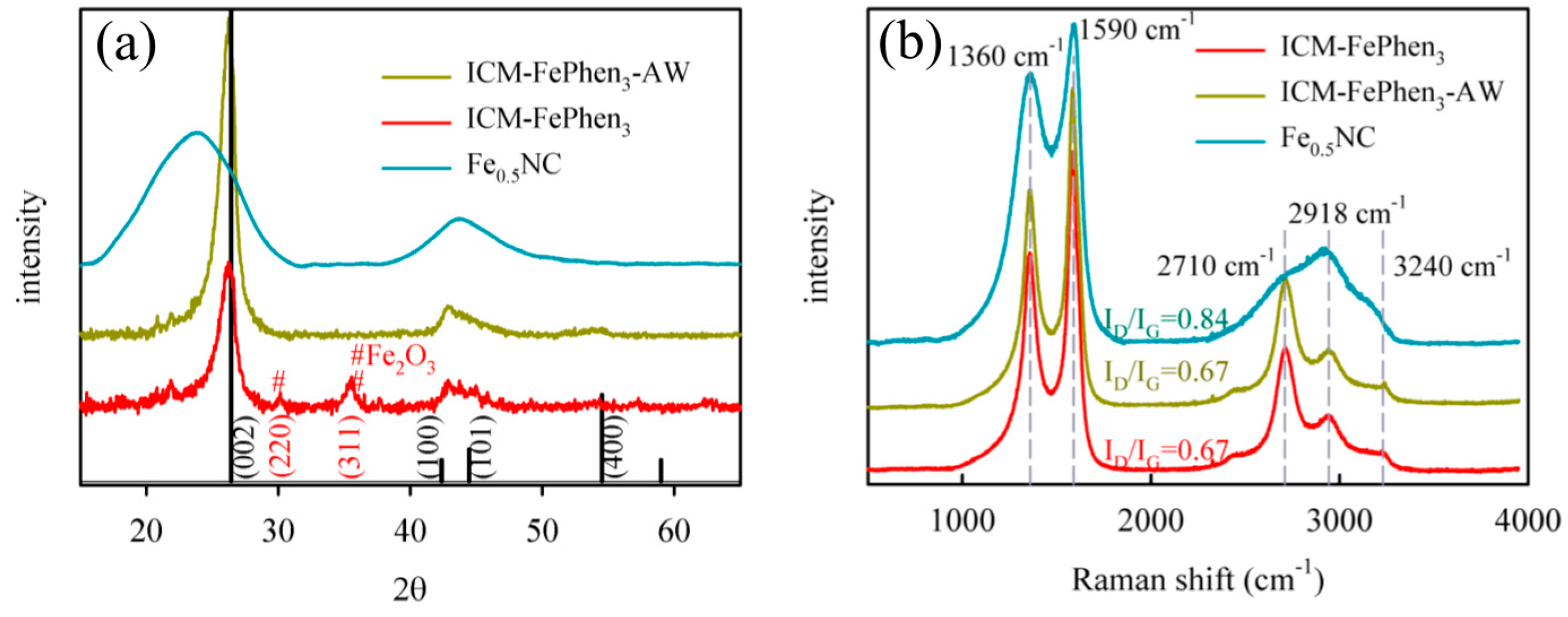
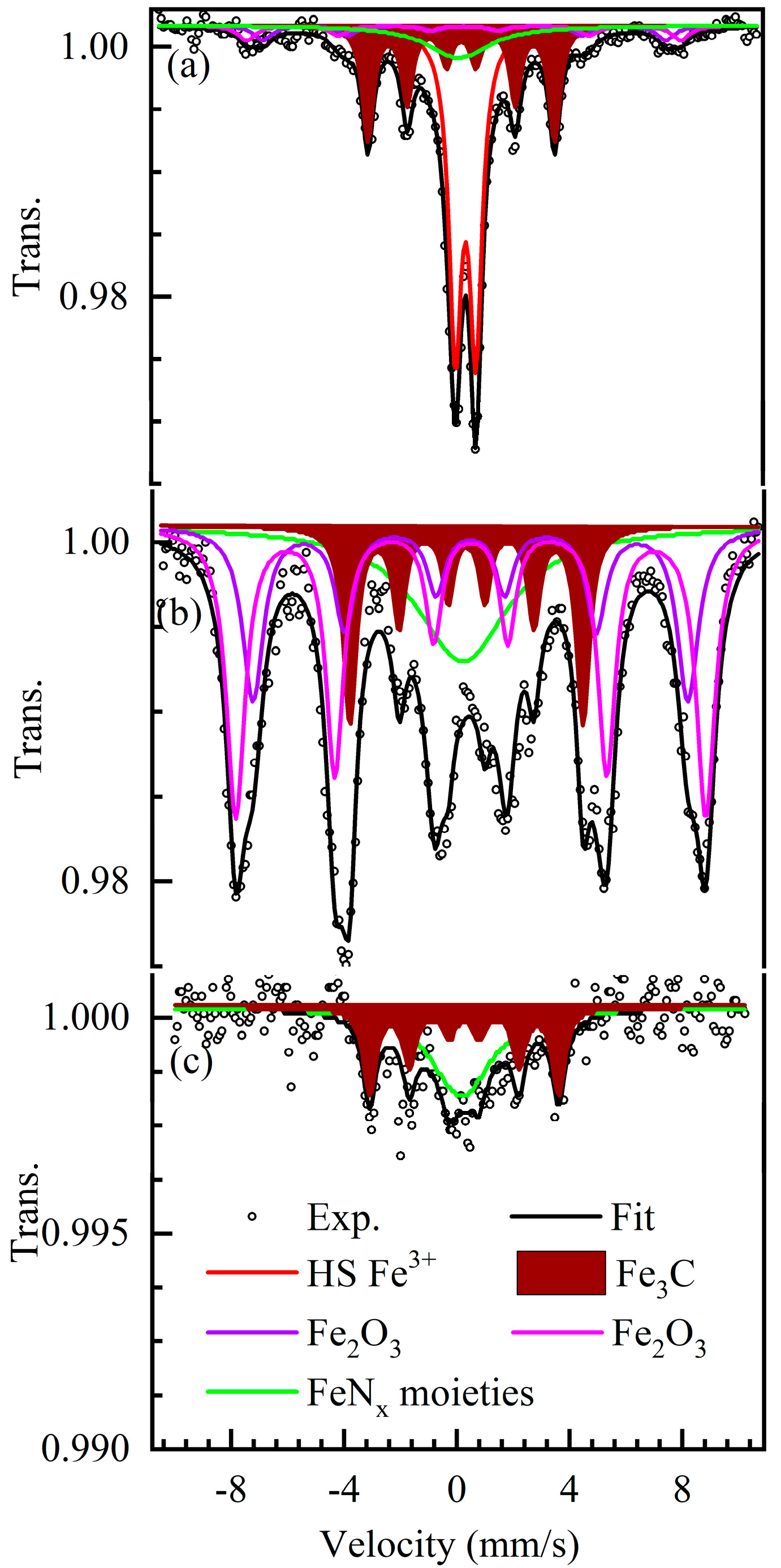
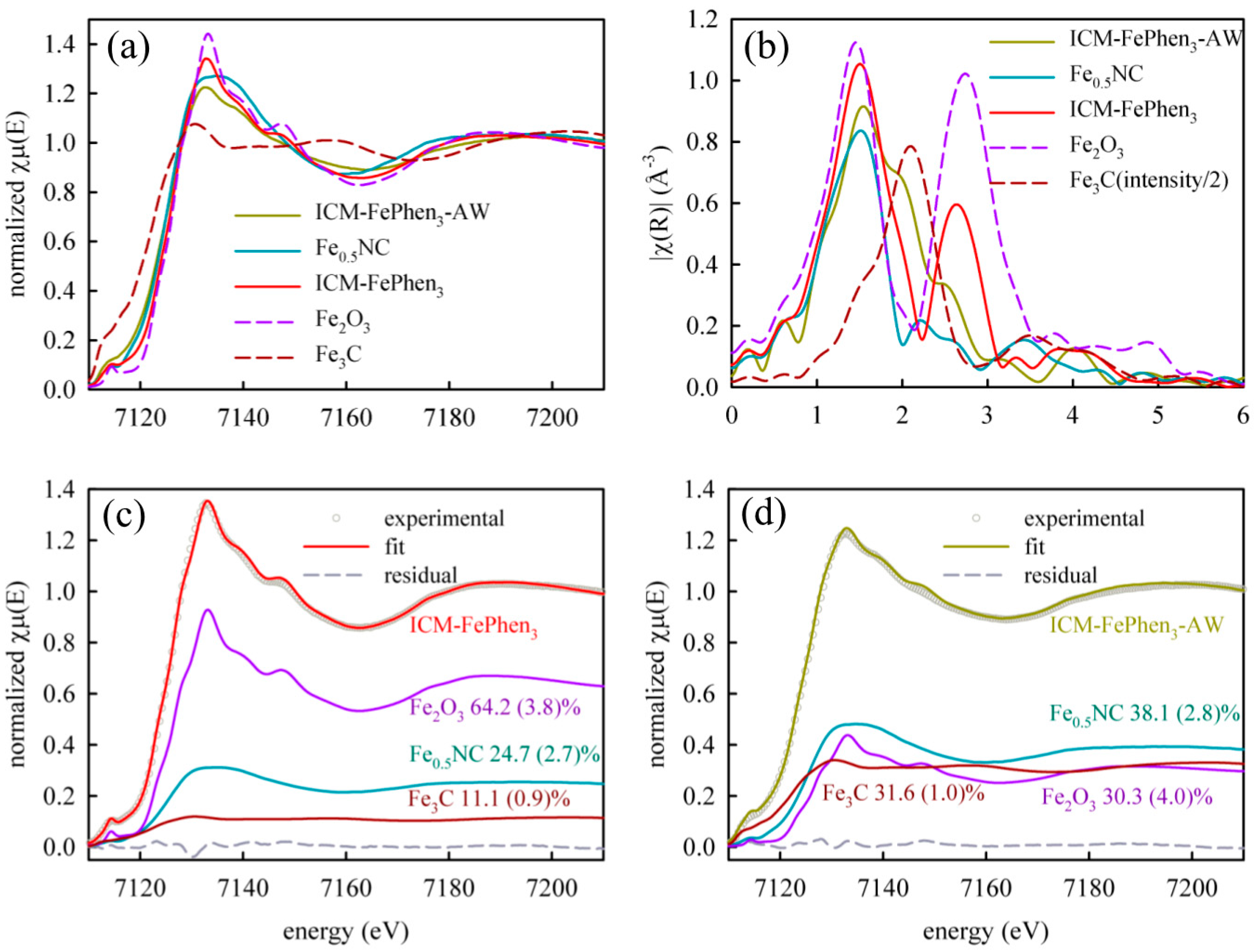
2.2. Iron Speciation
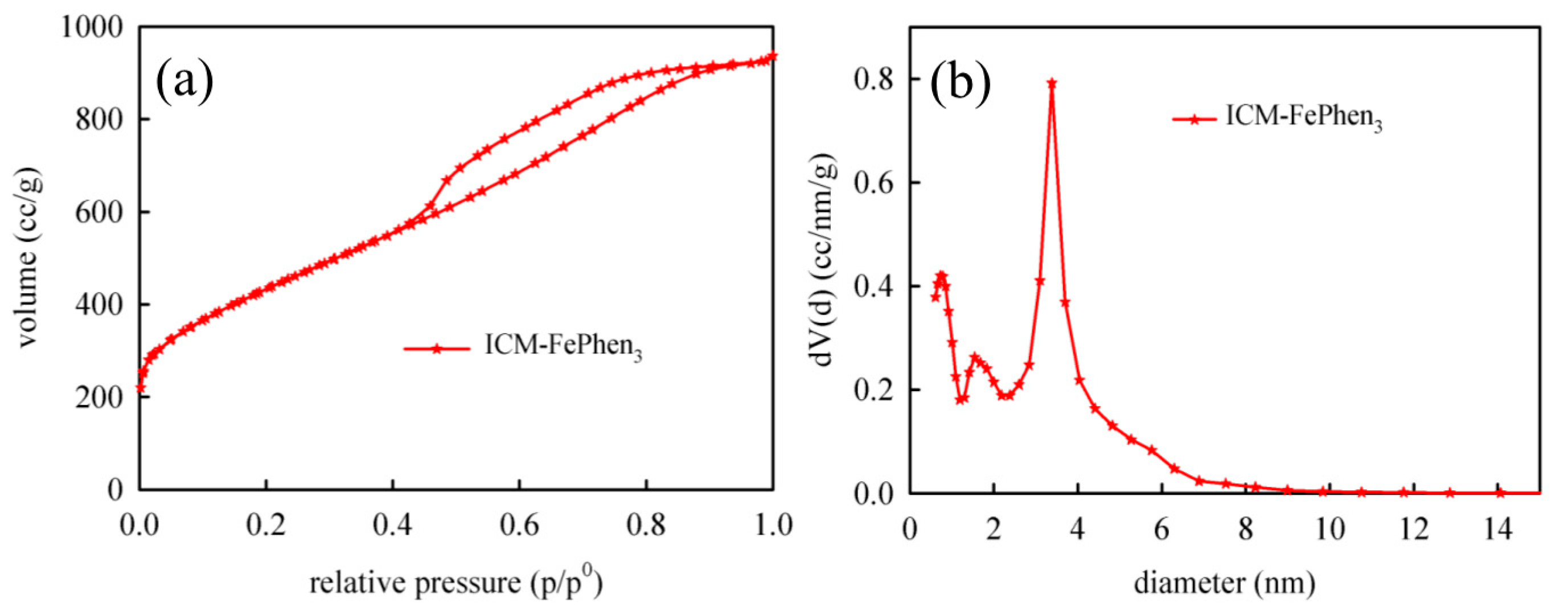
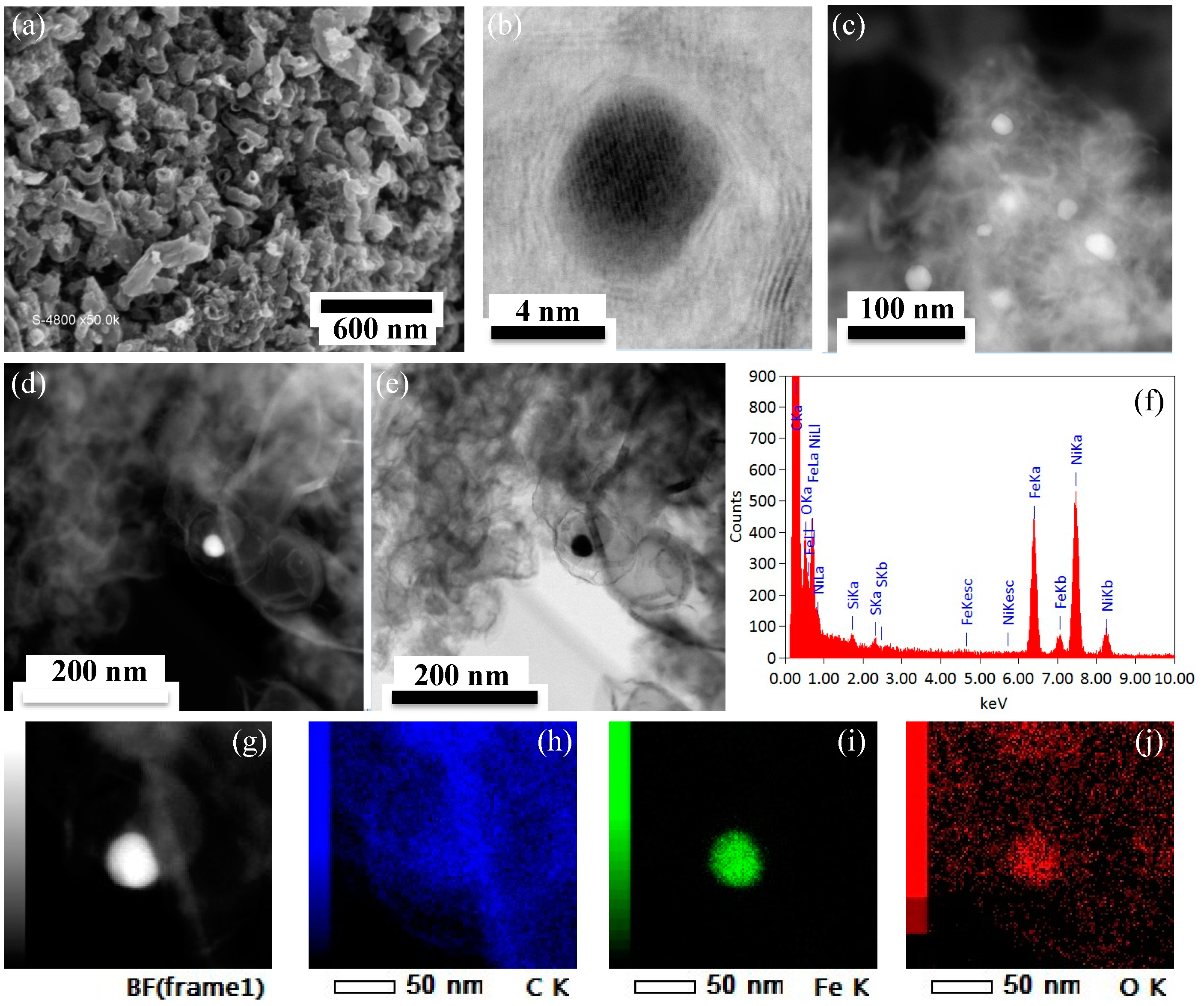
2.3. Porous Structure and Morphology
3. Discussion
4. Materials and Methods
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Kulkarni, A.; Siahrostami, S.; Patel, A.; Nørskov, J.K. Understanding catalytic activity trends in the oxygen reduction reaction. Chem. Rev. 2018, 118, 2302–2312. [Google Scholar] [CrossRef] [PubMed]
- Sui, S.; Wang, X.; Zhou, X.; Su, Y.; Riffat, S.; Liu, C.-J. A comprehensive review of Pt electrocatalysts for the oxygen reduction reaction: Nanostructure, activity, mechanism and carbon support in PEM fuel cells. J. Mater. Chem. A 2017, 5, 1808–1825. [Google Scholar] [CrossRef]
- Nie, Y.; Li, L.; Wei, Z. Recent advancements in Pt and Pt-free catalysts for oxygen reduction reaction. Chem. Soc. Rev. 2015, 44, 2168–2201. [Google Scholar] [CrossRef] [PubMed]
- Setzler, B.P.; Zhuang, Z.; Wittkopf, J.A.; Yan, Y. Activity targets for nanostructured platinum-group-metal-free catalysts in hydroxide exchange membrane fuel cells. Nat. Nanotechnol. 2016, 11, 1020–1025. [Google Scholar] [CrossRef] [PubMed]
- Shao, M.; Chang, Q.; Dodelet, J.-P.; Chenitz, R. Recent advances in electrocatalysts for oxygen reduction reaction. Chem. Rev. 2016, 116, 3594–3657. [Google Scholar] [CrossRef] [PubMed]
- Chattot, R.; Bacq, O.L.; Beermann, V.; Kühl, S.; Herranz, J.; Henning, S.; Kühn, L.; Asset, T.; Guétaz, L.; Renou, G.; et al. Surface distortion as a unifying concept and descriptor in oxygen reduction reaction electrocatalysis. Nat. Mater. 2018, 17, 827–833. [Google Scholar] [CrossRef]
- Huang, X.; Zhao, Z.; Cao, L.; Chen, Y.; Zhu, E.; Lin, Z.; Li, M.; Yan, A.; Zettl, A.; Wang, Y.M.; et al. High-performance transition metal–doped Pt3Ni octahedra for oxygen reduction reaction. Science 2015, 348, 1230–1234. [Google Scholar] [CrossRef]
- Stamenkovic, V.R.; Fowler, B.; Mun, B.S.; Wang, G.; Ross, P.N.; Lucas, C.A.; Marković, N.M. Improved oxygen reduction activity on Pt3Ni(111) via increased surface site availability. Science 2007, 315, 493–497. [Google Scholar] [CrossRef]
- Chong, L.; Wen, J.; Kubal, J.; Sen, F.G.; Zou, J.; Greeley, J.; Chan, M.; Barkholtz, H.; Ding, W.; Liu, D.-J. Ultralow-loading platinum-cobalt fuel cell catalysts derived from imidazolate frameworks. Science 2018, 362, 1276–1281. [Google Scholar] [CrossRef]
- Rosli, N.F.; Mayorga-Martinez, C.C.; Latiff, N.M.; Rohaizad, N.; Sofer, Z.; Fisher, A.C.; Pumera, M. Layered PtTe2 matches electrocatalytic performance of Pt/C for oxygen reduction reaction with significantly lower toxicity. ACS Sustain. Chem. Eng. 2018, 6, 7432–7441. [Google Scholar] [CrossRef]
- Zitolo, A.; Goellner, V.; Armel, V.; Sougrati, M.-T.; Mineva, T.; Stievano, L.; Fonda, E.; Jaouen, F. Identification of catalytic sites for oxygen reduction in iron-and nitrogen-doped graphene materials. Nat. Mater. 2015, 14, 937–942. [Google Scholar] [CrossRef] [PubMed]
- Zitolo, A.; Ranjbar-Sahraie, N.; Mineva, T.; Li, J.; Jia, Q.; Stamatin, S.; Harrington, G.F.; Lyth, S.M.; Krtil, P.; Mukerjee, S.; et al. Identification of catalytic sites in cobalt-nitrogen-carbon materials for the oxygen reduction reaction. Nat. Commun. 2017, 8, 957. [Google Scholar] [CrossRef]
- Wang, X.X.; Cullen, D.A.; Pan, Y.T.; Hwang, S.; Wang, M.; Feng, Z.; Wang, J.; Engelhard, M.H.; Zhang, H.; He, Y.; et al. Nitrogen-coordinated single cobalt atom catalysts for oxygen reduction in proton exchange membrane fuel cells. Adv. Mater. 2018, 30, 1706758–1706768. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Hwang, S.; Wang, M.; Feng, Z.; Karakalos, S.; Luo, L.; Qiao, Z.; Xie, X.; Wang, C.; Su, D.; et al. Single atomic iron catalysts for oxygen reduction in acidic media: Particle size control and thermal activation. J. Am. Chem. Soc. 2017, 139, 14143–14149. [Google Scholar] [CrossRef]
- Zhu, C.; Shi, Q.; Xu, B.Z.; Fu, S.; Wan, G.; Yang, C.; Yao, S.; Song, J.; Zhou, H.; Du, D.; et al. Hierarchically Porous M-N-C (M = Co and Fe) Single-atom electrocatalysts with robust MNx active moieties enable enhanced ORR performance. Adv. Energy Mater. 2018, 29, 1801956–1801963. [Google Scholar] [CrossRef]
- Li, J.; Jaouen, F. Structure and activity of metal-centered coordination sites in pyrolyzed metal-nitrogen-carbon catalysts for the electrochemical reduction of O2. Curr. Opin. Electrochem. 2018, 9, 198–206. [Google Scholar] [CrossRef]
- Li, J.; Ghoshal, S.; Liang, W.; Sougrati, M.-T.; Jaouen, F.; Halevi, B.; McKinney, S.; McCool, G.; Ma, C.; Yuan, X.; et al. Structural and mechanistic basis for the high activity of Fe-N-C catalysts toward oxygen reduction. Energy Environ. Sci. 2016, 9, 2418–2432. [Google Scholar] [CrossRef]
- Jia, Q.; Ramaswamy, N.; Tylus, U.; Strickland, K.; Li, J.; Serov, A.; Artyushkova, K.; Atanassov, P.; Anibal, J.; Gumeci, C.; et al. Spectroscopic insights into the nature of active sites in iron-nitrogen-carbon electrocatalysts for oxygen reduction in acid. Nano Energy 2016, 29, 65–82. [Google Scholar] [CrossRef]
- Jia, Q.; Ramaswamy, N.; Hafiz, H.; Tylus, U.; Strickland, K.; Wu, G.; Barbiellini, B.; Bansil, A.; Holby, E.F.; Zelenay, P.; et al. Experimental observation of redox-induced Fe-N switching behavior as a determinant role for oxygen reduction activity. ACS Nano 2015, 9, 12496–12505. [Google Scholar] [CrossRef]
- Choi, C.H.; Lim, H.-K.; Chung, M.W.; Chon, G.; Sahraie, N.R.; Altin, A.; Sougrati, M.-T.; Stievano, L.; Oh, H.S.; Park, E.S.; et al. The Achilles’ heel of iron-based catalysts during oxygen reduction in an acidic medium. Energy Environ. Sci. 2018, 11, 3176–3182. [Google Scholar] [CrossRef]
- Jaouen, F.; Lefèvre, M.; Dodelet, J.-P.; Cai, M. Heat-treated Fe/N/C catalysts for O2 electroreduction: Are active sites hosted in micropores? J. Phys. Chem. B 2006, 110, 5553–5558. [Google Scholar] [CrossRef] [PubMed]
- Proietti, E.; Jaouen, F.; Lefèvre, M.; Larouche, N.; Tian, J.; Herranz, J.; Dodelet, J.-P. Iron-based cathode catalyst with enhanced power density in polymer electrolyte membrane fuel cells. Nat. Commun. 2011, 2, 416. [Google Scholar] [CrossRef] [PubMed]
- Shui, J.; Chen, C.; Grabstanowicz, L.; Zhao, D.; Liu, D.-J. Highly efficient nonprecious metal catalyst prepared with metal–organic framework in a continuous carbon nanofibrous network. Proc. Natl. Acad. Sci. USA 2015, 112, 10629–10634. [Google Scholar] [CrossRef] [PubMed]
- Chang, Y.; Antonietti, M.; Fellinger, T.P. Synthesis of nanostructured carbon through ionothermal carbonization of common organic solvents and solutions. Angew. Chem. Int. Ed. 2015, 54, 5507–5512. [Google Scholar] [CrossRef] [PubMed]
- Pampel, J.; Denton, C.; Fellinger, T.-P. Glucose derived ionothermal carbons with tailor-made porosity. Carbon 2016, 107, 288–296. [Google Scholar] [CrossRef]
- Fellinger, T.-P. Sol-gel carbons from ionothermal syntheses. J. Sol-Gel Sci. Technol. 2017, 81, 52–58. [Google Scholar] [CrossRef]
- Pampel, J.; Mehmood, A.; Antonietti, M.; Fellinger, T.-P. Ionothermal template transformations for preparation of tubular porous nitrogen doped carbons. Mater. Horizons 2017, 4, 493–501. [Google Scholar] [CrossRef]
- Antonietti, M.; Fechler, N.; Fellinger, T.-P. Carbon aerogels and monoliths: Control of porosity and nanoarchitecture via sol-gel routes. Chem. Mater. 2013, 26, 196–210. [Google Scholar] [CrossRef]
- Li, J.; Chen, S.; Li, W.; Wu, R.; Ibraheem, S.; Li, J.; Ding, W.; Li, L.; Wei, Z. A eutectic salt-assisted semi-closed pyrolysis route to fabricate high-density active-site hierarchically porous Fe/N/C catalysts for the oxygen reduction reaction. J. Mater. Chem. A 2018, 6, 15504–15509. [Google Scholar] [CrossRef]
- Li, J.; Alsudairi, A.; Ma, Z.-F.; Mukerjee, S.; Jia, Q. Asymmetric volcano trend in oxygen reduction activity of Pt and non-Pt catalysts: In situ identification of the site-blocking effect. J. Am. Chem. Soc. 2017, 139, 1384–1387. [Google Scholar] [CrossRef]
- Webster, S.; Maultzsch, J.; Thomsen, C.; Liu, J.; Czerw, R.; Terrones, M.; Adar, F.; John, C.; Whitley, A.; Carroll, D.L. Raman characterization of nitrogen doped multiwalled carbon nanotubes. MRS Online Proc. Libr. Arch. 2003, 772. [Google Scholar] [CrossRef]
- Ferrari, A.C.; Basko, D.M. Raman spectroscopy as a versatile tool for studying the properties of graphene. Nat. Nanotechnol. 2013, 8, 235–246. [Google Scholar] [CrossRef] [PubMed]
- Wu, G.; Santandreu, A.; Kellogg, W.; Gupta, S.; Ogoke, O.; Zhang, H.; Wang, H.-L.; Dai, L. Carbon nanocomposite catalysts for oxygen reduction and evolution reactions: From nitrogen doping to transition-metal addition. Nano Energy 2016, 29, 83–110. [Google Scholar] [CrossRef]
- Jiao, L.; Wan, G.; Zhang, R.; Zhou, H.; Yu, S.-H.; Jiang, H.-L. From Metal-organic frameworks to single-atom Fe implanted N-doped porous carbons: Efficient oxygen reduction in both alkaline and acidic media. Angew. Chem. Int. Ed. 2018, 130, 8661–8665. [Google Scholar] [CrossRef]
- Chen, P.; Zhou, T.; Xing, L.; Xu, K.; Tong, Y.; Xie, H.; Zhang, L.; Yan, W.; Chu, W.; Wu, C.; et al. Atomically dispersed iron–nitrogen species as electrocatalysts for bifunctional oxygen evolution and reduction reactions. Angew. Chem. Int. Ed. 2017, 56, 610–614. [Google Scholar] [CrossRef] [PubMed]
- Yi, J.-D.; Xu, R.; Wu, Q.; Zhang, T.; Zang, K.-T.; Luo, J.; Liang, Y.-L.; Huang, Y.-B.; Cao, R. Atomically dispersed iron-nitrogen active sites within porphyrinic triazine-based frameworks for oxygen reduction reaction in both alkaline and acidic media. ACS Energy Lett. 2018, 3, 883–889. [Google Scholar] [CrossRef]
- Strickland, K.; Miner, E.; Jia, Q.; Tylus, U.; Ramaswamy, N.; Liang, W.; Sougrati, M.-T.; Jaouen, F.; Mukerjee, S. Highly active oxygen reduction non-platinum group metal electrocatalyst without direct metal–nitrogen coordination. Nat. Commun. 2015, 6, 7343. [Google Scholar] [CrossRef]
- Tuček, J.; Zbořil, R.; Namai, A.; Ohkoshi, S.-I. ε-Fe2O3: An advanced nanomaterial exhibiting giant coercive field, millimeter-wave ferromagnetic resonance, and magnetoelectric coupling. Chem. Mater. 2010, 22, 6483–6505. [Google Scholar] [CrossRef]
- Kramm, U.I.; Herrmann-Geppert, I.; Behrends, J.; Lips, K.; Fiechter, S.; Bogdanoff, P. On an easy way to prepare metal–nitrogen doped carbon with exclusive presence of MeN4-type sites active for the ORR. J. Am. Chem. Soc. 2016, 138, 635–640. [Google Scholar] [CrossRef]
- Chen, J.Y.; Dang, L.; Liang, H.; Bi, W.; Gerken, J.B.; Jin, S.; Alp, E.E.; Stahl, S.S. Operando analysis of NiFe and Fe oxyhydroxide electrocatalysts for water oxidation: Detection of Fe4+ by Mossbauer spectroscopy. J. Am. Chem. Soc. 2015, 137, 15090–15093. [Google Scholar] [CrossRef]
- Zboril, R.; Mashlan, M.; Petridis, D. Iron (III) oxides from thermal processes synthesis, structural and magnetic properties, Mössbauer spectroscopy characterization, and applications. Chem. Mater. 2002, 14, 969–982. [Google Scholar] [CrossRef]
- Kamali-M, S.; Ericsson, T.; Wäppling, R. Characterization of iron oxide nanoparticles by Mössbauer spectroscopy. Thin Solid Films 2006, 515, 721–723. [Google Scholar] [CrossRef]
- Shinjo, T.; Kiyama, M.; Sugita, N.; Watanabe, K.; Takada, T. Surface magnetism of α-Fe2O3 by Mössbauer spectroscopy. J. Magn. Magn. Mater. 1983, 35, 133–135. [Google Scholar] [CrossRef]
- Sougrati, M.T.; Goellner, V.; Schuppert, A.K.; Stievano, L.; Jaouen, F. Probing active sites in iron-based catalysts for oxygen electro-reduction: A temperature-dependent 57Fe Mössbauer spectroscopy study. Catal. Today 2016, 262, 110–120. [Google Scholar] [CrossRef]
- Kramm, U.I.; Herranz, J.; Larouche, N.; Arruda, T.M.; Lefèvre, M.; Jaouen, F.; Bogdanoff, P.; Fiechter, S.; Abs-Wurmbach, I.; Mukerjee, S.; et al. Structure of the catalytic sites in Fe/N/C-catalysts for O2-reduction in PEM fuel cells. Phys. Chem. Chem. Phys. 2012, 14, 11673–11688. [Google Scholar] [CrossRef] [PubMed]
- Liang, H.-W.; Wei, W.; Wu, Z.-S.; Feng, X.; Müllen, K. Mesoporous metal-nitrogen-doped carbon electrocatalysts for highly efficient oxygen reduction reaction. J. Am. Chem. Soc. 2013, 135, 16002–16005. [Google Scholar] [CrossRef] [PubMed]
- Chung, H.T.; Cullen, D.A.; Higgins, D.; Sneed, B.T.; Holby, E.F.; More, K.L.; Zelenay, P. Direct atomic-level insight into the active sites of a high-performance PGM-free ORR catalyst. Science 2017, 357, 479–484. [Google Scholar] [CrossRef]
- Li, Y.; Huang, J.; Hu, X.; Bi, L.; Cai, P.; Jia, J.; Chai, G.; Wei, S.; Dai, L.; Wen, Z. Fe vacancies induced surface FeO6 in nanoarchitectures of N-doped graphene protected β-FeOOH: Effective active sites for pH-universal electrocatalytic oxygen reduction. Adv. Funct. Mater. 2018, 28, 1803330–1803338. [Google Scholar] [CrossRef]
- Negro, E.; Nale, A.; Vezzù, K.; Pagot, G.; Polizzi, S.; Bertoncello, R.; Ansaldo, A.; Prato, M.; Bonaccorso, F.; Rutkowska, I.A.; et al. Hierarchical oxygen reduction reaction electrocatalysts based on FeSn0.5 species embedded in carbon nitride-graphene based supports. Electrochim. Acta 2018, 280, 149–162. [Google Scholar] [CrossRef]
- Negro, E.; Delpeuch, A.B.; Vezzu, K.; Nawn, G.; Bertasi, F.; Ansaldo, A.; Pellegrini, V.; Dembinska, B.; Zoladek, S.; Miecznikowski, K.; et al. Toward Pt-free anion-exchange membrane fuel cells: Fe–Sn carbon nitride–graphene core–shell electrocatalysts for the oxygen reduction reaction. Chem. Mater. 2018, 30, 2651–2659. [Google Scholar] [CrossRef]
- Kong, F.; Fan, X.; Kong, A.; Zhou, Z.; Zhang, X.; Shan, Y. Covalent phenanthroline framework derived FeS@Fe3C composite nanoparticles embedding in N-S-codoped carbons as highly efficient trifunctional electrocatalysts. Adv. Funct. Mater. 2018, 28, 1803973. [Google Scholar] [CrossRef]
- Fan, X.; Kong, F.; Kong, A.; Chen, A.; Zhou, Z.; Shan, Y. Covalent porphyrin framework-derived Fe2P@ Fe4N-coupled nanoparticles embedded in N-doped carbons as efficient trifunctional electrocatalysts. ACS Appl. Mater. Interfaces 2017, 9, 32840–32850. [Google Scholar] [CrossRef] [PubMed]
- Varnell, J.A.; Edmund, C.; Schulz, C.E.; Fister, T.T.; Haasch, R.T.; Timoshenko, J.; Frenkel, A.I.; Gewirth, A.A. Identification of carbon-encapsulated iron nanoparticles as active species in non-precious metal oxygen reduction catalysts. Nat. Commun. 2016, 7, 12582. [Google Scholar] [CrossRef] [PubMed]
- Reda, M.; Hansen, H.A.; Vegge, T. DFT Study of the oxygen reduction reaction on carbon-coated iron and iron carbide. ACS Catal. 2018, 8, 10521–10529. [Google Scholar] [CrossRef]
- Ravel, B.; Newville, M. ATHENA, ARTEMIS, HEPHAESTUS: Data analysis for X-ray absorption spectroscopy using IFEFFIT. J. Synchrotron Radiat. 2005, 12, 537–541. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | Comp. | RA % | IS mm/s | QS mm/s | LW mm/s | H Tesla | Assignment |
|---|---|---|---|---|---|---|---|
| ICM-FePhen3 300 K | Singlet | 122 | 0.1 | - | 2.7 | - | Unresolved (1) |
| Doublet | 464 | 0.34 | 0.741 | 0.54 | - | Nano-Fe2O3 | |
| Sextet 1 | 61 | 0.31 | - | 0.7 | 44.4 | Fe2O3 | |
| Sextet 2 | 61 | 0.31 | - | 0.7 | 47.8 | Fe2O3 | |
| Sextet 3 | 301 | 0.19 | - | 0.48 | 20.6 | Fe3C | |
| ICM-FePhen3 5 K | Singlet | 222 | 0.21 | - | 4.0 | - | Unresolved (1) |
| Sextet 1 | 232 | 0.48 | - | 0.77 | 47.9 | Fe2O3 | |
| Sextet 2 | 392 | 0.49 | - | 0.67 | 51.7 | Fe2O3 | |
| Sextet 3 | 162 | 0.33 | - | 0.49 | 25.7 | Fe3C | |
| ICM-FePhen3-AW 300 K | Singlet | 5512 (2) | 0.17 | - | 2.6 | - | Unresolved (1) |
| Sextet | 4512 (2) | 0.26 | - | 0.5 | 20.8 | Fe3C |
| Sample | Mössbauer Component | Mössbauer RA % (300 K) | Mössbauer RA % (5 K) | LCF XANES Reference | XANES % |
|---|---|---|---|---|---|
| ICM-FePhen3 | Singlet | 122 | 222 | Fe0.5NC | 253 |
| Fe2O3 (1) | 58 | 622 | Fe2O3 | 644 | |
| Fe3C | 301 | 162 | Fe3C | 111 | |
| ICM-FePhen3-AW | Singlet | 5512 (2) | NA | Fe0.5NC | 383 |
| Fe2O3 (1) | 0 | NA | Fe2O3 | 304 | |
| Fe3C | 4512 (2) | NA | Fe3C | 321 |
| Sample | BET Surface Area (m2/g) | Pore Volume (cc/g) | Pore Size (nm) | |
|---|---|---|---|---|
| Single Point | DFT | DFT | ||
| ICM-FePhen3 | 1546.0 | 1.5 | 1.4 | 3.4 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, J.; Jia, Q.; Mukerjee, S.; Sougrati, M.-T.; Drazic, G.; Zitolo, A.; Jaouen, F. The Challenge of Achieving a High Density of Fe-Based Active Sites in a Highly Graphitic Carbon Matrix. Catalysts 2019, 9, 144. https://doi.org/10.3390/catal9020144
Li J, Jia Q, Mukerjee S, Sougrati M-T, Drazic G, Zitolo A, Jaouen F. The Challenge of Achieving a High Density of Fe-Based Active Sites in a Highly Graphitic Carbon Matrix. Catalysts. 2019; 9(2):144. https://doi.org/10.3390/catal9020144
Chicago/Turabian StyleLi, Jingkun, Qingying Jia, Sanjeev Mukerjee, Moulay-Tahar Sougrati, Goran Drazic, Andrea Zitolo, and Frédéric Jaouen. 2019. "The Challenge of Achieving a High Density of Fe-Based Active Sites in a Highly Graphitic Carbon Matrix" Catalysts 9, no. 2: 144. https://doi.org/10.3390/catal9020144
APA StyleLi, J., Jia, Q., Mukerjee, S., Sougrati, M.-T., Drazic, G., Zitolo, A., & Jaouen, F. (2019). The Challenge of Achieving a High Density of Fe-Based Active Sites in a Highly Graphitic Carbon Matrix. Catalysts, 9(2), 144. https://doi.org/10.3390/catal9020144