1. Introduction
The continuous growth of global polymer production leads to the development of new methods and catalysts for obtaining of olefin monomers; for example, alkane dehydrogenation, methanol-to-olefins, metathesis and olefin oligomerization [
1,
2,
3,
4,
5]. In its turn, ethylene oligomerization aimed to obtain linear alpha-olefins (LAO), which are used as co-monomers in the production of the demanded types of polyethylene (linear low-density polyethylene, high-density polyethylene), as well as for the production of a wide range of unique chemicals, such as lubricants and additives, plasticizers, surfactants and so forth [
6].
From the development of the Ziegler–Natta catalytic system to the present time, zirconium organometallic compounds have been widely investigated in the context of their use as pre-catalysts (substances that acquire catalytic properties after activation) in the reactions of the oligomerization and polymerization of unsaturated hydrocarbons [
4,
7,
8,
9,
10,
11,
12]. For example, zirconium pre-catalysts are used in many industrial processes of homogeneous ethylene oligomerization, such as Alphaselect (IFP Energies Nouvelles, Rueil-Malmaison CEDEX, France), Alpha–Sablin (SABIC-Linde, Munich, Germany) and the process of Idemitsu Kosan Co. Ltd (Tokyo, Japan) [
10,
11,
12]. The products of these processes are even-numbered LAOs with a wide distribution regarding the number of carbon atoms (from 4 to 30–40).
Usually, the main precursor for zirconium catalysts is zirconium tetrachloride [
13,
14,
15,
16,
17]. However, the disadvantage of using this reagent is connected to its high hygroscopicity and, as a result, its low air resistance, as well as the release of hydrogen chloride during the preparation of the pre-catalyst, which negatively affects the nature environment. The use of the electrochemical synthesis of zirconium carboxylates will not only exclude the release of HCl but will also reduce the energy costs. Therefore, the elaboration of new methods for the preparation of Zr-based pre-catalysts is of high interest.
Herein we present the results of the electrochemical synthesis of zirconium carboxylates directly from a metal and the corresponding acid, which are efficient pre-catalysts for ethylene oligomerization.
2. Results and Discussion
The traditional method of zirconium carboxylate synthesis is highly energy-consuming. It involves the prolonged boiling of zirconium tetrachloride in carboxylic acid or its solution [
18,
19,
20,
21,
22]. A direct electrochemical method for the preparation of transition metal carboxylates in mild and ecologically safe conditions is currently known [
23]; however, this example is limited by di- and trivalent 3D-metals (Cr, Mn, Fe, Co, Ni, Cu). We have tried to use a similar approach for the synthesis of zirconium carboxylates with a variety of carboxylic acids bearing a wide range of carbon atoms in hydrocarbon chain (acetate, octanoate, laurate). The choice of these compounds was based on their usage as a pre-catalyst in an important industrial process of LAO synthesis—Alpha–Sablin (SABIC-Linde) [
11]—as well as due to their use for the synthesis of materials with unique properties, such as metal–organic frameworks and catalysts [
24,
25,
26].
2.1. Cyclic Voltammetry
The electrochemical system which was planned to be used in electrosynthesis was investigated by cyclic voltammetry (
Figure 1). The following electrodes were used: working electrode—zirconium plate; reference electrode—silver electrode Ag/AgNO
3 (0.01 mol⋅L
−1 solution in acetonitrile); auxiliary electrode—platinum wire.
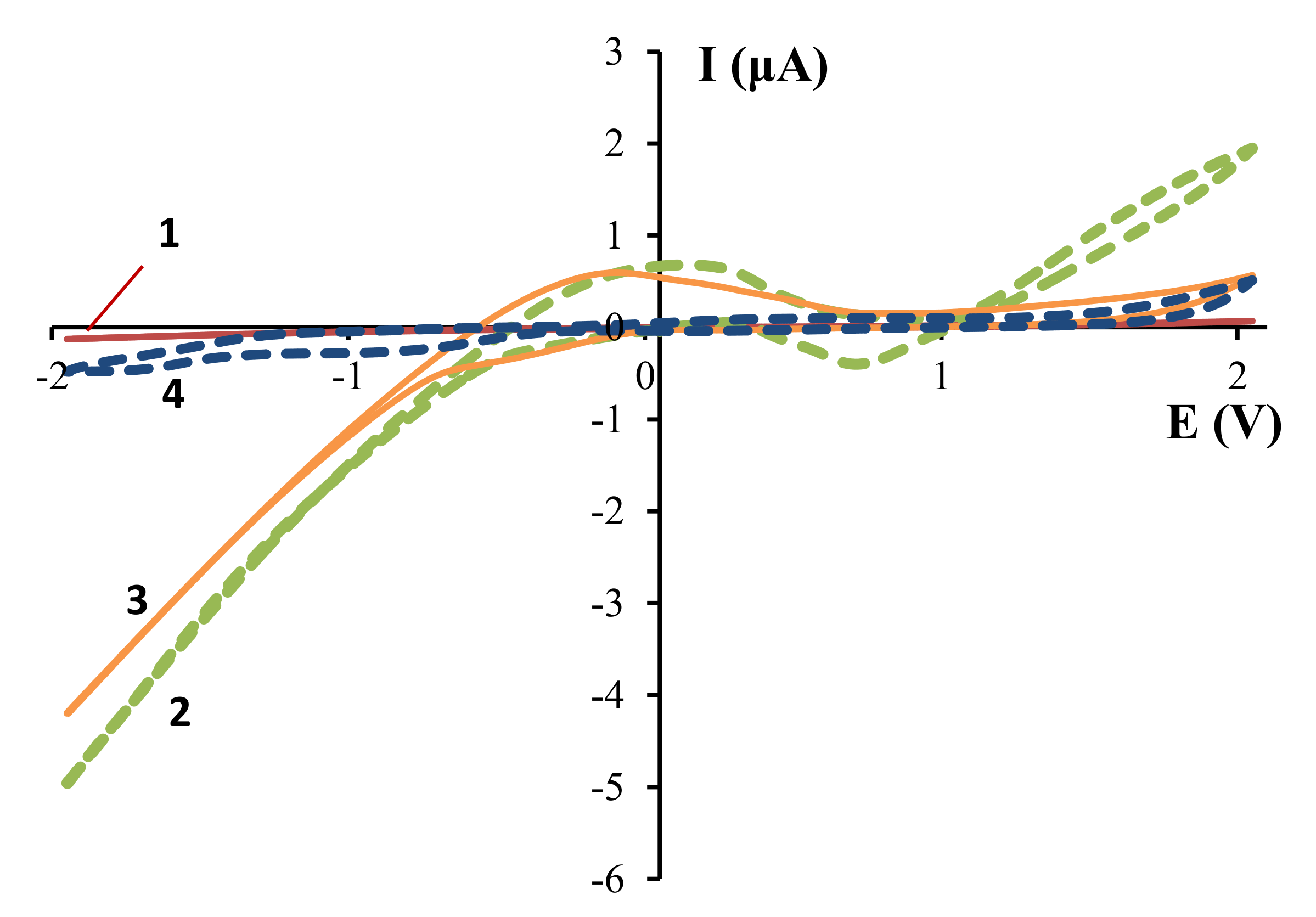
The voltammetric scanning of an acetic acid solution showed that the anodic and cathodic currents are close to 0 (curve 1,
Figure 1). With the addition of Et
4NCl-supporting salt, the anodic and cathodic current increased significantly (up to 5 and 2 μA, respectively) and a shoulder in the (–0.5–0.5 V) region and a negative signal in the (0.3–1.0 V) region appeared (curve 2,
Figure 1). The significant increase in the cathodic current is probably due to a shift in the equilibrium of the acetic acid dissociation to the right by Et
4N
+ ions, which accumulated near the cathode, and subsequent hydrogen sorption and reduction at the Zr-cathode. The increased anodic current indicates the surface interaction of zirconium with dissociated acetic acid.
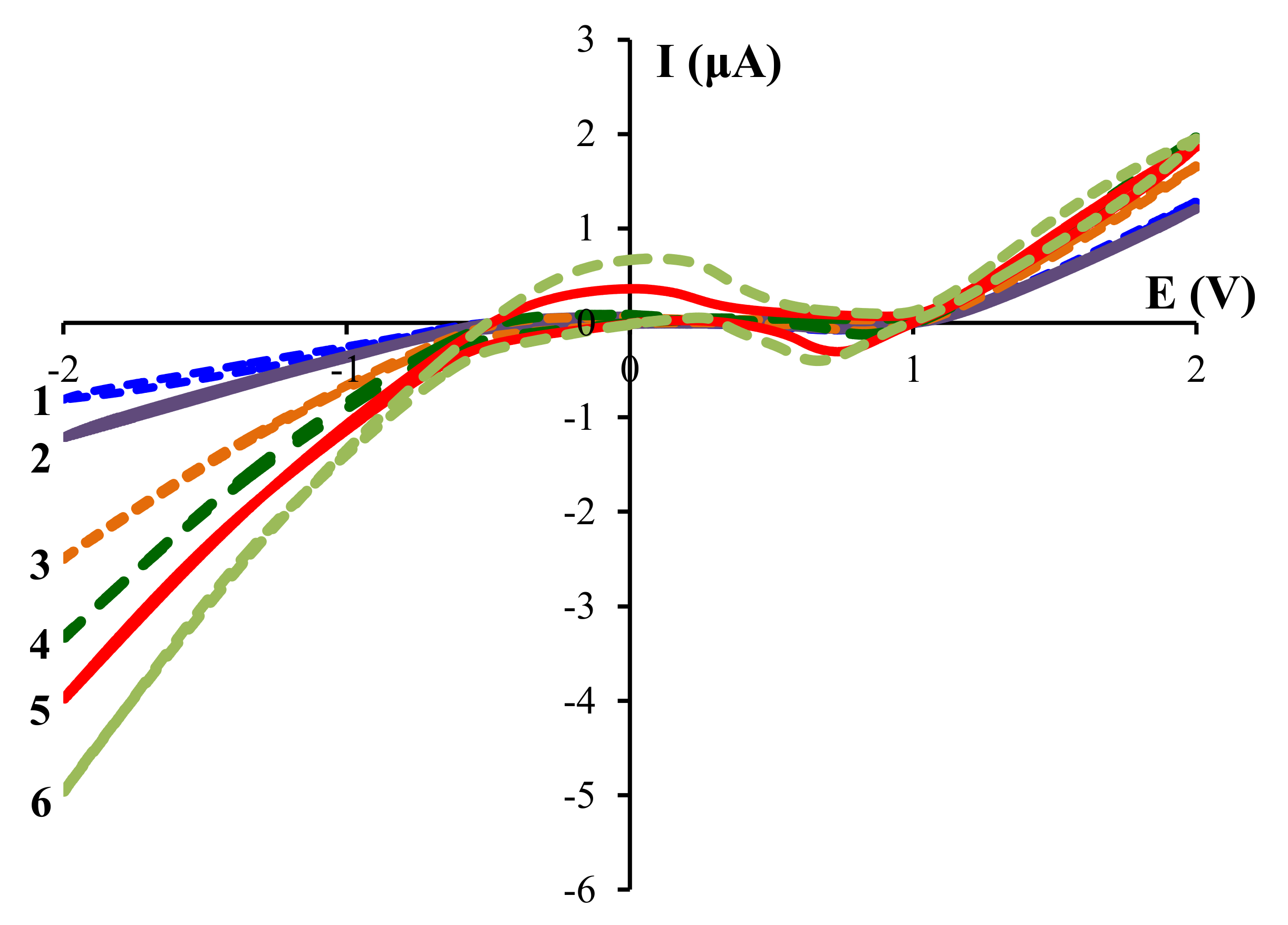
The shoulder in the transition (–0.5–0.5 V) region is probably due to desorption from the electrode of hydrogen, which reduced at more negative potentials. This assumption is confirmed by an increase in the intensity of this signal with acetic acid concentration (
Figure 2). The negative signal in the anodic (0.3–1.0 V) region also became more pronounced with acetic acid concentration. This signal is probably due to the product of the interaction between zirconium and acetic acid.
The replacement of the Et
4NCl-supporting salt by
nBu
4NBF
4 is accompanied by a decrease in the anode current (curve 3,
Figure 1). This may indicate that the electrochemical interaction of acetic acid with the zirconium anode is realized by the preliminary formation of the Zr–Cl bond (Equations (1)–(3)).
Equations (1)–(3). Proposed mechanism of interaction between the Zr-anode and carboxylate ions.
At the same time, the current at the zirconium cathode is nearly constant (curve 3,
Figure 1), which also may be due to the shift of the equilibrium of acetic acid dissociation in the presence of
nBu
4N
+.
The relatively high value of the anode current in the presence of the
nBu
4NBF
4 might be due to the anodic oxidation of BF
4– ions rather than the interaction of acetic acid with zirconium. This assumption is confirmed by the preservation of the anodic current in the absence of acetic acid (curve 4,
Figure 1). The oxidation of BF
4− ions to BF
3, F
2, B
2F
7− on a platinum anode was observed in Reference [
27].
It should be noted that, for solutions with octanoic (C
7H
15COOH) and lauric acid (C
11H
23COOH), similar voltammograms and regularities were also observed (see
Supplementary Materials).
Thus, cyclic voltammetry studies showed the possibility of the interaction between zirconium metal and carboxylic acid under electrochemical conditions in the presence of a supporting salt, which contains chloride anion.
2.2. Electrosynthesis
The electrochemical synthesis of zirconium carboxylates was carried out in an electrochemical cell without separation of the anodic and cathodic compartments, without the thermostating and stirring of the electrolyte. Zirconium plates were used as the anode and cathode. The conditions and characteristics of electrochemical synthesis are shown in
Table 1.
After 120 min of electrolysis of the solution containing 1.0 mL of acetic acid, 10 mL of acetonitrile and 16 mg of Et
4NCl, yellow coloration occurred (entry 1,
Table 1). A similar color change was observed when the current was passed through the electrolyte without acetic acid. This indicates the interaction of zirconium with acetonitrile or supporting salt, which may cause the contamination of the obtained carboxylate. To prevent the interaction, an excess of acid was used, which allowed us to carry out electrolysis until the precipitate was formed. Electrolysis was stopped when the current decreased to 10 mA due to the anode being covered by the precipitate. As a result of the electrolysis of acetic acid solution, a white powder was obtained; for octanoic acid solution, a yellow viscous liquid at the bottom of the cell was obtained (entries 2 and 3 respectively,
Table 1).
Lauric acid concentration in the electrolyte was limited by its solubility in acetonitrile. In this case, the least amount of product was formed (entry 4,
Table 1). The reason for this may be not only the low solubility of the starting acid but also the rapid covering of the anode by the product (white plastic mass) due to the large size of its molecules and therefore their low mobility.
To confirm the determining role of the halide anion in the interaction of zirconium with carboxylic acid, electrolysis of acetic acid solution with supporting salts containing halides (Br
–, I
–) and tetrafluoroborate (BF
4–) was performed. If Me
4NBr and Et
4NI were used as the supporting salt, the precipitate also formed during the electrolysis (entries 5 and 6,
Table 1). It should be noted that the resulting substances had a light beige color, probably due to the presence of residual bromine and iodine.
When
nBu
4NBF
4 was used, no precipitate formed (entry 7,
Table 1). The current gradually decreased as a result of the deposit formation on the electrodes. There was no precipitation from electrolyte even 12 h after the electrolysis. This confirms the crucial role of halogen in the electrosynthesis of zirconium carboxylates.
From the analysis of the electrochemical product composition (
Table 2) it can be concluded that the obtained substances are not stoichiometric zirconium carboxylates–[Zr(RCOO)
X] (3 < x < 4). This is indicated by the zirconium content, which is higher than the theoretical value for the Zr(RCOO)
4. This may be due to the formation of polymer structures with additional bonds between zirconium atoms [
20,
28]. Electrochemically synthesized zirconium acetate [Zr(Ac)
x] (Ac = CH
3COO) does not contain chlorine, while octanoate [Zr(Oct)
x] (Oct = C
7H
15COO) and laurate [Zr(Laur)
x] (Laur = C
11H
23COO) contain 0.4–0.5 wt% chlorine, which corresponds to a molar ratio of Zr/Cl = 16–18. This confirms that Cl
– ions play the role of mediators during electrosynthesis.
2.3. Nuclear Magnetic Resonance Spectroscopy
The products of electrosynthesis were studied by 1H and 13C{1H} NMR spectroscopy in CDCl3 as a solvent.
On the
1H NMR spectra of all samples (see
Supplementary Materials), there was no signal of the hydrogen of the carboxyl group in the region of ~11.5 ppm [
29], indicating the absence of residual acids in the obtained substances. On the spectrum of [Zr(Ac)
x], the hydrogen signal of the methyl group split to a multiplet. For all samples, a shift of the hydrogen signals to the high-field region is observed; this effect is more pronounced for hydrogen of methyl/methylenes adjacent to the COO
– group. The splitting and shifting of hydrogen signals indicate the coordination of carboxylate moieties to the metal, as well as the formation of metal carboxylate associates. A similar signal shifting is observed for other carboxylates (e.g., lithium acetate, sodium octanoate) [
29].
On the
13C{
1H} NMR spectrum of [Zr(Ac)
x], for carbon signals of COO
– and CH
3-groups, splitting and a shift to lower fields are observed. According to the Spectral Database for Organic Compounds [
29], the carbon signal of COO
– group in acetic acid is observed at 178.1 ppm. On the spectrum of the [Zr(Ac)
x], the carbon signals of the COO
– group are presented as a triplet at 177.7–177.9 ppm and singlets at 176.5 ppm and 181.2 ppm. The signal of the acetic acid CH
3-group is observed at 20.8 ppm [
29], while in the case of [Zr(Ac)
x], it splits into a quadruplet at 23.6–24.1 ppm, a singlet at 23.1 ppm and a shoulder at 21.0 ppm. Similar effects were observed for the carboxylate group and the methyl group adjacent to it in the case of [Zr(Oct)
x] and [Zr(Laur)
x] (see
Supplementary Materials).
The relative intensity of the COO– group carbon signal is reduced compared to the CH3/CH2 groups carbon signals (in the case of [Zr(Laur)x], the signal at ~180 ppm is not observed, probably due to its limited solubility in CDCl3).
The splitting and shifting of carbon signals of COO
–, CH
3- (for [Zr(Ac)
x]) and -αCH
2-groups (for [Zr(Oct)
x] and [Zr(Laur)
x]), as well as the decrease in their intensity, are due to a change in the electron density at these carbon atoms as a result of the coordination of carboxylate moieties to zirconium [
30,
31,
32]. Also, conformational changes in carboxylate moieties and the formation of their nonequivalence due to different interactions with the metal center may be the cause of the observed splitting and shift of the carbon signals [
33,
34]. A similar effect of the splitting of carbon signals of the COO
– and –αCH
2-groups is observed for lead(II) carboxylates [
35]. The shift of the COO
– group carbon signal to lower fields is also observed for carboxylates of other metals [
29].
2.4. IR Spectroscopy
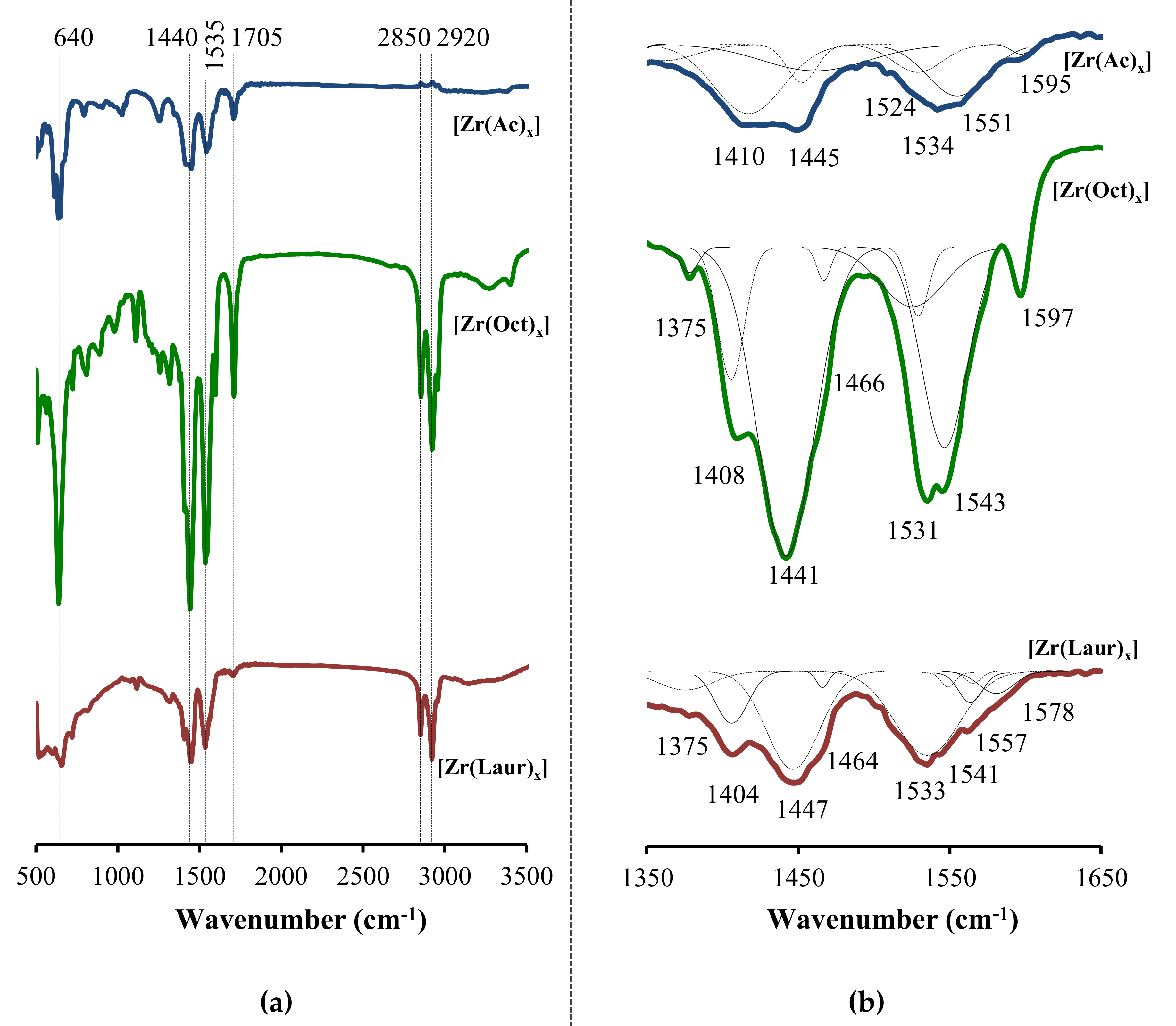
Electrochemical products were investigated by IR spectroscopy (
Figure 3).
The IR spectra of the samples have signals in the region of ~2900 cm
−1 corresponding to stretching vibrations of the CH
2- and CH
3-groups [
36]. Signals at ~640, ~1440, ~1530 cm
−1 are characteristic of the formation of metal carboxylates. Absorption at ~640 cm
−1 corresponds to the COO
– group bending mode [
37]. As a result of the formation of zirconium carboxylates, the absorption band at ~1700 cm
−1, which is assigned to antisymmetric stretching vibrations of the C=O bond in hydrogen-bonded dimers [
38], is converted into signals of symmetric (ν
sym(COO
–) at 1390–1460 cm
−1) and asymmetric (ν
asym(COO
–) at 1520–1590 cm
−1) stretching vibrations of the carboxylate group [
29,
36,
39] (
Figure 3 and
Supplementary Materials).
The ratio of intensities and the position of the signals ν
asym(COO
–) and ν
sym(COO
–) in the IR spectrum, as well as the difference of their frequencies Δν, are widely used to characterize the coordination mode of the carboxylate group to the metal [
40,
41]. For the studied samples, close intensities of ν
sym(COO
−) and ν
asym(COO
−) signals indicate the equivalence of C–O bonds in the carboxylate group [
41,
42]. The ν
asym(COO
−) signals have frequencies of 1530–1540 cm
−1, which are lower than corresponding signals for the ionic and monodentate coordinated carboxylates (1600–1630 cm
−1) [
40]. In addition, the frequency difference of Δν = 86–106 cm
−1 (
Figure 3) is significantly lower than the difference for the monodentate coordinated carboxylate (Δν = 230–300 cm
−1) [
40,
41] and corresponds to those for the chelate bidentate and bridge bidentate coordination [
40,
41,
43]. Therefore, in the electrochemically synthesized [Zr(Ac)
x], [Zr(Oct)
x] and [Zr(Laur)
x], both bidentate bridging and chelating coordination of COO
− groups to zirconium could be observed. The Gauss decomposition of IR spectra (
Figure 3b) confirms the variety of bidentate (Δν = 86–158 cm
−1) coordination modes of carboxylate moieties to zirconium. The lack of order in the coordination of carboxylate moieties to the metal center may be the reason of non-stoichiometric zirconium carboxylate formation [
18,
28].
As a result of the zirconium carboxylate formation, the signal at 920–940 cm
−1 (assigned to a C–C stretch adjacent to COO
–) disappeared and the intensity of the CH
2 rocking vibration signal at 720 cm
−1 decreased (see
Supplementary Materials) [
37,
43]. This is consistent with the results of
13C{
1H} NMR spectroscopy: a decrease in the intensity of signals of carbon atoms adjacent to the carboxylate group (see
Supplementary Materials).
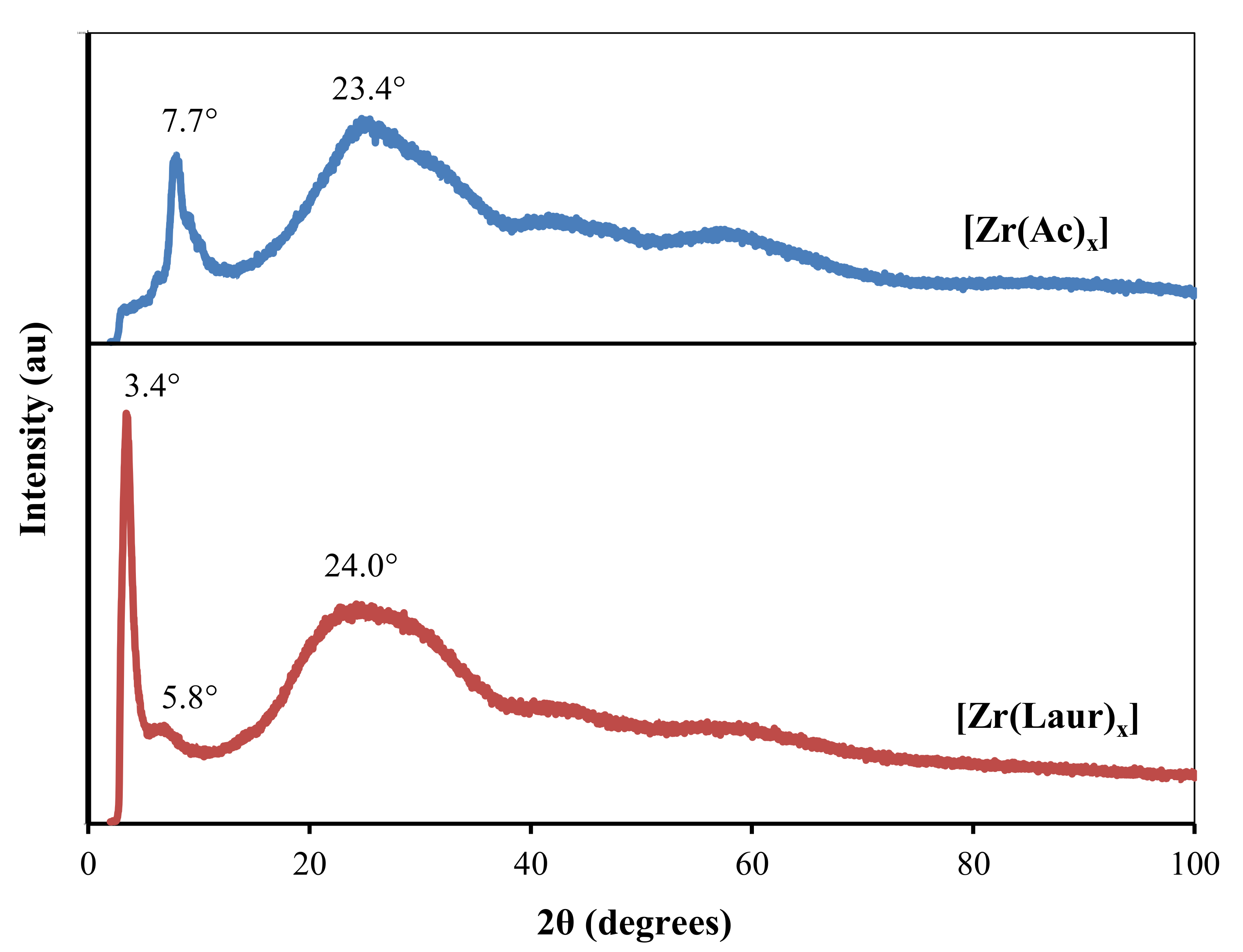
2.5. Powder X-Ray Diffraction (PXRD)
Solid products of electrosynthesis (based on acetic and lauric acids) were studied by PXRD. A high level of background in the diffractograms (
Figure 4) indicates that the products of electrosynthesis are amorphous substances. At the same time, they contain an ordered mesophase, which causes an intense reflection in the region of small angles (3.4–7.7°). With an increase in the number of carbon atoms in the carboxylate from 2 to 12 (with the transition from [Zr(Ac)
x] to [Zr(Laur)
x]), the d Bragg spacing value corresponding to this reflection increases from 11.5 Å (2θ = 7.7°) to 26.0 Å (2θ = 3.4°). A similar effect was observed in the work [
42] for cerium(III) carboxylates.
The ratio of d Bragg spacing values corresponding to neighboring reflections in the diffractogram of [Zr(Laur)
x] (at 2θ = 3.4° and 2θ = 5.8°) is 1:√3. This indicates the hexagonal packing of the mesophase. In addition, the d Bragg spacing value of 26.0 Å, which corresponds to the main reflection at 2θ = 3.4°, is more characteristic of the hexagonal packing than of the lamellar structure (typical for most carboxylates) with the same length of the alkyl chain [
37,
42,
43].
In the range 2θ = 20–40°, a wide halo is observed, which corresponds to diffraction from disordered alkyl chains [
37,
44].
2.6. Ethylene Oligomerization
Electrochemically synthesized [Zr(Ac)
x], [Zr(Oct)
x] and [Zr(Laur)
x] (3 < x < 4) were used as pre-catalysts in ethylene oligomerization. The results of the catalytic tests are shown in
Table 3. The product of oligomerization is the mixture of even-numbered olefins, predominantly LAOs, with a wide distribution on the carbon atoms, following Schulz–Flory law [
45,
46]. For the characterization of oligomer distribution on molecular weight, a Schulz–Flory parameter “α” was used, which is equal to the ratio of the propagation rate to the sum of propagation and chain transfer rates [
45]. Consequently, on the basis of the Schulz–Flory parameter, the molecular weight distribution of oligomers was calculated (
Table 3) [
45,
46]. In addition, in all oligomerization products, there were alkylbenzenes (mainly products of solvent (toluene) alkylation by C
4-C
8-olefins) in an amount of 0.3–1.3 wt%.
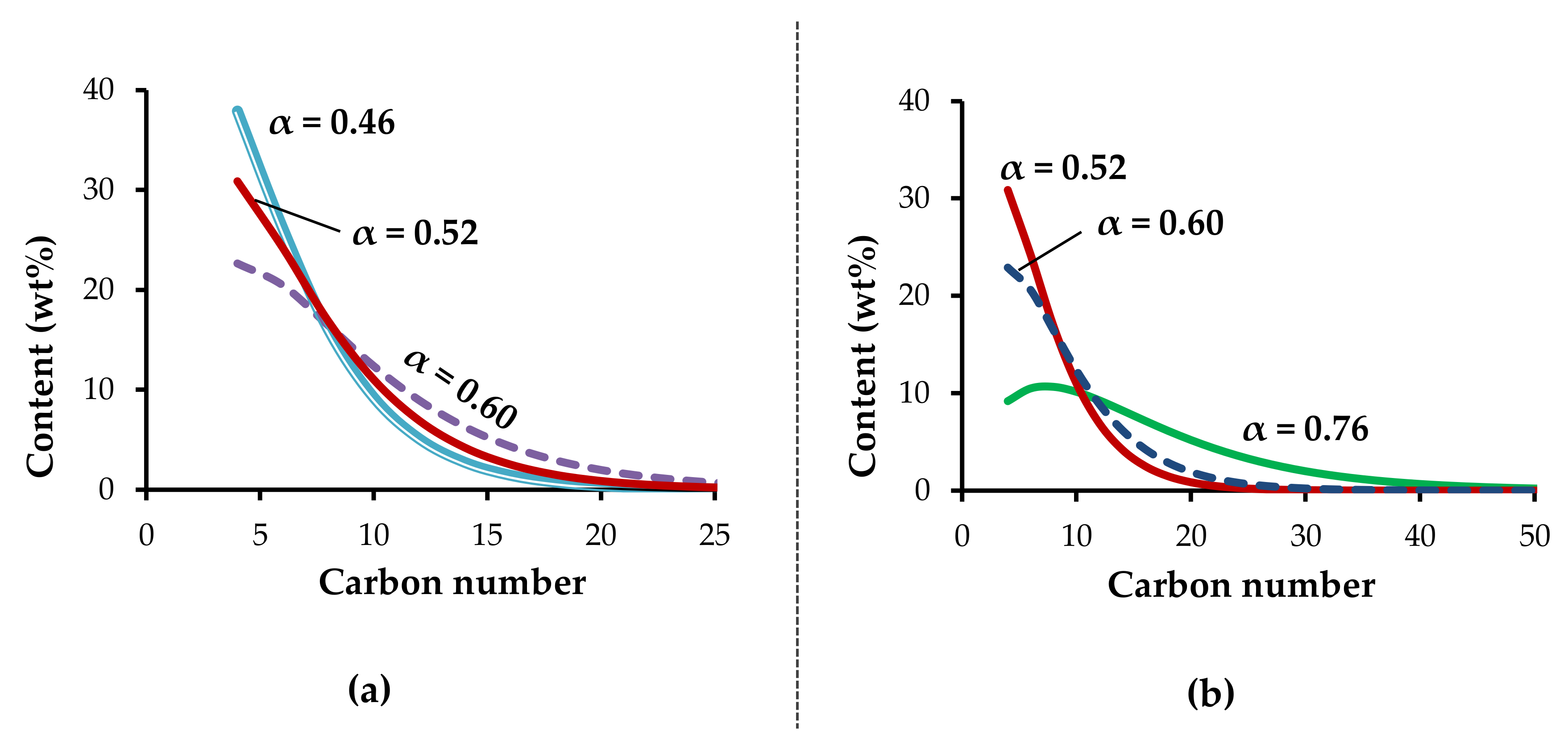
Zirconium acetates obtained by chemical and electrochemical methods have similar catalytic properties in ethylene oligomerization (entries 2 and 3,
Table 3). With the increasing length of the carboxylate moiety, the distribution of oligomers shifts to the higher molecular weights. This is indicated by the increase of the Schulz–Flory parameter from 0.46 (for acetate) to 0.52 (for octanoate) and 0.60 (for laurate) (entries 3–5,
Table 3,
Figure 5a). At the same time, the activity of the catalytic system increased from 7.6 × 10
3 to 9.9 × 10
3 and 9.5 × 10
3 mol
C2H4⋅mol
Zr−1⋅h
−1, respectively. The formation of longer chain oligomers may indicate the existence of a certain similarity between the length of the alkyl chain in Zr–R catalytic intermediate and the length of the carboxylate moiety in the initial pre-catalyst. The increase in activity upon transition from [Zr(Ac)
x] to [Zr(Oct)
x] and [Zr(Laur)
x] may be due to the increase of their solubility in toluene [
47]. The electrochemically synthesized zirconium carboxylates are superior in activity to ZrCl
4 and provide the formation of a smaller amount of polymer (entries 1 and 3-5,
Table 3).
Using [Zr(Oct)x], the effects of nature and the amount of co-catalyst (Al/Zr ratio), as well as of process parameters (temperature, pressure), on catalytic properties were investigated.
The decrease of thermostat temperature from 80 °C to 60 °C led to the decrease of activity from 9.9 × 10
3 to 6.7 × 10
3 mol
C2H4⋅mol
Zr−1⋅h
−1 and to the increase of the selectivity to LAO (entry 9,
Table 3). The latter is probably due to a decrease in the rate of isomerization. At the same time, the length of the oligomers formed increased; α increased from 0.52 to 0.57 due to the decrease of chain transfer rate with temperature [
48,
49,
50]. Therefore, chain transfer and chain isomerization have higher activation energies than chain propagation. With the increasing of oligomerization temperature to 100 °C, opposite effects were observed—the activity increased to 13.6 × 10
3 mol
C2H4⋅mol
Zr−1⋅h
−1, α decreased to 0.48 and selectivity to LAO decreased to 75% (entry 10,
Table 3).
The increase of the ethylene pressure to 30 bar resulted in an increase of productivity to 14.0 × 10
3 mol
C2H4⋅mol
Zr−1⋅h
−1 (entry 11,
Table 3). At the same time, the molecular weight of oligomers increased (as indicated by the growth of α to 0.60), probably due to the intensification of the ethylene insertion to the Zr–C bond at the chain propagation stage of the catalytic cycle [
50].
The characteristics of oligomerization products are strongly dependent on the nature and the amount of co-catalyst used. A decrease in the acidity of aluminum in the co-catalyst causes an increase in the amount of polymer formed. For example, in the transition from Et
3Al
2Cl
3 to Et
2AlCl, the polymer content increased from 0.3 to 0.9 wt% (entry 7,
Table 3). In the case of AlEt
3 as a co-catalyst, predominant polymerization was observed (entry 8,
Table 3). In turn, in the presence of the co-catalyst with higher aluminum acidity—EtAlCl
2—along with oligomerization, toluene alkylation could take place (entry 6,
Table 3).
The decrease of the Al/Zr ratio from 20 to 10 led to a decrease in activity and to a significant increase in the chain length of oligomers (entry 12,
Table 3;
Figure 5b). At the same time, selectivity to LAO increased to 97% as a result of the decrease in the number of aluminum acid centers which are active in olefin isomerization [
51,
52]. In turn, the increase of the Al/Zr ratio from 20 to 40 resulted in the shortening of oligomers (α decreased to 0.45) and decrease in activity to 7.6 × 10
3 mol
C2H4⋅mol
Zr−1⋅h
−1 (entry 13,
Table 3). An increase in the content of short-chain oligomers, which resulted from the increase in the Al/Zr ratio, is consistent with ethylene oligomerization kinetic studies using the Zr(OC
3H
7)
4-Et
3Al
2Cl
3 system [
53].
A twofold decrease in the amount of pre-catalyst and reaction time, as well as an increase in ethylene pressure to 30 bar, led to an increase in the activity to 31.3 × 10
3 mol
C2H4⋅mol
Zr−1⋅h
−1 (entry 14,
Table 3).
Thus, electrochemically synthesized zirconium carboxylates, in combination with Et
3Al
2Cl
3, oligomerize ethylene with moderate activity (7.6 × 10
3–9.9 × 10
3 mol
C2H4⋅mol
Zr−1⋅h
−1) to even-numbered linear alpha-olefins (78–85%) with Schulz–Flory distribution, predominantly to C
4–C
10 olefins (72–89%). The performance of catalytic systems investigated is in accordance with the literature data on ethylene oligomerization using zirconium carboxylates [
16,
17,
54,
55,
56].
3. Materials and Methods
In electrosynthesis processes, a power source AKIP-1137-200-1 (AKIP, Russia) was used. Commercial acetonitrile (analytical grade, EKOS-1, Russia), zirconium(IV) chloride (98%, anhydrous, ACROS Organics), acetic acid (reagent grade, EKOS-1, Russia), octanoic acid (99%; ACROS Organics), lauric (dodecanoic) acid (99%; ACROS Organics), tetraethylammonium chloride Et4NCl (≥98%; Sigma-Aldrich, St. Louis, Missouri, USA) and tetra-n-butylammonium tetrafluoroborate nBu4NBF4 (99%; Alfa Aesar) were used as received without further purification.
Metal zirconium, obtained by iodide technology (99.8 wt%; Chepetsk Mechanical Plant, Russia), was used for the anode and cathode (plates, S = 510 mm2) during the electrosynthesis procedures.
Voltammetric studies were performed using an EC Epsilon potentiostat with a C3 module (BASi, West Lafayette, IN, USA). Metal zirconium was used as the working electrode (plate, S = 90 mm2), a platinum wire was used as the auxiliary electrode and Ag/Ag+ (0.01 mol⋅L−1 AgNO3 in acetonitrile, (E°(Fc/Fc+) = +0.20 V) served as the reference electrode. All curves were recorded at a potential sweep rate of 100 mV × s−1.
The content of the zirconium in the products of electrochemical synthesis was determined by the burning of the sample at 1000 °C in an oxygen flow followed by weighing the residue (ZrO2). The residue mass was converted to metal.
Carbon and hydrogen content was determined using the high-temperature single-reactor analyzer CHN-3 (Chimavtomatika, Russia) by burning the sample at 950 °C in an oxygen flow and the chromatographic determination of oxidation products (CO2 and H2O).
The content of chlorine was determined by the Sheniger procedure [
57].
NMR spectra were collected at room temperature using an Avance III 400 MHz spectrometer (Bruker, Germany) operating at 400 MHz (1H) and 100.6 MHz (13C). Typically, 10,000–30,000 scans were collected with a sweep width of 24038 Hz (238.869 ppm) and relaxation delay of 0.63 s.
IR spectra were recorded using an FT-801 FT-IR spectrometer (SIMEX, Russia) and an NPVO-ZDO single-reflectance ATR accessory (SIMEX, Russia) with ZnSe crystal. Spectra were recorded over the range 4000–500 cm−1, with a resolution of 2 cm−1 and averaged over 64 scans. To obtain good contact with ATR crystal the samples were clamped by a swivel tip press.
Powder X-ray diffraction (PXRD) measurements were carried out using a MiniFlex 600 diffractometer (Rigaku, Japan). The patterns were collected using CuKα radiation (40 kV, 15 mA) at room temperature in the range of 2θ from 2 to 100° with 0.02° steps and a 0.24 s exposure time at each point.
GC-MS analyses were performed on a GC 7890–MSD 5977B system (Agilent, USA) equipped with capillary column HP-5MS Ultra Inert (30 m length, 0.25 mm diameter, 0.50 µm film of (5%-phenyl)-methylpolysiloxane; Agilent, USA). For chromatography, the following temperature program was used—thermostating at 35 °C for 10 min; heating to 120 °C with a rate of (10° × min−1), thermostating at 120 °C for 3 min; and heating to 280 °C with a rate of (20° × min−1) and thermostating at 280 °C for 10 min. Substances were identified using the NIST Mass Spectral Search Program (National Institute of Standards and Technology, USA), version 2.2.
3.1. Cyclic Voltammetry
In cyclic voltammetric studies, flat metal zirconium with a surface of 90 mm
2 was used as a working electrode, a silver electrode Ag/AgNO
3 (0.01 mol⋅L
−1 solution in acetonitrile) was used as a reference electrode and a platinum wire served as an auxiliary electrode. The electrolyte was a solution of a carboxylic acid and a supporting salt (0.01 mol⋅L
−1) in acetonitrile (5 mL). Voltammograms were recorded according to the following order of voltage change—from 0 to –2.0 V, from –2.0 to 2.0 V and from 2.0 to 0 V. For each system, three continuous scanning cycles were performed; the third cycles are shown in
Figure 1 and
Figure 2 and in
Supplementary Materials.
3.2. Preparative Electrolysis
The electrochemical synthesis was carried out in an electrochemical cell without separation of the anodic and cathodic compartments. A working solution for electrolysis was prepared by dissolving the supporting salt (1×10−4 mol) and the corresponding carboxylic acid in acetonitrile (10 mL). A flat zirconium anode with a working surface area of 510 mm2 and an identical zirconium cathode were used. The electrolysis was carried out in a potentiostatic mode, without stirring and thermostating of the electrolyte. The process was completed when the current decreased to 10 mA as a result of partially coating the anode surface by product. To calculate the current efficiency, the amount of transmitted electricity was integrated.
3.2.1. Electrosynthesis of [Zr(Ac)x]
In total, 16 mg (1 × 10−4 mol) of Et4NCl and 2.0 mL (35 × 10−3 mol) of acetic acid were dissolved in 10 mL of acetonitrile. Zirconium electrodes were immersed in the electrolyte. To prevent short circuits, the electrodes were separated by a PTFE plate.
Electrolysis was carried out at a constant voltage of 80 V. The current was stable at ~50 mA. During the initial 45 min of electrolysis, the solution temperature increased to 60 °C and was kept constant. After 110 min of electrolysis, the precipitation of white solid started, which led to a decrease in current. The total electrolysis time was 160 min. The working solution was left for 120 min, during which an additional amount of the product was precipitated from the solution. Then, the electrolyte was poured out of the cell. The resulting precipitate was washed three times with 10 mL of fresh acetonitrile and subsequently dissolved in 30 mL of acetone. The resulting solution was filtered from metal particles (scree from anode) using a PTFE filter with a pore size of 0.45 μm (MACHEREY-NAGEL, Germany). Acetone was removed and the product was dried under reduced pressure and 80 °C. The product was obtained as white flakes. The yield was 0.29 g.
The conditions of the electrosynthesis procedure in the case of Me4NBr (99%, Sigma-Aldrich) and Et4NI (98%, Sigma-Aldrich) supporting salts were the same.
3.2.2. Electrosynthesis of [Zr(Oct)x]
In total, 16 mg (1 × 10−4 mol) of Et4NCl and 2.0 mL (13 × 10−3 mol) of octanoic acid were dissolved in 10 mL of acetonitrile.
Electrolysis was carried out at a constant voltage of 70 V. The current was stable at ~40 mA. During the initial 25 min of electrolysis, the working solution was heated to 55 °C and became turbid. The total electrolysis time was 320 min. In the last 120 min, the current gradually decreased. The working solution after electrolysis was left for 180 min to sediment. Then, the electrolyte was poured out of the cell; a yellow viscous liquid was obtained at its bottom. The liquid was dissolved in CCl4 and the obtained solution was passed through a 0.45 μm PTFE filter. CCl4 was removed and the product was dried under reduced pressure and 80 °C. The yield was 0.70 g.
3.2.3. Electrosynthesis of [Zr(Laur)x]
In total, 16 mg (1 × 10−4 mol) of Et4NCl and 110 mg (0.6 × 10−3 mol) of lauric acid were dissolved in 10 mL of acetonitrile.
Electrolysis was carried out at a constant voltage of 80 V. During the initial 10 min of electrolysis, the working solution was heated to 45 °C and became turbid. After 15 min of electrolysis, the current started to decrease. The total electrolysis time was 35 min. The product was obtained as a white plastic mass on the surface of the anode. The yield was 70 mg.
3.3. Chemical Synthesis of Zirconium Acetate
In total, 2.33 g of ZrCl
4 (10 × 10
−3 mol) was dissolved in 20 mL (350 × 10
−3 mol) of acetic acid [
58]. The mixture was refluxed for 5 h. Zirconium acetate formed as a white precipitate. Then it was dried under reduced pressure and 80 °C. The yield was 2.98 g.
Calc. for Zr(CH3COO)4 (M = 327.4 g⋅mol−1), wt%: C, 29.4, H, 3.7, Zr, 27.9; found: C, 26.9, H, 3.4, Zr, 30.5, Cl, 1.2.
1H NMR and
13C{
1H} NMR spectra of the chemically synthesized zirconium acetate are shown in
Supplementary Materials.
3.4. Ethylene Oligomerization
Ethylene oligomerization was carried out in a 75 mL stainless steel autoclave with a magnetic stirrer inside and equipped with an electronic pressure sensor. Before the oligomerization process, the autoclave was dried under reduced pressure, purged and filled with nitrogen. During the oligomerization process, ethylene (≥99.95 vol%, Linde Gas, Munich, Germany) was continuously fed into the autoclave to maintain a constant pressure. A constant temperature in the autoclave was provided by a silicon oil bath.
Toluene (reagent grade) was used as a solvent; it was previously purified from traces of water by distillation in the presence of sodium and benzophenone. Ethylaluminium sesquichloride Et3Al2Cl3 (0.4M solution in hexane, ACROS Organics), ethylaluminum dichloride EtAlCl2 (1.0 M solution in hexanes, Sigma-Aldrich), diethylaluminum chloride Et2AlCl (1.0 M solution in hexanes, Sigma-Aldrich) and triethylaluminum AlEt3 (1.0 M solution in hexanes, Sigma-Aldrich) were used as co-catalysts.
All manipulations for the preparation of solutions (V = 25 mL) of pre-catalyst (20-40 µmol) and co-catalyst were carried out using the Schlenk system. After the addition of solution with a pre-catalyst and co-catalyst, the autoclave was weighed.
The oligomerization of ethylene was carried out for 30-60 min. After the oligomerization process, the autoclave was cooled to –20 °C (to condensate butenes) and unreacted ethylene was released. Then, the catalytic system was deactivated by the hydrolysis of aluminum co-catalyst with 2 mL of 5 vol% HCl solution in methanol. The autoclave was weighed to determine the amount of oligomers formed. The oligomerization activity A was calculated on the basis of the weight of oligomers formed using Equation (4):
where
moligomer is the weight of oligomers, g;
MC2H4 is the molar weight of ethylene, 28 g⋅mol
−1;
nZr is the amount of pre-catalyst, (20–40) × 10
−6 mol; and
τ is the reaction time, 0.5–1.0 h. The oligomerization activity A is expressed in units [mol
C2H4⋅mol
Zr−1⋅h
−1]–(mol of ethylene converted) × (mol of Zr)
−1 × h
−1.
The reaction mixture was analyzed by GC-MS. According to the results of the GC-MS-analysis, in particular on the basis of the signals of 1-hexene, 1-octene and 1-dodecene and using calibrations with the standard mixtures of 1-hexene (97%, ACROS Organics), 1-octene (99+%, ACROS Organics) and 1-dodecene (93–95%, ACROS Organics), the average Schulz–Flory parameter “α” was calculated and consequently the molecular weight distribution of oligomers was determined.
The amount of alkylbenzenes was calculated using results of GC-MS and calibrations with standard mixtures of 1-octene (99+%, ACROS Organics) and ethylbenzene (laboratory reagent grade, Chemsnab-SPB, Russia).
4. Conclusions
The process of the electrochemical synthesis of zirconium carboxylates was studied. Using cyclic voltammetry, it was found that the halide anion acts as a mediator in the interaction of zirconium metal and carboxylic acid.
The electrochemical synthesis of zirconium carboxylates was carried out. The electrochemical method is effective for producing zirconium acetate and octanoate (the productivity is 0.11–0.13 g per hour). In the case of lauric acid, its low solubility and the rapid covering of the anode surface by the product during electrolysis do not allow satisfactory yields of zirconium laurate to be obtained.
Using element analysis, NMR and IR spectroscopy and PXRD techniques, it was found that the obtained substances were amorphous non-stoichiometric zirconium carboxylates [Zr(Ac)x], [Zr(Oct)x], [Zr(Laur)x] (3 < x < 4) with a bidentate coordination of carboxylate moieties to zirconium.
The zirconium carboxylates in combination with an organoaluminium co-catalyst catalyze the homogeneous oligomerization and polymerization of ethylene. The characteristics of the product obtained strongly depend on the aluminum acidity, as well as on the Al/Zr ratio. The zirconium carboxylates, which are activated by 10 eq. of Et3Al2Cl3 (Al/Zr = 20), have an activity of (7.6–9.9) × 103 molC2H4⋅molZr−1⋅h−1 and selectivity to LAO of 78–85% (T = 80 °C; p = 20 bar; τ = 60 min); finally, the C4–C10 fraction content in the oligomerization products is 72–89 wt%.

 ,
, 






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}